
Abstract
Background
Several recently completed large-scale enviromental sequencing projects produced a large amount of genetic information about microbial communities ('metagenomes') which is not biased towards cultured organisms. It is a good source for estimation of the abundance of genes and regulatory structures in both known and unknown members of microbial communities. In this study we consider the distribution of RNA regulatory structures, riboswitches, in the Sargasso Sea, Minnesota Soil and Whale Falls metagenomes.
Results
Over three hundred riboswitches were found in about 2 Gbp metagenome DNA sequences. The abundabce of riboswitches in metagenomes was highest for the TPP, B12 and GCVT riboswitches; the S-box, RFN, YKKC/YXKD, YYBP/YKOY regulatory elements showed lower but significant abundance, while the LYS, G-box, GLMS and YKOK riboswitches were rare. Regions downstream of identified riboswitches were scanned for open reading frames. Comparative analysis of identified ORFs revealed new riboswitch-regulated functions for several classes of riboswitches. In particular, we have observed phosphoserine aminotransferase serC (COG1932) and malate synthase glcB (COG2225) to be regulated by the glycine (GCVT) riboswitch; fatty acid desaturase ole1 (COG1398), by the cobalamin (B12) riboswitch; 5-methylthioribose-1-phosphate isomerase ykrS (COG0182), by the SAM-riboswitch. We also identified conserved riboswitches upstream of genes of unknown function: thiamine (TPP), cobalamine (B12), and glycine (GCVT, upstream of genes from COG4198).
Conclusion
This study demonstrates applicability of bioinformatics to the analysis of RNA regulatory structures in metagenomes.
Similar content being viewed by others


Background
Recent advances in sequencing technologies have led to a significant progress in studies of organisms in their natural habitats [1, 2]. While a vast majority of currently sequenced prokaryotic organisms are culturable, they constitute less than 1% of all microbial species [3]. New sequencing methods allow one to extract and clone DNA directly from environmental samples. It makes it possible to sequence uncultured microbes, obligate parasites and symbionts. Genomic libraries created by new methods may contain DNA from many different species. This opens a new direction in sequencing, called metagenomics, which provides an opportunity for studies of microbial communities with applications in ecology, biotechnology and medicine. To date, several large-scale environmental sequencing projects have been completed. The first large project was the metagenomic sequencing project of the surface-water microbial community of the Sargasso Sea [4]. This microbial community was found to be extremely complex and diversified: the analysis of 1.6 Gbp of DNA sequence revealed over 1.2 millions genes from more than 1800 species, including 148 new species. Two other metagenomic projects [5], completed in early 2005, considered microbial communities from the surface soil of a Minnesota farm and from whale skeletons found at 500 m water depth in two different oceans. While the surface water of Sargasso Sea represents a nutrient-poor environment, agricultural soil and deep-sea whale skeletons, also known as 'whale falls', are nutrient-rich environments. The two latter projects together produced about 300 Mbp DNA sequence from more than 3000 genomes. Thus, all datasets from these projects, referred to as 'metagenomes', represent genetic information about microbial communities from different environments, including numerous known and unknown species.
In this study we consider the distribution of riboswitches in microbial communities. The riboswitches are highly conserved RNA structures that regulate gene expression without involvement of protein factors [6–9]. The riboswitch structure can be divided logically and structurally into sensory and regulatory domains. The sensory domain is a natural aptamer that selectively recognizes a target metabolite and thus indirectly estimates its concentration. After binding of an effector molecule, the sensory domain undergoes structural changes that cause simultaneous restructuring of the regulatory domain. In most cases these changes lead to repression of gene expression by transcription termination or inhibition of translation initiation. The known riboswitches are involved in regulation of numerous fundamental metabolic pathways [6, 10] and have been found not only in bacterial genomes, but also in archaeal [11] and eukaryotic [12] genomes. In addition to transcription and translation, riboswitches regulate splicing [13] and RNA cleavage [14]. All that suggests that riboswitches represent one of the oldest regulatory systems [6]. On the other hand, recently discovered new classes of riboswitches [15, 16] with a narrow taxonomic distribution likely have emerged a relatively short time ago.
As mentioned above, sequencing of environmental samples yields DNA fragments from both known and unknown members of microbial communities. Thus, metagenomes are an appropriate source for estimation of the riboswitch abundance in microbial communities and the diversity of their functions. The average length of DNA fragments from the three analyzed metagenomes is about 1000 bp. The riboswitch length (≈100–200 bp) and conservation level are sufficient for their reliable identification. On the other hand, in most cases the DNA fragments include both a riboswitch and a considerable part of the regulated gene. Here we present the analysis of most known types of riboswitches in DNA sequences from three large-scale environmental sequencing projects (further referred to as Sargasso Sea, Minnesota Soil and Whale Falls). We annotated functions of genes located downstream of identified riboswitches by comparative analysis. We also predicted the taxonomy origin of these DNA fragments and thus estimated the abundance of riboswitches in various taxonomic groups.
