
Abstract
Background
Lungworms of the genus Dictyocaulus (family Dictyocaulidae) are parasitic nematodes of major economic importance. They cause pathological effects and clinical disease in various ruminant hosts, particularly in young animals. Dictyocaulus viviparus, called the bovine lungworm, is a major pathogen of cattle, with severe infections being fatal. In this study, we provide first insights into the transcriptome of the adult stage of D. viviparus through the analysis of expressed sequence tags (ESTs).
Results
Using our EST analysis pipeline, we estimate that the present dataset of 4436 ESTs is derived from 2258 genes based on cluster and comparative genomic analyses of the ESTs. Of the 2258 representative ESTs, 1159 (51.3%) had homologues in the free-living nematode C. elegans, 1174 (51.9%) in parasitic nematodes, 827 (36.6%) in organisms other than nematodes, and 863 (38%) had no significant match to any sequence in the current databases. Of the C. elegans homologues, 569 had observed 'non-wildtype' RNAi phenotypes, including embryonic lethality, maternal sterility, sterility in progeny, larval arrest and slow growth. We could functionally classify 776 (35%) sequences using the Gene Ontologies (GO) and established pathway associations to 696 (31%) sequences in Kyoto Encyclopedia of Genes and Genomes (KEGG). In addition, we predicted 85 secreted proteins which could represent potential candidates for developing novel anthelmintics or vaccines.
Conclusion
The bioinformatic analyses of ESTs data for D. viviparus has elucidated sets of relatively conserved and potentially novel genes. The genes discovered in this study should assist research toward a better understanding of the basic molecular biology of D. viviparus, which could lead, in the longer term, to novel intervention strategies. The characterization of the D. viviparus transcriptome also provides a foundation for whole genome sequence analysis and future comparative transcriptomic analyses.
Similar content being viewed by others

Background
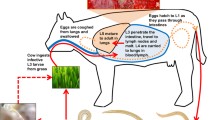
Parasitic nematodes of livestock cause substantial economic losses due to poor productivity, failure to thrive and deaths [1, 2]. The financial losses associated with these endoparasites are estimated at billions of dollars per annum [3]. Lungworms of the genus Dictyocaulus (family Dictyocaulidae) are key parasitic nematodes which cause pathological effects and clinical disease in different ruminant hosts, particularly in young animals [4, 5]. Dictyocaulus viviparus, the bovine lungworm, causes a severe and frequently fatal bronchitis (known colloquially as 'husk') which is of major importance in many countries [6]. Severe cases of dictyocaulosis lead to emphysema and pneumonia – heavy infections can cause a mortality rate of >20% among affected cattle [1, 2].
Dictyocaulus viviparus has a direct life cycle [7]. The adult stages (females and males) live in the bronchi, where the ovoviviparous females produce eggs from which first-stage larvae (L1) usually hatch rapidly whilst in the lung or the intestinal tract. The L1s are then shed in the faeces of the bovine host. Under favourable environmental conditions, L1s develop through to the infective third-stage larvae (L3s) during a period of ~4–6 days. After ingestion by the host, L3s migrate through the gut wall to the mesenteric lymph nodes, moult, and, as fourth-stage larvae (L4s), are transported to the lungs. L4s penetrate the alveoli, moult and then develop into adults. However, larval stages can remain inhibited in the lungs for up to 5 months. In cattle, the period from ingestion of L3s to reproductive maturity of the adult worms is 3–4 weeks.
While there is considerable knowledge of the morphological changes taking place during the life cycle of D. viviparus, very little is understood about the fundamental molecular and biochemical processes underlying the development and survival of this parasite and the parasite-host interplay. Insights into such processes are fundamentally important and could provide a basis for the identification of molecular targets for the rational design of nematocidal compounds, vaccines or/and for diagnosis. To date, studies of D. viviparus have been limited to individual genes and proteins. For instance, before the present study (March 2007), 323 gene sequences, 221 protein sequences and 229 research articles relating to D. viviparus were available in public databases.
Current technological advances in genomics provide exciting opportunities for exploring basic molecular biological and biochemical aspects of D. viviparus and related nematodes. For instance, expressed sequence tag (EST) data sets facilitate the prediction and categorization of key molecules, particularly those linked to development (both in pre-parasitic and parasitic stages), sexual differentiation and maturation, based on comparisons with other organisms for which sequence and functional genomic data sets are available. Also, the complete genome sequence of the free-living nematode Caenorhabditis elegans and the wealth of information on gene expression and function for this nematode [8, 9] provide a means of evaluating homologues and orthologues [10], since D. viviparus cannot be maintained or propagated effectively in vitro for the functional testing of genes and gene products. Also, the potential of gene silencing techniques [11, 12] provides a prospect for the functional analysis of molecules in this and other parasitic nematodes.
In the present study, we provide a first insight into the transcriptome of the adult stage of D. viviparus via EST sequencing and apply a newly established computational platform [13] for the clustering and comparative analyses of the data set against data available for a range of organisms, with an emphasis on the best characterized nematode, C. elegans. The representative ESTs from this dataset have been annotated functionally at the gene and protein levels to aid in assigning, in the main, gene ontologies, protein families and biochemical pathways. Such annotation techniques have enabled us to pin-point genes that could be considered in the development of intervention strategies. The present data provide a foundation for future investigations in areas, such as the stage-, sex- and tissue-specific gene transcription or expression, whole genome sequencing and proteomics of D. viviparus.
