
Abstract
Background
Mycobacterium smegmatis is fast growing non-pathogenic mycobacteria. This organism has been widely used as a model organism to study the biology of other virulent and extremely slow growing species like Mycobacterium tuberculosis. Based on the homology of the N-terminal DNA binding domain, the recently sequenced genome of M. smegmatis has been shown to possess several putative GntR regulators. A striking characteristic feature of this family of regulators is that they possess a conserved N-terminal DNA binding domain and a diverse C-terminal domain involved in the effector binding and/or oligomerization. Since the physiological role of these regulators is critically dependent upon effector binding and operator sites, we have analysed and classified these regulators into their specific subfamilies and identified their potential binding sites.
Results
The sequence analysis of M. smegmatis putative GntRs has revealed that FadR, HutC, MocR and the YtrA-like regulators are encoded by 45, 8, 8 and 1 genes respectively. Further out of 45 FadR-like regulators, 19 were classified into the FadR group and 26 into the VanR group. All these proteins showed similar secondary structural elements specific to their respective subfamilies except MSMEG_3959, which showed additional secondary structural elements. Using the reciprocal BLAST searches, we further identified the orthologs of these regulators in Bacillus subtilis and other mycobacteria. Since the expression of many regulators is auto-regulatory, we have identified potential operator sites for a number of these GntR regulators by analyzing the upstream sequences.
Conclusion
This study helps in extending the annotation of M. smegmatis GntR proteins. It identifies the GntR regulators of M. smegmatis that could serve as a model for studying orthologous regulators from virulent as well as other saprophytic mycobacteria. This study also sheds some light on the nucleotide preferences in the target-motifs of GntRs thus providing important leads for initiating the experimental characterization of these proteins, construction of the gene regulatory network for these regulators and an understanding of the influence of these proteins on the physiology of the mycobacteria.
Similar content being viewed by others
Background
Being a fast growing, non-pathogenic mycobacteria, Mycobacterium smegmatis has been widely used as a model organism to study the biology of other virulent and extremely slow growing species like M. tuberculosis. The genome of M. smegmatis, as listed at the TIGR site, contains a large number of putative GntR-like regulators. These regulators play an important role in the cellular physiology. Many such regulators are involved in regulation of gene expression in response to various oxidized substrates related to either amino acid metabolism or at the branch points of various other metabolic pathways.
The GntR family of bacterial regulators is named after the Bacillus subtilis transcription regulator- GntR- a repressor of the gluconate operon [1]. Regulators of this family possess a conserved N-terminal domain that is involved in the DNA binding. Based on this conservation, these proteins can easily be recognized by a Conserved Domain Database (CDD) search [2]. However, the C-terminal domain, which is involved in the effector binding and/or oligomerization (E-b/O), is quite diverse and heterogeneous. As a consequence of this heterogeneity, the GntR regulators have been further classified into six subfamilies (FadR, HutC, MocR, YtrA, AraR and PlmA) [3, 4]. The members of subfamilies possess conserved secondary structural features specific to their subfamily and interact with a limited number of molecules [5]. Considering these conserved secondary structural features in sequence analysis, GntR regulators are defined as a part of specific subfamily [6]. Earlier, we have characterized GntR regulators from M. tuberculosis [7]. In present study putative GntR regulators from M. smegmatis are classified into their specific subfamilies. Further, suitable orthologs of the M. smegmatis GntRs were also identified using reciprocal BLAST searches in M. tuberculosis, M. avium, M. bovis, M. ulcerans, M. sp. KMS, M. sp. MCS, M. vanbaalenii PYR-1 and B. subtilis. To identify the DNA targets of these regulators, we utilized the information about the nucleotide preferences for regulators of a given subfamily. All the upstream DNA sequences of the GntR coding genes were scanned to locate potential palindromes that matched the nucleotide preference criteria [5].