Results and discussion
Scanning of metagenomes with patterns describing eleven riboswitch classes resulted in 311 candidate riboswitches (the corresponded patterns and alignments of identified riboswitches are presented in Additional files 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19). The fraction of riboswitch-regulated genes in all three metagenomes was 0.03 respectively. The distribution of candidate riboswitches and riboswitch classes identified in the three metagenomes is shown in Table 1.
The distribution of the riboswitch classes was as follows: the thiamine (THI-element), cobalamin (B12-element) and glycine (GCVT) riboswitches were the most abundant; the methionine (S-box), riboflavin (RFN-element), YKKC/YXKD and YYBP/YKOY riboswitches were less abundant; and only a few cases of the lysine (L-box or LYS-element), purine, GLMS and YKOK riboswitches were observed. Table 2 shows the distribution of identified riboswitches in taxonomic groups. This table also includes the values that were normalized by the total size of fragments from the taxonomic groups. The normalized frequencies of riboswitch occurrence are expressed as the number of riboswitches per 105 bp. One can see that riboswitches are abundant in the α-,β-,γ-Protebacteria, Firmicutes and Bacteroidetes/Chlorobi. In other taxonomic groups present in the three considered metagenomes the riboswitches are rare. The distribution of particular riboswitch types is not uniform: the THI-element is relatively more abundant in γ-Proteobacteria and Firmicutes, the B12-box, in β/γ-Proteobacteria and Bacteroidetes/Chlorobi, and the glycine riboswitch, in α/β-Proteobacteria and Firmicutes. These observations are described below in more detail.
S-box (SAM-riboswitch)
The S-adenosylmethionine-dependent riboswitches [17–21] identified in the analyzed metagenomes are presented in Additional file 1. All annotated proteins possibly regulated by S-boxes, i.e. located downstream of the identified riboswitches, except one (COG0182) were observed in B. subtilis and Escherichia coli earlier [21]. One protein remains unannotated because the search against the Genbank database retrieved no result. The taxonomy of seven out of twelve riboswitch-containing DNA fragments was successfully predicted. The methionine riboswitch occurs not only in Firmicutes and Actinobacteria groups, as previously thought [21], but also in the Bacteroidetes/Chlorobi and Cyanobacteria groups. Only one out of seven classified DNA fragments was assigned to the Actinobacteria group, while other were assigned to the Bacteroidetes/Chlorobi and Cyanobacteria groups despite the fact that the estimated total size of the Firmicutes and Actinobacteria groups in the Sargasso Sea metagenome is approximately equal to the estimated total size of the Bacteroidetes/Chlorobi and Cyanobacteria groups [4].
One protein was significantly similar to the predicted translation initiation factor 2B subunit from the eIF-2B α/β/δ family (COG0182). However, recently the YkrS protein from B. subtilis belonging to this family was characterized as a 5-methylthioribose-1-phosphate isomerase [22]. This enzyme participates in the methionine salvage pathway, and this is consistent with its regulation by S-boxes.
RFN-element (FMN-riboswitch)
The distribution of FMN-dependent riboswitches (RFN-element) [23–26] (Additional file 2) is consistent with results obtained for complete bacterial genomes [26]. In two out of seven cases, FMN-riboswitches were identified upstream of a single gene ribB not belonging to a riboflavin biosynthesis operon. Based on the gene taxonomic affinity these riboswitches belong to the Proteobacteria. Three times, an FMN riboswitch was identified upstream of the gene ribH, and it could not be determined whether this gene belongs to a riboflavin biosynthesis operon because of the small fragment size. These DNA fragments, except one unclassified case, were assigned to the α-Proteobacteria. Two remaining FMN riboswitches were observed upstream of the genes ribD and ribC. These genes may be the first genes of riboflavin operons [26], but again, whether this is the case could not be determined because of insufficient size of DNA fragments.