Results and discussion
General EST analysis
Of 5271 clones sequenced, a total of 4436 quality ESTs were obtained (Phase I, Figure 1), achieving a sequencing success of 84% (Table 1), which is consistent with previous studies [14, 15]. These pre-processed ESTs ranged from 80–1164 bp, with a mean of 730 bp and a standard deviation (S.D.) of 258 bp. After clustering, the mean length of the contigs (or consensus sequences) increased to 787 (+/- 313) bp. The G+C content of the coding sequences was 43.5%, consistent with other nematodes from clade V [16, 17] and slightly more than C. elegans (37%) and its congener, C. briggsae (38%) [18]. Under the assumption that the D. viviparus genome codes for ~22,000 proteins [19], the EST clusters were predicted to represent ~10–15% of the proteins encoded by this genome.

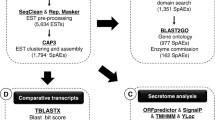
Bioinformatics analysis of D. viviparus ESTs. ESTExplorer analysis comprising Phases I (pre-processing), II (DNA-level annotation) and III (protein-level annotation), were augmented by homologue identification from nematodes as well as parasitic nematodes, using specialized databases.
The cluster analysis of the 4436 ESTs from adult D. viviparus yielded 2258 representative ESTs (rESTs; 458 contig and 1800 singleton sequences; see Table 1), of which 1685 (74.6%) had open reading frames (ORFs). All rESTs were then subjected to analyses using ESTExplorer [13], a semi-automated bioinformatics pipeline (Fig. 1). Also, we queried rESTs (at the amino acid level) against three databases containing protein sequences from different organisms, in order to categorize the molecules from D. viviparus. Data were compared with protein sequences available for (i) C. elegans (from WORMPEP v.167 [20]), (ii) parasitic nematodes (available protein sequences and peptides from conceptually translated ESTs), and (iii) organisms other than nematodes (from NCBI non-redundant protein database) [21]. Three-way comparison of D. viviparus rESTs with homologues from C. elegans, WORMPEP and parasitic nematodes have been figuratively presented using SimiTri [22] (Figure 2). For this comparison, similarity searches of the 2258 representative sequences resulted in 1159 (51.3%) homologues to C. elegans, 1174 (51.9%) to those from other parasitic nematodes (including some strongylids), 827 (36.6%) homologues in organisms other than nematodes, and 39.5% had no significant similarity to any other organism (employing a cut-off of 1e-05) for which sequence data are currently available (Figure 2, Additional File 1). The SimiTri plot (Figure 2) shows that, given the current database contents, the sampled transcriptome from D. viviparus is equally close to available C. elegans and parasitic nematode sequences, compared with non-nematodes.

Comparison of D. viviparus rESTs with C. elegans , parasitic nematode and non-nematode protein sequence databases using SimiTri. The numbers at each vertex indicate the number of rESTs matching only that specific database. The numbers on the edges indicate the number of rESTs matching the two databases linked by that edge. The number within the triangle indicates the number of D. viviparus genes with matches to all three databases.
Comparative analyses with C. elegans data sets
The comparative analysis to identify homologues in C. elegans is important because D. viviparus and this free-living nematode are both considered to belong to clade V of the Nematoda [16, 17], and because C. elegans also represents the best characterized nematode in many respects, particularly in terms of its genome, genetics, biology, physiology, biochemistry, as well as the localization and functions of molecules [20, 23]. Specifically, the comparative analysis (at the amino acid level) of all rESTs with C. elegans data (see Additional File 1) revealed 1159 (51.3%) key, well-characterized molecules associated with various biological processes (n = 540), including development, regulation of biological processes, response to abiotic and biotic stimuli and reproduction. 'Non-wildtype' RNAi phenotypes in C. elegans (such as embryonic lethality, maternal sterility, sterility in progeny, larval arrest and slow growth) were associated with 569 (48.7%) of these 1159 molecules (Table 3). Of the 1159 C. elegans homologues, the functions for 776 (66%) rESTs could be inferred using Gene Ontologies (GO) [24], with 696 (41%) sequences being mapped to key biological pathways (including signal transduction mechanisms, antigen processing and presentation, regulation of actin cytoskeleton, ribosomal proteins and translation factors). Overall, the functional classification revealed that approximately half of the rESTs had homologues in C. elegans and parasitic nematodes, one tenth were specific to parasitic nematodes, and one third of the rESTs did not match any sequence in current databases, possibly representing novel genes.
As multiple ESTs can be derived from the same gene, it was important to predict how many unique genes were represented by the rESTs. Mapping the 2258 D. viviparus rESTs to C. elegans revealed that, of the 1159 D. viviparus rESTs with similarities to 927 C. elegans genes, the majority of these (798/1159 or 68.8%) had a one-to-one relationship to their C. elegans homologue. The remaining 369 rESTs mapped to multiple non-overlapping regions from 129 C. elegans genes (with an estimated fragmentation rate of 25%). After discounting for fragmentation, we estimated that 1694 unique D. viviparus genes were identified, with a suggested new gene discovery rate of 38.2% (1694/2258).