Results and discussion
Classification of the putative M. smegmatis GntRs into subfamilies
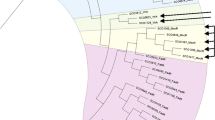
Unrooted tree of the M. smegmatis GntRs was constructed with the classified representatives of all subfamilies (Table 1) [5]. Among all putative M. smegmatis GntRs two proteins (MSMEG_1043 and MSMEG_2323) were found to be identical in sequence, hence only one of them MSMEG_1043 was taken for the classification. Each branch of the constructed tree represents a subfamily. Bootstrapping, involving 1000 replicates, shows all subfamily branches clustered with high bootstrap values. FadR subfamily is divided into two groups, FadR and VanR (Figure 1).

Unrooted tree of the proteins of GntR family regulators of M. smegmatis including representatives of all subfamily regulators from different Bacterial Genomes with 1000 bootstrap replicates. All the GntR regulators are clustered into six subfamilies. FadR subfamily is branched again into two groups (FadR and VanR). (Abbreviations are as indicated in Table 1 and Table 2).
FadR-like proteins of M. smegmatis
Of all the putative GntRs, 45 proteins were classified as the FadR-like regulators. These subfamily members are further classified into two groups FadR and VanR where the C-terminal effector binding and/or oligomerization domain length is about 170 and 150 amino acid residues respectively comprising all α-helices [5]. Among all FadR-like regulators, 19 regulators were clustered as members of the FadR group while 26 for the VanR group (Table 2). To study secondary structural features both the group members were dealt with separately. C-terminal domain of all the members of FadR group were predicted with seven α-helices except MSMEG_2599. All the regulators showed distinguishable predicted secondary structural features specific to this subfamily (Figure 2 and Figure 3) [5]. Secondary structural patterns of the regulator MSMEG_3959 revealed an extra secondary structural element, which could be significant in studying protein family evolution. FadR-like regulators are known to be involved in the regulation of gene expression in response to oxidized substrates related to either amino acid metabolism or at the branch point in various metabolic pathways such as glycolate [8], pyruvate [9], lactate [10], malonate [11] or gluconate [12]. One of FadR-like classified transcriptional regulator MSMEG_6700 is known to be involved in the regulation of piperidine and pyrrolidine metabolism [13]. These results provide a starting point for a detailed biochemical and genetic characterization of M. smegmatis FadR-like regulators.

Structure based sequence analysis of M. smegmatis GntR-like regulators by the multiple sequence alignment of the C-terminal domains of GntR regulators belonging to FadR Subfamily (FadR group). Abbreviations are as indicated in Table 1. Consensus sequence from the multiple sequence alignment has been drawn. High and low consensus levels were fixed arbitrarily at 80% and 40% of identity and are represented respectively by the capital and lowercase letters. Consensus symbol ! used for anyone of IV; $ is anyone of LM; % is anyone of FY; # is anyone of NDQEBZ. In graphical representation α-helix region and β-sheet regions are highlighted with light and dark gray background.

Structure based sequence analysis of M. smegmatis GntR-like regulators by the multiple sequence alignment of C-terminal domains of GntR regulators belonging to FadR Subfamily (VanR group). Abbreviations are as indicated in Table 1. Consensus sequence from the multiple sequence alignment has been drawn. High and low consensus levels were fixed arbitrarily at 80% and 40% of identity and are represented respectively by the capital and lowercase letters. Consensus symbol ! used for anyone of IV; $ is anyone of LM; % is anyone of FY; # is anyone of NDQEBZ. In graphical representation α-helix region and β-sheet regions are highlighted with light and dark gray background.
HutC-like proteins of M. smegmatis
Contrary to the FadR-like regulators, the regulators of this subfamily consist of both α-helices and β-sheet structures in the C-terminal domain. We identified eight GntRs as members of this subfamily (Table 2). All these members showed distinguishable predicted secondary structural features specific to this subfamily (Figure 4) [5]. These regulators are known to acquire the same protein fold as Escherichia coli UbiC; hence it is also named as UbiC transcription regulator-associated (UTRA) domain [14]. This effector-binding domain responds to various ligands like histidine (HutC) [15], long chain fatty acids [16], trehalose 6-phosphate [17] or alkylphosphonate [18]. A range of known ligands, specific to many HutC-like regulators, will help in characterizing the classified M. smegmatis regulators.