B12-element
Cobalamin riboswitches (B12) [27–31] observed in three metagenomes are presented in Additional file 3. The genes preceded by identified B12-elements could be divided by function into three groups. The first group contains cobalamin biosynthesis genes cobW, hupE, cobU. The second group, which is the most numerous one, are cobalt and cobalamin transporters btuB, btuC, fecB. The third group if formed by metE, a B12-independent isozyme of a B12-dependent enzyme. It was shown [31] that if both isozymes are encoded in one genome, then the expression of the B12-independent isozyme is repressed in the presence of cobalamin. The fatty-acid desaturase gene ole1 that was for the first time observed to be regulated by a cobalamin riboswitch, may also belong to such a pair of isozymes. If this hypothesis is correct, there must exist a corresponding B12-dependent isozyme. For 24 identified B12-elements the regulated gene could not be determined: in three cases because riboswitches were located at the end of DNA fragments, in six cases because of the absence of open reading frames in the downstream region, and in fifteen cases because of the absence of similar genes in the database. Two of the latter fifteen genes are significantly similar to each other.
The distribution of B12-elements over taxonomic groups is slightly different from that observed for complete bacterial genomes [31]. In the latter, B12-elements were found mainly in the Proteobacteria, Firmicutes and Actinobacteria, and the number of B12-elements in the Firmicutes and Actinobacteria was only slightly less than the number of B12-elements in the Proteobacteria. Although the percentage of the Firmicutes and Actinobacteria groups is not negligible (about 10% in the Sargasso Sea metagenome), none of 32 cobalamin riboswitch-containing DNA fragments belonged to the Firmicutes or Actinobacteria.
THI-element (TPP-riboswitch)
Thiamine pyrophosphate (TPP)-dependent riboswitches [11–13, 32, 33] identified in three metagenomes are presented in Additional file 4. Among the thiamine biosynthesis genes, most TPP-riboswitches were found upstream of the thiC and thiM genes. These genes are known to occur as first genes of THI-element-regulated thiamine biosynthesis operons [11], but we were able to confirm this only in one case because of the small size of DNA fragments. One THI-element was found upstream of another thiamine biosynthesis gene, thiD. This gene did not seem to be a part of an operon.
Another group of TPP-riboswitch-regulated genes are thiamine transporters. Most TPP-riboswitches were found upstream of the thiB gene. We were able to examine whether these genes belong to operons, as found earlier [11], in only three cases, where it was a part of the thiBP operon. This agrees with the analysis of complete bacterial genomes [11]. Other identified transporters regulated by THI-elements were omr (homolog of the outer membrane receptor btuB), ABC-type transporter thiXYZ from the nitrate/sulfonate/bicarbonate transporter family, and thiV, homologous to the Na+/panthothenate symporter gene panF [11].
Functions of 23 other TPP-riboswitches were not determined: in one case because of the absence of open reading frames in the downstream region, in ten cases because riboswitches were located at the end of DNA fragments, and in twelve cases because of the absence of similar genes in the databases. Two of the latter twelve genes were significantly similar to each other.
GCVT
The glycine riboswitch [34, 35] is highly abundant and, as expected [34, 35], was mainly identified in upstream regions of genes encoding the glycine cleavage system (Additional file 5). In 70 out of 119 cases (≈59%) the GCVT riboswitch occurred upstream of the gcvT gene. In 25 out of these 70 cases this gene was a part of the operon gcvTHP, whereas for other DNA fragments the operon structure was not discernible because of their small size. The second frequently observed (≈20%) glycine-riboswitch regulated gene was the malate synthase gene glcB involved in the pyruvate metabolism. This study provides the first such example. Most of these genes belonged to DNA fragments from the Sargasso Sea metagenome, moreover, 78 out of 94 (≈83%) DNA fragments apparently belonged to one specie Candidatus Pelagibacter ubique from the α-Proteobacteria. Other annotated genes form ≈7% of all identified genes downstream of glycine riboswitches. All of them, except serC, already have been observed under glycine riboswitch regulation [34] and are involved in the glycine or pyruvate metabolism. All these glycine riboswitches had tandem aptamer domains, as in [35]. All DNA fragments containing these riboswitches (with one exception) presumably belong to the Proteobacteria.
For remaining ≈14% of the identified glycine riboswitches, the regulated functions could not be identified. The search against the COG database showed that five genes were significantly similar to genes from the COG4198 cluster, described as a group of uncharacterized conserved proteins. Interestingly, the structure of riboswitches for these seven cases had only one aptamer domain. The DNA fragments of these proteins presumably belong to the Firmicutes. In twelve remaining cases protein sequences could not be determined because riboswitches were located at the end of DNA fragments.
YKKC/YXKD
The search for the YKKC/YXKD riboswitches (Additional file 6) confirmed known regulated functions of these elements [34]. Four out of five identified riboswitches were upstream of two components of an ABC-type transport system from the nitrate/sulfonate/bicarbonate family. One YKKC/YXKD riboswitch was identified upstream of the amino acid transporter potE. All YKKC/YXKD riboswitches were found in DNA fragments from the Proteobacteria.