C. elegans (non-wild-type) RNAi phenotypes can provide some indication of the relevance and functions of orthologous genes in other nematodes, particularly in parasitic nematodes of clade V, for which the complexity of an obligate parasitic life cycle and the lack of an effective (long-term) laboratory culture system make high-throughput functional screening impractical [25]. We retrieved C. elegans RNAi data representing D. viviparus homologues. Of 1159 D. viviparus rESTs, 569 had homologues in C. elegans which could be silenced by RNAi (Additional File 1). The RNAi phenotypes (as described by Wormbase) included Adl (adult lethal), Age (ageing alteration), Bmd (body morphology defect), Dpy (dumpy), Egl (egg laying defect), Emb (embryonic lethal), Gro (slow growth), Let (larval lethal), Lvl (larval lethal), Lva (larval arrest) and Unc (uncoordinated), whereas 590 homologues had no observable RNAi phenotype in C. elegans. We also found that 23% of the most abundant (Table 2) and 22% of the transcripts predicted to represent secreted proteins of D. viviparus (Additional File 2) had C. elegans homologues with non-wildtype RNAi phenotypes.
Comparative analysis with data for other nematodes within clade V
D. viviparus is a member of clade V of the phylum Nematoda [16, 17], which comprises members of the orders Strongylida, Rhabditida and Diplogasterida. We conducted a comparative analysis of all 2258 rESTs with data [17, 26] for various nematodes (including Ancylostoma caninum, Ancylostoma ceylanicum, Necator americanus, Nippostrongylus brasiliensis, Haemonchus contortus, Ostertagia ostertagi, Teladorsagia circumcincta and Pristionchus pacificus) belonging to clade V (Additional File 3), to examine gene conservation within this clade. The analysis revealed 601 (26.6%) rESTs to have significant similarity to molecules from the members of clade V. These rESTs represented house-keeping (including cathepsin B-like cysteine proteases 1 and 2 and serine-threonine protein kinase) as well as nematode-specific (vitellogenin structural and major sperm protein) genes. We could assign GO terms to 461 of these 601 rEST sequences, associated with 118 different biological processes, such as embryonic development, intracellular protein transport, protein metabolism and responses to abiotic and/or biotic stimuli. Furthermore, 404 sequences could be mapped to biological pathways predicted to be associated with ribosomal proteins and the proteasome system, including 23 predicted secreted proteins mapping to cysteine proteinases, secreted protein 5 precursor and parasite pepsinogen.
Abundant transcripts in adult D. viviparus
A high level of representation in a cDNA library usually correlates with high transcript abundance in the original biological sample [27], although artefacts of library construction can result in a selection for or against representation of some transcripts. The D. viviparus clusters were ranked according to the number of contributing ESTs, and the top 30 clusters, which represented 1261 (24%) of the total number of rESTs (2258) obtained, were investigated in detail (Table 2). A number of clusters had significant alignments to known proteins, the majority of which were house-keeping genes, such as elongation factors, ribosomal proteins, aldolases, kinases, proteases and actins. Some of these genes have been identified in a number of other nematodes, including Ancylostoma caninum, Ancylostoma ceylanicum, Dirofilaria immitis, Strongyloides ratti and Meloidogyne incognita [28–32], requiring detailed characterisation to understand their functions in D. viviparus.
We analysed the most abundantly expressed transcripts from D. viviparus and found that 12 of the 30 (40%) contigs had no significant similarity to any sequence in the non-redundant protein database. As most of the nematode data are available only as ESTs and not included in the BLAST databases, we further compared these 12 contigs with sequences against Parasite Genomes using WU-Blast2 and BLASTN against the NCBI 'other-ESTs' database. We found that five sequences did not match any sequence in the databases, whereas seven entries had similarity to ESTs from other nematodes. The other 18 (60%) contigs were assigned functionality based on BLASTP against the NR database, and all of them had homologues in either non-parasitic (C. elegans and/or C. briggsae) or parasitic nematodes. A summary of these findings is provided in Table 2. Most of the homologues were found to be house-keeping or structural genes (including aldolase, histone family members, lectin, collagen and cytochrome oxidase), and four contigs were represented by molecules specific to nematodes, such as the major sperm proteins.
Comparison with cDNAs from third-stage larvae of D. viviparus
Recently, Strube et al. [33] identified and characterized 28 cDNAs differentially transcribed between experimentally induced hypobiotic and infective third-stage larvae (L3) of D. viviparus using a suppressive-subtractive hybridization (SSH) approach. We compared these 28 sequences against our dataset of 2258 rESTs, using BLASTN, to identify whether any of them were represented in the adult stage, with only one match. The sole sequence common to both datasets was L3ni 18 (accession number EG374523), which matched EST D.viviparus_42_A11 in the present dataset, being a homologue of the C. elegans hypothetical protein C05D11.10 (mitochondrial/chloroplast ribosomal S17-like protein [code KOG3447]). Thus, this EST study of adult D. viviparus represents a novel dataset for parasitic nematodes in clade V, representing the Metastrongyloidea (cf. [47]).
Functional classification of rESTs from adult D. viviparus
We annotated all 2258 rESTs systematically using a range of bioinformatic tools (details available in Figure 1). This annotation included functional classifications of rESTs using Gene Ontologies (GO) [24], pathway mapping using KEGG (Kyoto Encyclopedia of Genes and Genomes) [34], visualisation of EST data comparisons using SimiTri [22] and analyses of the D. viviparus secretome using SignalP [35], TMHMM [36] and PSORTII [37]. Results of these analyses are described in the following two sections
a. Gene Ontologies
Gene Ontology (GO) has been used widely to predict gene function and classification. GO provides a dynamic vocabulary and hierarchy that unifies descriptions of biological, cellular and molecular functions across genomes. We used BLAST2GO [38], a sequence-based tool to assign GO terms, extracting them for each BLAST hit obtained by mapping to extant annotation associations. We found that 776 (31%) of 2258 rESTs could be functionally assigned to biological processes (n = 540), cellular components (n = 328) and molecular functions (n = 457). A summary GO representation (using GO Slim) of the D. viviparus rESTs is given in Table 3.