Structure based sequence analysis of M. smegmatis GntR-like regulators by the multiple sequence alignment of C-terminal domains of GntR regulators belonging to the HutC Subfamily. Abbreviations are as indicated in Table 1. Consensus sequence from the multiple sequence alignment has been drawn. High and low consensus levels were fixed arbitrarily at 80% and 40% of identity and are represented respectively by the capital and lowercase letters. Consensus symbol ! used for anyone of IV; $ is anyone of LM; % is anyone of FY; # is anyone of NDQEBZ. In graphical representation α-helix region and β-sheet regions are highlighted with light and dark gray background.
MocR-like protein of M. smegmatis
Among all the putative GntR regulators, eight were classified as members of the MocR subfamily (Table 2). All the eight regulators showed distinguishable predicted secondary structural features specific to this subfamily (Figure 5) [5]. MocR-like regulators show homology to the class I aminotransferase proteins [19], which requires pyridoxal 5'-phosphate (PLP) as a co-factor. All MocR-like regulators exhibit a PLP attachment site with a conserved lysine residue, which is also evident in the classified MocR-like regulators (Figure 5). It would thus be interesting to study the role of pyridoxal phosphate regulation in the classified regulators. [20].

Structure based sequence analysis of M. smegmatis GntR-like regulators by the multiple sequence alignment of C-terminal domains of GntR regulators belonging to the MocR Subfamily. Abbreviations are as indicated in Table 1. Consensus sequence from the multiple sequence alignment has been drawn. High and low consensus levels were fixed arbitrarily at 80% and 40% of identity and are represented respectively by the capital and lowercase letters. Consensus symbol ! used for anyone of IV; $ is anyone of LM; % is anyone of FY; # is anyone of NDQEBZ. In graphical representation α-helix region and β-sheet regions are highlighted with light and dark gray background.
YtrA-like protein of M. smegmatis
The YtrA subfamily is the least represented GntR-like regulator in the bacterial genomes. Among all M. smegmatis GntR regulators, only one regulator MSMEG_5174, showed the signatures of the YtrA subfamily member (Table 2, Figure 6). YtrA possesses a reduced C-terminal domain with only two α-helices. The average length of the putative effector binding and/or oligomerization domain is about 50 amino acids [5]. YtrA from B. subtilis is an experimentally explored regulator, which is part of a large self-regulated operon. This operon consists of genes encoding the ATP binding cassette (ABC) transport systems in addition to the YtrA [21]. It would be interesting to study further, whether MSMEG_5174 has any role in modulating such an operon.

Structure based sequence analysis of M. smegmatis GntR-like regulators by the multiple sequence alignment of the C-terminal domains of GntR regulators belonging to YtrA Subfamily. Abbreviations are as indicated in Table 1. Consensus sequence from the multiple sequence alignment has been drawn. High and low consensus levels were fixed arbitrarily at 80% and 40% of identity and are represented respectively by the capital and lowercase letters. Consensus symbol ! used for anyone of IV; $ is anyone of LM; % is anyone of FY; # is anyone of NDQEBZ. In graphical representation α-helix region and β-sheet regions are highlighted with light and dark gray background.
Operator/binding site analysis
We have tabulated a list of potential operator sites near the perfect palindrome sequence with conserved residues, which are found to be specific for most of the subfamily members (Table 3) [5]. We did not find an operator sequence in the upstream sequences of all the remaining regulators. All the predicted sites were found to be in the upstream region from the translation start site except MSMEG_2599. Identification of these sites is an important step to understand the GntR associated regulon or the gene regulatory network in the genome [22–25].