YYBP/YKOY
The YYBP/YKOY riboswitch was less abundant than the GCVT element in metagenomes (Additional file 7) unlike the situation in complete genomes [34]. On the other hand, the regulated functions were essentially the same as in the latter [34]. All annotated genes observed downstream of the identified riboswitches could be classified into three groups: two encoding predicted membrane proteins, and one, the terC genes. In three cases no open reading frames were found in the downstream region. Most YYBP/YKOY riboswitch-containing DNA fragments belong to the Proteobacteria.
L-box (LYS-element), G-box, GLMS, YKOK
The lysine (L-box) [36–40] and purine riboswitch (G-box) [41–45] as well as the GLMS [14, 34] and YKOK [34] riboswitches were rare in metagenomes. The only lisine riboswitch were identified in the Sargasso Sea metagenome and presumably belongs to the γ-Proteobacteria. This riboswitch was located upstream of the predicted lysine transporter from the Na+:H+ antiporter superfamily [40]. The function of the only purine riboswitch-regulated gene was not recognized because of the absence of similar genes in the databases. Two GLMS riboswitches were observed: one upstream of the glmS gene, and one at the end of a DNA fragment. Of two observed YKOK riboswitches, one was upstream of a gene similar to the Mg/Co/Ni transporter mgtE (COG2239), and one, upstream of a gene with unknown function.
Conclusion
The riboswitch counts in the Sargasso Sea, Minessota Soil and Whale Falls bacterial communities estimated here generally agree with the riboswitch abundance in complete bacterial genomes [11, 21, 26, 31, 34, 40]. In the bacterial communities, the THI-elements, B12-elements and GCVT riboswitches are the most abundant. The two former riboswitches are highly abundant in complete bacterial genomes as well [11, 31], whereas the GCVT riboswitch was not the most frequent one among computationally discovered riboswitches GLMS, GCVT, YKKC/YXKD, YKOK and YYBP/YKOY [34]. The YYBP/YKOY riboswitch was characterized as the most abundant one among these new riboswitches [34], however it occurs in metagenomes less frequently than the THI, B12 and GCVT elements. The S-boxes, RFN-elements, YYBP/YKOY and YKKC/YXKD riboswitches demonstrated lower but still significant abundance, whereas the GLMS, YKOK, lysine and purine riboswitches were rare. In general, the riboswitch frequencies weakly depend on a particular metagenome, however they are slightly higher in the Minnesota Soil and Whale Falls metagenomes then in the Sargasso Sea metagenome. The glycine riboswitch (GCVT) is an exception: its frequency in the Sargasso Sea metagenome was the highest, about fourfold higher than in the Minnesota Soil metagenome and 1.2-fold higher than in the Whale Falls metagenome. However, ≈81% of glycine riboswitches coming from the Sargasso Sea metagenome presumably belong to a single specie Candidatus Pelagibacter ubique from the SAR11 clade, which is known to be abundant in marine surface waters [46, 47]. This example and the discrepancies between the riboswitch abundance in metagenomes and complete genomes demonstrate the influence of species frequencies in the communities on the gene and riboswitch contents of the latter.
The riboswitch-regulated functions in metagenomes in most cases coincide with those observed in complete bacterial genomes [11, 21, 26, 31, 34, 40]. However, several new regulated functions were recognized for some riboswitch classes. The new functions regulated by the glycine riboswitch, phosphoserine aminotransferase (COG1932) and malate synthase (COG2225), are involved in the glycine and pyruvate metabolism, respectively. Fatty-acid desaturase (COG1398) was recognized as a new regulated function of the cobalamin riboswitch (B12-element). We suggest that this fatty-acid desaturase gene belongs to a pair of B12-dependend and B12-independed isozymes [31]. One more new riboswitch-regulated gene is under methionine (SAM) regulation and shows a significant similarity with the translation initiation factor 2B subunit from the eIF-2B α/β/δ family (COG0182); however, according to recent studies [22] the real function of this protein is 5-methylthioribose-1-phosphate isomerase.
Sometimes different riboswitches were found upstream of homologous genes. For example, components of ABC-type nitrate/sulfonate/bicarbonate transport systems homologous to tauA and tauC were found downstream of thiamine and YKKC/YXKD riboswitches. One other example is provided by btuB homologs from the outer membrane receptor proteins family regulated by thiamine and B12 riboswitches.
In addition to genes with known (or reliably predicted) functions, this study revealed several hypothetical riboswitch-regulated genes. Leaving aside relatively less reliable "orphans", that is, open reading frames preceded by riboswitches, but having no homologs, we have observed several groups of homologous genes preceded by riboswitches. The examples are two pairs of homologous proteins regulated by B12 and thiamine riboswitches, respectively, and five uncharacterized conserved proteins regulated by the GCVT riboswitch and belonging to COG4198 cluster.