Amongst the most common GO categories representing biological processes were: binding (GO: 0005488), catalytic activity (GO: 0003824) and structural molecule activity (GO: 0005198); development (GO: 0007275), metabolism (GO: 0008152), reproduction (GO:0000003) and growth (GO:0040007). The largest number of GO terms in cellular components was for cell part (GO:0044464), membrane-bound organelle (GO:0043227) and non-membrane-bound organelle (GO:0043228). A complete listing of GO mappings assigned for rESTs is provided in Additional File 4.
b. Pathway analysis using KEGG assignments
Biochemical functionality was predicted by mapping all 2258 rESTs to pathways, using KOBAS implemented within ESTExplorer [13], with an E-value cut-off of 1.0e-5. Enzyme commission (EC) numbers were used to appraise which sequences pertained to a specific pathway. A total of 696 (31%) sequences were mapped to 139 KEGG pathways, with 453 sequences representing metabolic enzymes characterized by unique EC numbers. The top 30 (highly represented) pathways are shown in Table 4.
Molecules involved in signal transduction mechanisms (n= 26) and glycolysis/gluconeogenesis (h= 24) had the highest representation amongst the sequences mapped to KEGG pathways. We also identified 50 predicted proteins with potential roles in host-parasite interactions, with 11 molecules predicted to be involved in antigen processing and/or presentation, six in T-cell receptor signalling pathway and seven CD molecules. Although, at this stage, the precise role of such molecules in the parasite-host interplay is unclear, they could be involved in manipulating or evading the host's immune response(s) or associated with the parasite's innate immune response. Extensive experimental work would be required to test these proposals. Furthermore, we identified families of proteins representing serine, cysteine and metallo-proteinases as well as proteinase inhibitors (e.g., cystatins). While these enzymes are inferred to mediate or modulate proteolytic functions, which, in turn, may facilitate tissue migration and other interactions with host cells, the proteinase inhibitors may protect the parasite against digestion by host or endogenous proteinases excreted/secreted in the lung of the mammalian host [39]. A complete listing of the KEGG mappings is available as supplementary data (Additional File 5).
Secretome analysis
An important starting point in the identification of potential novel drug or vaccine candidates in parasites is the prediction of molecules that are secreted or excreted in or around the host- parasite interface [40–42]. Examples of such proteins are the aspartyl protease inhibitor (API-1) [43], mi-msp-1 (similar to a venom allergen antigen AG5-like) [44] and the Ancylostoma-secreted protein (ASP) [45]. In the present data set (= 2258 rESTs), we identified 85 putatively secreted proteins representing a non-redundant catalogue of D. viviparus molecules (Additional 2). Of these, 26 (30.5%) sequences had no significant similarity to any sequence available in current databases, whereas 59 (69.4%) had homologues in nematodes, with 43 (50.5%) C. elegans and/or C. briggsae matches, and 16 (18.8%) homologues in various other parasitic nematodes, including the blood-feeding nematodes Ancylostoma ceylanicum, Necator americanus and Haemonchus contortus.
The secretome analysis (Additional File 2) revealed a number of unique features. Firstly, seven of the putative secreted protein entries were homologous to either sperm-specific family members [41] or major sperm proteins (MSP), consistent with the mass spectrometric analysis of secreted molecules from D. viviparus [41]. Secondly, acetylcholinesterase (AChE) has been identified as an important enzyme secreted by adult D. viviparus, thought to be involved in parasite survival in the host and, therefore, being a vaccine candidate [46]. We now report an entry for secreted acetylcholinesterase (D.viviparus_8_A01 of 332 amino acids, with a signal peptide length of 21) in the secretome analysis of the current EST dataset (Additional File 2), which may also represent a target for further characterization.
Conclusion
The present study has given us a first glimpse of the transcriptome of the adult stage of the bovine lungworm, D. viviparus, and represents a starting point for studies in a number of different fundamental and applied areas. We used a comprehensive EST analysis pipeline, ESTExplorer, for this purpose, for functional annotation at the DNA and protein levels [13]. From this single study of 5271 ESTs, we have identified 55 novel sequences (1.4%), with high confidence, with no known homologue in any other nematode or mammal for which sequence data are presently available in public databases. These molecules are particularly interesting, as they may represent genes that may be specific to parasitism or to the species. However, such molecules are very challenging to work on, as their potential functions cannot be predicted using current bioinformatic approaches. However, there is considerable scope in exploring such molecules in the future, using a combination of genomic and proteomic approaches. Insights into such molecules and/or their interaction with the bovine host could provide opportunities for developing novel intervention approaches.