When this study had been completed, a much larger metagenomic collectioin was published [48], and several new riboswitches were identified [[49], D. Rodionov personal communication]. This study demonstrates that metagenomics and bioinformatics can be applied to the analysis not only of genes, proteins, and metabolic pathways [50–52], but regulatory structures in natural environments not biased towards cultured organisms. We expect that the new datasets may contain not only new examples of functions regulated by known riboswiches, but new types of riboswitches as well.
Methods
The RNA-PATTERN program [53] was used to search for RNA regulatory elements in all three metagenomes. The input RNA pattern described the RNA secondary structure and the sequence consensus motifs as a set of the following parameters: the number of helices, the length of each helix, the loop lengths and the topology of helix pairs. The appropriate patterns were created for the analyzed riboswitches and used in search procedure, see Additional files 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19.
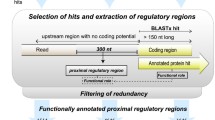
For the automated analysis we developed a specialized software based on the Relational Database Management System (RDBMS) Oracle 10g Express Edition [54]. It was used to load the metagenomic data, execute the RNA-PATTERN program, and support the functional annotation of regulated genes and taxonomic annotation of riboswitch-containing DNA fragments. The data processing flow is shown schematically in Additional file 8. At the first step, metagenome DNA sequence files were loaded into the database from GenBank [55]. We excluded the metagenome data belonging to the first of the seven Sargasso sea samples, because it seems to be contaminated by Shewanella and Burkholderia species [48, 56]. Then, automated search for all riboswitch classes in each file was performed by invoking the RNA-PATTERN program. Search results were loaded into the database and could be viewed in an ad-hoc user interface. To simplify functional annotation of genes located downstream of identified riboswitches, the automated search of similar sequences was performed by the Entrez Programming Utilities interface [57] to the BLAST program [58]. The resulting lists of similar protein sequences were also loaded into the database and could be viewed using the same user interface. The functional annotation of proteins was performed by comparison with the COG database [59]. The organism names were extracted from each BLAST hit and the complete taxonomy of the organism was requested from the NCBI Taxonomy Browser [60]. The retrieved complete taxonomy of organisms was linked in the database to the associated BLAST hit and used to predict the taxonomy of riboswitch-containing DNA fragments. To do that, we compared the taxonomy of the several most similar hits and extracted the maximum level of the taxonomic hierarchy that was common for the considered hits. If these hits had at least one common taxonomic level of hierarchy, then we considered that taxonomy of DNA fragment was successfully predicted and assigned this taxonomic level to the DNA fragment itself.
Sequence alignments of identified riboswitches were prepared for publishing using the TEXshade package [61] and an ad hoc unpublished program (MK).
References
Shendure J, Mitra RD, Varma C, Church GM: Advanced sequencing technologies: methods and goals. Nature Rev Genet. 2004, 5: 335-344. 10.1038/nrg1325.
Tringe SG, Rubin EM: Metagenomics: DNA sequencing of environmental samples. Nature Rev Genet. 2005, 6: 335-344.
Hugenholtz P: Exploring prokaryotic diversity in the genomic era. Genome Biol. 2002, 2 (3): reviews0003.1-reviews0003.8. 10.1186/gb-2002-3-2-reviews0003.
Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu D, Paulsen I, Nelson KE, Nelson W, Fouts DE, Levy S, Knap AH, Lomas MW, Nealson K, White O, Peterson J, Hoffman J, Parsons R, Baden-Tillson H, Pfannkoch C, Rogers YH, Smith HO: Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004, 304: 66-74. 10.1126/science.1093857.
Tringe SG, von Mering C, Kobayashi A, Salamov AA, Chen K, Chang HW, Podar M, Short JM, Mathur EJ, Detter JC, Bork P, Hugenholtz P, Rubin EM: Comparative metagenomics of microbial communities. Science. 2005, 308: 554-557. 10.1126/science.1107851.
Vitreschak AG, Rodionov DA, Mironov AA, Gelfand MS: Riboswitches: the oldest mechanism for the regulation of gene expression?. Trends in Genetics. 2004, 20: 44-50. 10.1016/j.tig.2003.11.008.
Gelfand MS: Bacterial cis-regulatory RNA structures. Mol Biol (Mosk). 2006, 40: 541-550. 10.1134/S0026893306040066.
Mandal M, Breaker RR: Gene regulation by riboswitches. Nature Rev Mol Cell Biol. 2004, 5: 451-463. 10.1038/nrm1403.
Grundy FJ, Henkin TM: Regulation of gene expression by effectors that bind to RNA. Curr Opin Microbiol. 2004, 7: 126-131. 10.1016/j.mib.2004.02.013.
Mandal M, Boese B, Barrick JE, Winkler WC, Breaker RR: Riboswitches control fundamental biochemical pathways in Bacillus subtilis and other bacteria. Cell. 2003, 113: 577-586. 10.1016/S0092-8674(03)00391-X.
Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS: Comparative genomics of thiamin biosynthesis in procaryotes. New genes and regulatory mechanisms. J Biol Chem. 2002, 277: 48949-48959. 10.1074/jbc.M208965200.
Sudarsan N, Barrick JE, Breaker RR: Metabolite binding RNA domains are present in the genes of eukaryotes. RNA. 2003, 9: 644-647. 10.1261/rna.5090103.
Kubodera T, Watanabe M, Yoshiuchi K, Yamashita N, Nishimura A, Nakai S, Gomi K, Hanamoto H: Thiamine-regulated gene expression of Aspergillus oryzae thiA requires splicing of the intron containing a riboswitch-like domain in the 5'-UTR. FEBS Lett. 2003, 555: 516-520. 10.1016/S0014-5793(03)01335-8.
Winkler WC, Nahvi A, Roth A, Collins JA, R BR: Control of gene expression by a natural metabolite-responsive ribozyme. Nature. 2004, 428: 263-264. 10.1038/nature02362.
Fuchs RT, Grundy FJ, Henkin TM: The S(MK) box is a new SAM-binding RNA for translational regulation of SAM synthetase. Nat Struct Mol Biol. 2006, 13 (3): 226-233. 10.1038/nsmb1059.
Corbino KA, Barrick JE, Lim J, Welz R, Tucker BJ, Puskarz I, Mandal M, Rudnick ND, Breaker RR: Evidence for a second class of S-adenosylmethionine riboswitches and other regulatory RNA motifs in alpha-proteobacteria. Genome Biol. 2005, 6 (8): R70-10.1186/gb-2005-6-8-r70.
Grundy FJ, Henkin TM: The S box regulon: a new global transcription termination control system for methionine and cysteine biosynthesis genes in gram-positive bacteria. Mol Microbiol. 1998, 30: 737-749. 10.1046/j.1365-2958.1998.01105.x.
McDaniel BA, Grundy FJ, Artsimovitch I, Henkin TM: Transcription termination control of the S box system: direct measurement of S-adenosylmethionine by the leader RNA. Proc Natl Acad Sci USA. 2003, 100: 3083-3088. 10.1073/pnas.0630422100.
Epshtein V, Mironov AS, Nudler E: The riboswitch-mediated control of sulfur metabolism in bacteria. Proc Natl Acad Sci USA. 2003, 100: 5052-5056. 10.1073/pnas.0531307100.
Winkler WC, Nahvi A, Sudarsan N, Barrick JE, Breaker RR: An mRNA structure that controls gene expression by binding S-adenosylmethionine. Nature Structural Biol. 2003, 10: 701-707. 10.1038/nsb967.
Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS: Comparative genomics of the methionine metabolism in Gram-positive bacteria: a variety of regulatory systems. Nucleic Acids Res. 2004, 32: 3340-3353. 10.1093/nar/gkh659.
Ashida H, Saito Y, Kojima C, Kobayashi K, Ogasawara N, Yokota A: A functional link between RuBisCO-like protein of Bacillus and photosynthetic RuBisCO. Science. 2003, 302: 286-290. 10.1126/science.1086997.
Gelfand MS, Mironov AA, Jomantas J, Kozlov YI, Perumov DA: A conserved RNA structure element involved in the regulation of bacterial riboflavin synthesis genes. Trends Genet. 1999, 15: 439-442. 10.1016/S0168-9525(99)01856-9.
Mironov AS, Gusarov I, Rafikov R, Lopez LE, Shatalin K, Kreneva RA, Perumov DA, Nudler E: Sensing small molecules by nascent RNA: a mechanism to control transcription in bacteria. Cell. 2002, 111: 747-756. 10.1016/S0092-8674(02)01134-0.
Winkler WC, Cohen-Chalamish S, Breaker RR: An mRNA structure that controls gene expression by binding FMN. Proc Natl Acad Sci. 2002, 99: 15908-15913. 10.1073/pnas.212628899.
Vitreschak AG, Rodionov DA, Mironov AA, Gelfand MS: Regulation of riboflavin biosynthesis and transport genes in bacteria by transcriptional and translational attenuation. Nucleic Acids Res. 2002, 30: 3141-3151. 10.1093/nar/gkf433.