From a systematic viewpoint, D. viviparus, belongs to the Metastrongyloidea ("metastrongyles" or "lungworms") based on nuclear ribosomal DNA sequence data [47], as distinct from the Trichostrongyloidea (mainly in the stomach and small intestine), Strongyloidea (mostly in the large intestine) and Ancylostomatoidea (small intestine), and thus represents, from biological, host-parasite relationship and molecular evolutionary perspectives, a very interesting species for comparative genomic analysis with nematodes from these superfamilies. Therefore, this nematode brings a number of important benefits for future investigations, particularly for genome sequencing and for subsequent comparative evolutionary analyses. Indeed, D. viviparus, among other strongylid nematodes, has recently been selected for whole genome sequencing, to be carried out at the Genome Sequencing Center of Washington University in St Louis, USA [48]. For D. viviparus, the genomic information from the present study underpins future microarray analyses, focused on exploring the transcriptional profiles among different stages (e.g., larval versus adult stages; hypobiotic stages versus those which are not arrested; free-living (L1s and L2s) versus infective versus late larval stages), sexes (female versus male) and tissues (e.g., germline versus neural versus musculature versus intestine) of the parasite. Such studies, particularly those of the molecules differentially transcribed and expressed during the transition to parasitism, the invasion of the host and hypobiosis, could provide unique insights into such key molecular developmental and reproductive processes. While there is some controversy regarding the applicability and usefulness of RNAi to some parasitic nematodes, such as the Strongylida [11, 12], comparative studies of gene-silencing and transgenesis in C. elegans are considered useful for exploring the function and regulation of some relatively conserved parasite genes, provided data are interpreted with caution [10]. This is particularly the case with the continued increase in genome sequence information.
With the future availability of whole genome sequence data for D. viviparus, it will also be possible to carry out meaningful mass spectroscopic analyses of differentially expressed proteins [49, 50], allowing large-scale analysis of proteins from small amounts of parasite material. Such analyses will enable the link to be made between the regulation of transcription and translation and, importantly, in the study of parasites, will allow the analysis of proteins expressed within short time frames within or external to the host animal, or within organs or micro-environments within the parasite [51, 52]. Hence, the application of an integrated bioinformatic-genomic-phenomic-proteomic ("systems biology") approach, focusing on developmental processes and mechanisms, could enhance our understanding of the molecular biology of moulting, invasion of and establishment in the host, hypobiosis (arrested development), and sexual differentiation, maturation and behaviour of D. viviparus. Clearly, progress in such fundamental areas could lead to the development of exciting new ways of treating, controlling or preventing this lungworm and other parasitic nematodes, by blocking or disrupting key biological pathways in them.
Methods
Parasite material
Adults of D. viviparus (strain HannoverDv2000) were produced in helminth-free male Holstein-Friesian calves (five months of age). Four weeks after oral inoculation with 3300 L3, the calves with patent D. viviparus infection were euthanized and the worms collected from the lungs as described by Wood et al. [53]. The worms were washed extensively (five times) in large volumes (100 ml) of fresh diethyl-pyrocarbonate (DEPC)-treated saline (22°C), transferred to sterile, RNase-free screw-top cryovials® (4 ml; Roth, Karlsruhe, Germany) and frozen as 200 μl pellets in a minimal amount of saline at -75°C or -196°C until RNA isolation.
Isolation of total RNA, cDNA synthesis and cDNA library construction
Total RNA was extracted from the adults of D. viviparus (under liquid nitrogen, employing a sterile mortar and pestle) using the TriPure isolation reagent® (Roche Molecular Biochemicals). Integrity and yields of RNA were verified and estimated, respectively, using the Bioanalyzer 2100 (Agilent). Each RNA sample (~10 μg) was treated with 2 U of DNase I (Promega), incubated at 37°C for 30 min prior to heat denaturation of the enzyme (75°C for 5 min) and then stored at -70°C until use. The cDNA was produced from 10 μg of total RNA from D. viviparus, using an oligo (dT) primer and Superscript II reverse transcriptase (Invitrogen) and then purified over DyeEx columns (Qiagen).
A non-directional cDNA library was constructed in the plasmid vector pGEM-T (Promega) by T-ended cloning, according to the manufacturer's protocol. Colonies were screened using blue-white selection. Clones (n = 5271) were picked randomly and patched on to grided Luria Bertani (LB) agarose plates containing 100 mg/ml ampicillin. Single-pass sequencing (using the T7 primer) was performed employing BigDye Chemistry (v3.1) in a 3730 × l DNA analyser (Applied Biosystems).
EST analysis
The ESTs were initially analysed and annotated using ESTExplorer, an automated EST analysis platform [13, 54]. In brief, the analyses comprised three phases (see Figure 1). In phase I, all ESTs were pre-processed (SeqClean, RepeatMasker), aligned/clustered using the Contig Assembly Program CAP3, employing a minimum sequence overlap length "cut-off" of 30 bases and an identity threshold of 95% (in Phase I) for the removal of flanking vector and adapter sequences, followed by assembly. Phase II of the ESTExplorer led to gene ontology inference, at the DNA-level annotation, using BLAST2GO (V 1.6.2) [38], using Gene Ontology (MySQL-DB-data release go_200609). In Phase III, rESTs were then conceptually translated into peptides (using ESTScan), which were further characterized via InterProScan (domain/motifs), and peptides mapped to respective pathways in C. elegans using KOBAS (KEGG Orthology-Based Annotation System, KEGG data release 40.0). We also retrieved KO data from KEGG and then classified enzymes from non-enzymes based on our pathway mapping results. Peptides predicted from rEST were also compared, using BLASTP, with the non-redundant protein sequence database from National Centre for Biotechnology Information (NCBI), as part of the generic ESTExplorer pipeline for systematic EST analysis and annotation.