Lundrigan MD, Koster W, Kadner RJ: Transcribed sequences of the Escherichia coli btuB gene control its expression and regulation by vitamin B12. Proc Natl Acad Sci USA. 1991, 88: 1479-1483. 10.1073/pnas.88.4.1479.
Ravnum S, Andersson DI: Vitamin B12 repression of the btuB gene in Salmonella typhimurium is mediated via a translational control which requires leader and coding sequences. Mol Microbiol. 1997, 23: 35-42. 10.1046/j.1365-2958.1997.1761543.x.
Nou X, Kadner RJ: Adenosylcobalamin inhibits ribosome binding to btuB. RNA Proc Natl Acad Sci USA. 2000, 97: 7190-7195. 10.1073/pnas.130013897.
Nahvi A, Sudarsan N, Ebert MS, Zou X, Brown KL, Breaker RR: Genetic control by a metabolite binding mRNA. ChemBiol. 2002, 9: 1043-1049.
Vitreschak AG, Rodionov DA, Mironov AA, Gelfand MS: Regulation of the vitamin B12 metabolism and transport in bacteria by a conserved RNA structural element. RNA. 2003, 9: 1084-1097. 10.1261/rna.5710303.
Miranda-Rios J, Morera C, Taboada H, Davalos A, Encarnacion S, Mora J, Soberon M: Expression of thiamin biosynthetic genes (thiCOGE) and production of symbiotic terminal oxidase cbb3 in Rhizobium etli. J Bacteriol. 1997, 179: 6887-6893.
Winkler W, Nahvi A, Breaker R: Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature. 2002, 419: 952-956. 10.1038/nature01145.
Barrick JE, Corbino KA, Winkler WC, Nahvi A, Mandal M, Collins J, Lee M, Roth A, Sudarsan N, Jona I, Wickiser JK, Breaker RR: New RNA motifs suggest an expanded scope for riboswitches in bacterial genetic control. Proc Natl Acad Sci USA. 2004, 101: 6421-6426. 10.1073/pnas.0308014101.
Mandal M, Lee M, Barrick JE, Weinberg Z, Emilsson GM, Ruzzo WL, Breaker RR: A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science. 2004, 306: 275-279. 10.1126/science.1100829.
Kochhar S, Paulus H: Lysine-induced premature transcription termination in the lysC operon of Bacillus subtilis. Microbiology. 1996, 142: 1635-1639.
Patte JC, Akrim M, V M: The leader sequence of the Escherichia coli lysC gene is involved in the regulation of the LysC synthesis. FEMS Microbiol Lett. 1998, 169: 165-170. 10.1111/j.1574-6968.1998.tb13313.x.
Grundy FJ, Lehman SC, Henkin TM: The L box regulon: lysine sensing by leader RNAs of bacterial lysine biosynthesis genes. Proc Natl Acad Sci USA. 2003, 100: 12057-12062. 10.1073/pnas.2133705100.
Sudarsan N, Wickiser JK, Nakamura S, Ebert MS, Breaker RR: An mRNA structure in bacteria that controls gene expression by binding lysine. Genes Dev. 2003, 17: 2688-2697. 10.1101/gad.1140003.
Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS: Regulation of lysine biosynthesis and transport genes in bacteria: yet another RNA riboswitch?. Nucleic Acids Res. 2003, 31: 6748-6757. 10.1093/nar/gkg900.
Christiansen LC, Schou S, Nygaard P, Saxild HH: Xanthine metabolism in Bacillus subtilis: characterization of the xpt-pbuX operon and evidence for purine- and nitrogen-controlled expression of genes involved in xanthine salvage and catabolism. J Bacteriol. 1997, 179: 2540-2550.
Mandal M, Boese B, Barrick JE, Winkler WC, Breaker RR: Riboswitches control fundamental biochemical pathways in Bacillus subtilis and other bacteria. Cell. 2003, 113: 577-586. 10.1016/S0092-8674(03)00391-X.
Mandal M, Breaker RR: Adenine riboswitches and gene activation by disruption of a transcription terminator. Structural & Molecular Biology. 2004, 11: 29-35. 10.1038/nsmb710.
Batey RT, Gilbert SD, Montange RK: Structure of a natural guanine responsive riboswitch complexed with the metabolite hypoxanthine. Nature. 2004, 432: 411-415. 10.1038/nature03037.
Serganov A, Yuan YR, Pikovskaya O, Polonskaia A, Malinina L, Phan AT, Hobartner C, Micura R, Breaker RR, Patel DJ: Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem Biol. 2004, 11: 1729-1741. 10.1016/j.chembiol.2004.11.018.