Protein databases for 'parasitic nematodes' and 'non-nematodes' were generated in-house for similarity searches. The 'parasitic nematodes' group contains all available protein sequences for parasitic nematodes and ESTs from GenBank (17 February 2007), translated into peptide sequences, whereas the 'non-nematodes' database comprises amino acid sequences from the complete non-redundant protein database NR (17 February 2007) excluding those from nematodes. Homologues to rESTs were identified via comparisons against WormBase using BLASTX and the Parasite genome WU-BLAST2 Nematoda database (from the European Bioinformatics Institute) using BLASTN. Each EST of D. viviparus was assigned a 'statistically significant' gene homologue if the E-value from the BLAST output of the sequence alignment was <1e-05. The program SimiTri [22] was used for the comparison (at the amino acid sequence level) of D. viviparus rESTs with data in C. elegans, parasitic nematode and non-nematode protein sequence databases. SimiTri provides a two-dimensional display of relative similarity relationships among three different datasets.
From the peptides inferred from rESTs, secreted proteins were predicted using a combination of three programs, to minimize the number of false positive predictions. Firstly, SignalP 3.0 [35] was used to predict the presence of secretory signal peptides and signal anchors for each predicted rEST proteins. A signal sequence was considered present when it was predicted both by the artificial neural network and the hidden Markov model prediction approaches (SignalPNN and SignalP-HMM, available as options within SignalP). In order to exclude the erroneous prediction of putative transmembrane (TM) sequences as signal sequences, TMHMM [36], a membrane topology prediction program, was then applied. We further validated the list of secreted proteins, using extracellular localization, employing PSORT [37].
Note: The final set of quality ESTs reported in this paper 15 are available in the EMBL, GenBank and DDJB databases under accession numbers EV849926 – EV854361.
References
Coles GC: The future of veterinary parasitology. Vet Parasitol. 2001, 98: 31-39. 10.1016/S0304-4017(01)00421-6.
Panuska C: Lungworms of ruminants. Vet Clin North Am Food Anim Pract. 2006, 22: 583-593. 10.1016/j.cvfa.2006.06.002.
Newton SE, Munn EA: The development of vaccines against gastrointestinal nematode parasites, particularly Haemonchus contortus. Parasitol Today. 1999, 15: 116-122. 10.1016/S0169-4758(99)01399-X.
David GP: Survey on lungworm in adult cattle. Vet Rec. 1997, 141: 343-344.
Eysker M, van Miltenburg L: Epidemiological patterns of gastrointestinal and lung helminth infections in grazing calves in The Netherlands. Vet Parasitol. 1988, 29: 29-39. 10.1016/0304-4017(88)90005-2.
Corwin RM: Economics of gastrointestinal parasitism of cattle. Vet Parasitol. 1997, 72: 451-457. 10.1016/S0304-4017(97)00110-6.
Anderson RC: Nematode Parasites of Vertebrates: Their Development and Transmission. 2001, Wallingford: CABI Publishing
Gilleard JS: The use of Caenorhabditis elegans in parasitic nematode research. Parasitology. 2004, 128 (Suppl): S49-S70.
Gilleard JS, Woods DJ, Dow JA: Model-organism genomics in veterinary parasite drug-discovery. Trends Parasitol. 2005, 21: 302-305. 10.1016/j.pt.2005.05.007.
Britton C, Murray L: Using Caenorhabditis elegans for functional analysis of genes of parasitic nematodes. Int J Parasitol. 2006, 36: 651-659. 10.1016/j.ijpara.2006.02.010.
Zawadzki JL, Presidente PJ, Meeusen EN, De Veer MJ: RNAi in Haemonchus contortus: a potential method for target validation. Trends Parasitol. 2006, 22: 495-499. 10.1016/j.pt.2006.08.015.
Geldhof P, Murray L, Couthier A, Gilleard JS, McLauchlan G, Knox DP, Britton C: Testing the efficacy of RNA interference in Haemonchus contortus. Int J Parasitol. 2006, 36: 801-810. 10.1016/j.ijpara.2005.12.004.
Nagaraj S, Deshpande N, Gasser R, Ranganathan S: ESTExplorer: an expressed sequence tag (EST) assembly and annotation platform. Nucleic Acids Res. 2007, 35: W143-W147. 10.1093/nar/gkm378.
Cottee PA, Nisbet AJ, Abs El-Osta YG, Webster TL, Gasser RB: Construction of gender-enriched cDNA archives for adult Oesophagostomum dentatum by suppressive-subtractive hybridization and a microarray analysis of expressed sequence tags. Parasitology. 2006, 132: 691-708. 10.1017/S0031182005009728.
Nisbet AJ, Gasser RB: Profiling of gender-specific gene expression for Trichostrongylus vitrinus (Nematoda: Strongylida) by microarray analysis of expressed sequence tag libraries constructed by suppressive-subtractive hybridisation. Int J Parasitol. 2004, 34: 633-643. 10.1016/j.ijpara.2003.12.007.
Blaxter M: Caenorhabditis elegans is a nematode. Science. 1998, 282: 2041-2046. 10.1126/science.282.5396.2041.
Parkinson J, Mitreva M, Whitton C, Thomson M, Daub J, Martin J, Schmid R, Hall N, Barrell B, Waterston RH, McCarter JP, Blaxter ML: A transcriptomic analysis of the phylum Nematoda. Nat Genet. 2004, 36: 1259-1267. 10.1038/ng1472.
Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N, Chinwalla A, Clarke L, Clee C, Coghlan A, Coulson A, D'Eustachio P, Fitch DH, Fulton LA, Fulton RE, Griffiths-Jones S, Harris TW, Hillier LW, Kamath R, Kuwabara PE, Mardis ER, Marra MA, Miner TL, Minx P, Mullikin JC, Plumb RW, Rogers J, Schein JE, Sohrmann M, Spieth J: The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 2003, 1: E45-10.1371/journal.pbio.0000045.
Consortium. CeS: Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998, 282: 2012-2018. 10.1126/science.282.5396.2012.
Wombase. [http://wormbase.org/]
Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL: GenBank. Nucleic Acids Res. 2006, 34: D16-D20. 10.1093/nar/gkj157.
Parkinson J, Blaxter M: SimiTri – visualizing similarity relationships for groups of sequences. Bioinformatics. 2003, 19: 390-395. 10.1093/bioinformatics/btf870.
Wormbook. [http://www.wormbook.org/]
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000, 25: 25-29. 10.1038/75556.
Geldhof P, Visser A, Clark D, Saunders G, Britton C, Gilleard J, Berriman M, Knox D: RNA interference in parasitic helminths: current situation, potential pitfalls and future prospects. Parasitology. 2007, 134: 609-619. 10.1017/S0031182006002071.
Mitreva M, Wendl MC, Martin J, Wylie T, Yin Y, Larson A, Parkinson J, Waterston RH, McCarter JP: Codon usage patterns in Nematoda: analysis based on over 25 million codons in thirty-two species. Genome Biol. 2006, 7: R75-10.1186/gb-2006-7-8-r75.
Audic S, Claverie JM: The significance of digital gene expression profiles. Genome Res. 1997, 7: 986-995.
Yin Y, Martin J, McCarter JP, Clifton SW, Wilson RK, Mitreva M: Identification and analysis of genes expressed in the adult filarial parasitic nematode Dirofilaria immitis. Int J Parasitol. 2006, 36: 829-839. 10.1016/j.ijpara.2006.03.002.
McCarter JP, Mitreva MD, Martin J, Dante M, Wylie T, Rao U, Pape D, Bowers Y, Theising B, Murphy CV, Kloek AP, Chiapelli BJ, Clifton SW, Bird DM, Waterston RH: Analysis and functional classification of transcripts from the nematode Meloidogyne incognita. Genome Biol. 2003, 4: R26-10.1186/gb-2003-4-4-r26.
Thompson FJ, Mitreva M, Barker GL, Martin J, Waterston RH, McCarter JP, Viney ME: An expressed sequence tag analysis of the life-cycle of the parasitic nematode Strongyloides ratti. Mol Biochem Parasitol. 2005, 142: 32-46. 10.1016/j.molbiopara.2005.03.006.
Geldhof P, Whitton C, Gregory WF, Blaxter M, Knox DP: Characterisation of the two most abundant genes in the Haemonchus contortus expressed sequence tag dataset. Int J Parasitol. 2005, 35: 513-522. 10.1016/j.ijpara.2005.02.009.
Mitreva M, McCarter JP, Arasu P, Hawdon J, Martin J, Dante M, Wylie T, Xu J, Stajich JE, Kapulkin W, Clifton SW, Waterston RH, Wilson RK: Investigating hookworm genomes by comparative analysis of two Ancylostoma species. BMC Genomics. 2005, 6: 58-10.1186/1471-2164-6-58.
Strube C, Schnieder T, von Samson-Himmelstjerna G: Differential gene expression in hypobiosis-induced and non-induced third-stage larvae of the bovine lungworm Dictyocaulus viviparus. Int J Parasitol. 2007, 37: 221-231. 10.1016/j.ijpara.2006.09.014.
Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M: From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006, 34: D354-D357. 10.1093/nar/gkj102.
Bendtsen JD, Nielsen H, von Heijne G, Brunak S: Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004, 340: 783-795. 10.1016/j.jmb.2004.05.028.
Krogh A, Larsson B, von Heijne G, Sonnhammer EL: Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001, 305: 567-580. 10.1006/jmbi.2000.4315.
Nakai K, Horton P: PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem Sci. 1999, 24: 34-36. 10.1016/S0968-0004(98)01336-X.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M: Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005, 21: 3674-3676. 10.1093/bioinformatics/bti610.
Knox DP: Proteinase inhibitors and helminth parasite infection. Parasite Immunol. 2007, 29: 57-71. 10.1111/j.1365-3024.2006.00913.x.
Harcus YM, Parkinson J, Fernandez C, Daub J, Selkirk ME, Blaxter ML, Maizels RM: Signal sequence analysis of expressed sequence tags from the nematode Nippostrongylus brasiliensis and the evolution of secreted proteins in parasites. Genome Biol. 2004, 5: R39-10.1186/gb-2004-5-6-r39.
Matthews JB, Davidson AJ, Beynon RJ: The application of mass spectrometry to identify immunogenic components of excretory/secretory products from adult Dictyocaulus viviparus. Parasitology. 2004, 128 (Suppl): S43-S47.
Vanholme B, De Meutter J, Tytgat T, Van Montagu M, Coomans A, Gheysen G: Secretions of plant-parasitic nematodes: a molecular update. Gene. 2004, 332: 13-27. 10.1016/j.gene.2004.02.024.
Delaney A, Williamson A, Brand A, Ashcom J, Varghese G, Goud GN, Hawdon JM: Cloning and characterisation of an aspartyl protease inhibitor (API-1) from Ancylostoma hookworms. Int J Parasitol. 2005, 35: 303-313. 10.1016/j.ijpara.2004.11.014.