Giovannoni SJ, Britschgi TB, Moyer CL, Field KG: Genetic diversity in Sargasso Sea bacterioplankton. Nature. 1990, 345: 60-63. 10.1038/345060a0.
Rappe MS, Giovannoni SJ: The uncultured microbial majority. Annu Rev Microbiol. 2003, 57: 369-394. 10.1146/annurev.micro.57.030502.090759.
Rusch DB, et al: The Sorcerer II Global Ocean Sampling Expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS Biology. 2007, 5 (3): 398-431. 10.1371/journal.pbio.0050077.
Roth A, Winkler WC, Regulski EE, Lee BW, Lim J, Jona I, Barrick JE, Ritwik A, Kim JN, Welz R, Iwata-Reuyl D, Breaker RR: A riboswitch selective for the queuosine precursor preQ(1) contains an unusually small aptamer domain. Nat Struct Mol Biol. 2007, 14 (4): 308-317. 10.1038/nsmb1224.
Johnston AW, Li Y, Ogilvie L: Metagenomic marine nitrogen fixation-feast or famine?. Trends Microbiol. 2005, 13: 416-420. 10.1016/j.tim.2005.07.002.
Zhang Y, Fomenko DE, Gladyshev VN: The microbial selenoproteome of the Sargasso Sea. Genome Biol. 2005, 6: R37-10.1186/gb-2005-6-4-r37.
Yooseph S, et al: The Sorcerer II Global Ocean Sampling Expedition: Expanding the Universe of Protein Families. PLoS Biology. 2007, 5 (3): 432-466. 10.1371/journal.pbio.0050016.
Vitreschak AG, Mironov AA, Gelfand MS: RNApattern program: searching for RNA secondary structure by the pattern rule. Proceedings of the 3rd International Conference on 'Complex Systems: Control and Modeling Problems', September 4–9 2001, Samara, Russia, The Institute of Control of Complex Systems. 2001, 623-625.
Oracle 10g Express Edition. [http://www.oracle.com/technology/products/database/xe/index.html]
Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL: GenBank. Nucleic Acids Res. 2006, 34: D16-20. 10.1093/nar/gkj157.
DeLong EF: Microbial community genomics in the ocean. Nature Rev Microbiol. 2005, 3: 459-469. 10.1038/nrmicro1158.
Entrez Programming Utilities. [http://eutils.ncbi.nlm.nih.gov/entrez/query/static/eutils_help.html]
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410.
Tatusov RL, Koonin EV, Lipman DJ: Basic local alignment search tool. Science. 1997, 278: 631-637. 10.1126/science.278.5338.631.
NCBI Taxonomy Browser. [http://www.ncbi.nlm.nih.gov/Taxonomy/taxonomyhome.html]
Beitz E: TEXshade: shading and labeling multiple sequence alignments using LATEX 2ε. Bioinformatics. 2000, 16: 135-139. 10.1093/bioinformatics/16.2.135.
Acknowledgements
The authors are grateful to Dmitry Rodionov for useful discussions and sharing unpublished data and to Eric Beitz for the enhancements in the TEXshade package. This study was partially supported by grants from the Howard Hughes Medical Institute (55005610), INTAS (05-1000008-8028) and the Russian Academy of Science (program "Molecular and Cellular Biology").
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declares that there are no competing interests.
Authors' contributions
MG conceived the project. MK performed the computational analysis of the metagenome data. LV provided the program for the identification of riboswitches. MK and LV performed functional annotation. MK and MG wrote the paper. All the authors have read and approved the final manuscript.
Electronic supplementary material
12864_2007_1060_MOESM1_ESM.pdf
Additional file 1: S-boxes (SAM-riboswitches) and their regulated functions identified in three metagenomes. New functions are set in boldface. (PDF 35 KB)
12864_2007_1060_MOESM2_ESM.pdf
Additional file 2: RFN-elements (FMN-riboswitches) and their regulated functions identified in three metagenomes. (PDF 23 KB)
12864_2007_1060_MOESM3_ESM.pdf
Additional file 3: B12-elements and their regulated functions identified in three meta genomes. New functions are set in boldface. (PDF 42 KB)
12864_2007_1060_MOESM5_ESM.pdf
Additional file 5: Glycine riboswitches and their regulated functions identified in three metagenomes. New functions are set in boldface. (PDF 39 KB)
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kazanov, M.D., Vitreschak, A.G. & Gelfand, M.S. Abundance and functional diversity of riboswitches in microbial communities. BMC Genomics 8, 347 (2007). https://doi.org/10.1186/1471-2164-8-347
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-8-347