Ding X, Shields J, Allen R, Hussey RS: A secretory cellulose-binding protein cDNA cloned from the root-knot nematode (Meloidogyne incognita). Mol Plant Microbe Interact. 1998, 11: 952-959. 10.1094/MPMI.1998.11.10.952.
Zhan B, Liu Y, Badamchian M, Williamson A, Feng J, Loukas A, Hawdon JM, Hotez PJ: Molecular characterisation of the Ancylostoma-secreted protein family from the adult stage of Ancylostoma caninum. Int J Parasitol. 2003, 33: 897-907. 10.1016/S0020-7519(03)00111-5.
McKeand JB: Vaccine development and diagnostics of Dictyocaulus viviparus. Parasitology. 2000, 120 (Suppl): S17-S23. 10.1017/S0031182099005727.
Chilton NB, Huby-Chilton F, Gasser RB, Beveridge I: The evolutionary origins of nematodes within the order Strongylida are related to predilection sites within hosts. Mol Phylogenet Evol. 2006, 40: 118-128. 10.1016/j.ympev.2006.01.003.
NHGRI sequencing for 10 parasitic nematode species, members of the order Strongylida. [http://www.genome.gov/11007951]
von Eggeling F, Davies H, Lomas L, Fiedler W, Junker K, Claussen U, Ernst G: Tissue-specific microdissection coupled with ProteinChip array technologies: applications in cancer research. Biotechniques. 2000, 29: 1066-1070.
Yatsuda AP, Krijgsveld J, Cornelissen AW, Heck AJ, de Vries E: Comprehensive analysis of the secreted proteins of the parasite Haemonchus contortus reveals extensive sequence variation and differential immune recognition. J Biol Chem. 2003, 278: 16941-16951. 10.1074/jbc.M212453200.
Barrett J, Brophy PM: Ascaris haemoglobin: new tricks for an old protein. Parasitol Today. 2000, 16: 90-91. 10.1016/S0169-4758(99)01613-0.
Pernthaner A, Cole SA, Morrison L, Green R, Shaw RJ, Hein WR: Cytokine and antibody subclass responses in the intestinal lymph of sheep during repeated experimental infections with the nematode parasite Trichostrongylus colubriformis. Vet Immunol Immunopathol. 2006, 114: 135-148. 10.1016/j.vetimm.2006.08.004.
Wood IB, Amaral NK, Bairden K, Duncan JL, Kassai T, Malone JB, Pankavich JA, Reinecke RK, Slocombe O, Taylor SM, Vercruysse J: World Association for the Advancement of Veterinary Parasitology (W.A.A.V.P.) second edition of guidelines for evaluating the efficacy of anthelmintics in ruminants (bovine, ovine, caprine). Vet Parasitol. 1995, 58: 181-213. 10.1016/0304-4017(95)00806-2.
ESTExplorer. [http://estexplorer.biolinfo.org]
Acknowledgements
This work was supported by grants from the Australian Research Council (LP0667795 and DP0665230), Genetic Technologies Limited and Meat and Livestock Australia. Thanks to Dr Gary Cobon, Stephen Page and Joan Lloyd for continued support. SHN is grateful to Macquarie University for the award of iMURS research scholarships and an MUPGR travel grant. Funding to pay the Open Access publication charges for this article was provided by Macquarie University.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Authors' contributions
SR, SHN and RBG conceived and designed the research plan and participated in all aspects of data collection and analysis. SHN and SR analysed, and SHN, SR, MH and RBG interpreted the data. CS and TS provided adult worms of D. viviparus. RBG and MH isolated and purified RNA for cDNA library construction and supervised and coordinated the sequencing. All authors contributed to the analyses and the writing of the manuscript. All authors read and approved the final manuscript.
Shoba Ranganathan, Shivashankar H Nagaraj contributed equally to this work.
Electronic supplementary material
12864_2007_1024_MOESM1_ESM.xls
Additional file 1: Comparison of 2258 rESTs from Dictyocaulus viviparus with Caenorhabditis elegans data (Wormpep v 167). The table also provides corresponding RNAi phenotypes in C. elegans and information on homologues in parasitic nematodes. (XLS 418 KB)
12864_2007_1024_MOESM3_ESM.xls
Additional file 3: Dictyocaulus viviparus homologues in nematodes in clade V, including Ancylostoma caninum, Ancylostoma ceylanicum, Necator americanus, Nippostrongylus brasiliensis, Haemonchus contortus, Ostertagia ostertagi, Teladorsagia circumcincta and Pristionchus pacificus. (XLS 336 KB)
12864_2007_1024_MOESM4_ESM.xls
Additional file 4: Mapped metabolic pathways in adult Dictyocaulus viviparus based on Kyoto Encyclopedia of Genes and Genomes (KEGG). The pathway mapping was carried out using the program KOBAS. (XLS 29 KB)
12864_2007_1024_MOESM5_ESM.xls
Additional file 5: Gene Ontology mappings (using GO slim terms) for Dictyocaulus viviparus clusters generated using BLAST2GO program. Note that individual GO categories can have multiple mappings. (XLS 50 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ranganathan, S., Nagaraj, S.H., Hu, M. et al. A transcriptomic analysis of the adult stage of the bovine lungworm, Dictyocaulus viviparus. BMC Genomics 8, 311 (2007). https://doi.org/10.1186/1471-2164-8-311
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-8-311