Ortholog prediction
We have found a number of M. smegmatis GntR regulators that are orthologs of proteins from the other species of mycobacteria and B. subtilis (Table 4). As orthologs typically share the same function, these regulators could serve as a model to study homologues from the other species of mycobacteria. These characterized orthologs may provide clues for initiating detailed biochemical characterization of M. smegmatis proteins. Many putative orthologs were experimentally known like Rv0165c that is involved in regulation of mce1 operon [6]; GntR, a transcriptional repressor of gluconate operon [12]; YcbG, involved in utilization of D-glucarate and D-galactarate [26]; YcnF, involved in utilization of gamma-aminobutyrate [27]. However, we did not find the orthologs for all M. smegmatis GntRs in other pathogenic species.
Our results help in extending the annotation of GntRs encoded in the M. smegmatis genome. We have classified putative M. smegmatis GntRs into four subfamilies. Though in the present study, we have made an attempt to explore M. smegmatis GntR regulators, this approach could also be effectively employed to extend the GntR family classification in other bacterial species as well.
Conclusion
This analysis has shown that M. smegmatis is equipped with large number of GntR-like regulators, belonging to four subfamilies. It further suggests that the GntR regulatory repertoires of M. smegmatis are far more complex than in M. tuberculosis. Indeed, additional GntR regulators possibly control a subset of genes required for adapting to a range of environmental conditions. One of the FadR-like regulators shows additional secondary structural elements, suggesting a possible origin of a new group within the FadR subfamily. Identified orthologs from M. smegmatis could serve as a model to decipher molecular regulation events taking place in the pathogenic mycobacteria. Potential operator sites were also identified based on the nucleotide recognition preferences of GntR-like regulators.
Methods
Selection of GntR-like Members
The sequences of M. smegmatis MC2 were downloaded from the Institute for Genomic Research Comprehensive Microbial Resource [28]. Apart from classified GntR regulators or proteins annotated as GntR-like regulator, other putative GntRs from M. smegmatis proteome were selected using GntR Pfam profile [29]. Among all predicted GntRs one protein (MSMEG_3400) was discarded for this study because of its unusual size (741 amino acid) and its annotation as glutamyl-tRNA(Gln) amidotransferase subunit A. Rest of the GntR regulators were retrieved from the SWISS-PROT/TrEMBL sequence database as per their Swiss-Prot ID (Table 1). Additionally published and annotated genome sequences of M. tuberculosis, M. avium subsp. paratuberculosis, M. bovis, M. ulcerans, M. sp KMS, M. sp. MCS, M. vanbaalenii PYR-1 and Bacillus subtilis were downloaded from the NCBI ftp site [30].
Secondary structure prediction
The secondary structural features of all bacterial GntR regulators including the M. smegmatis GntRs were analyzed (Table 1 and Table 2). Secondary structure predictions were made using Jpred [31], SsPro [32] and 3DPSSM [33]. A consensus of all the secondary structure predictions was considered for a better validity.
Multiple sequence alignments and Phylogenetic tree construction
Multiple sequence alignment was generated with MULTIALIN [34]. Distances between aligned proteins were computed with the PROTDIST program using the Dayhoff PAM matrix [35]. The FITCH program estimated phylogenies from distances in the matrix data using the Fitch-Margoliash algorithm [36]. The phylogenetic tree was drawn using the TREEVIEW program with incorporation of bootstrap values that were obtained involving 1000 replicates [37]. PROTDIST and FITCH programs are included in the PHYLIP package developed by Felsenstein [38].
Operator site analysis
To study the upstream region of GntR-like regulators, we considered sequences from 400 bases upstream to 50 bases downstream from the translation start site. As many GntR regulators are reported to recognize palindromes and also exhibit nucleotide recognition preferences among the same subfamily [5], we utilised these clues to scan the upstream sequences.
Reciprocal BLAST
Reciprocal BLAST hits are frequently utilized to identify the orthologs in two species [39, 40]. In this method we searched for the best reciprocal BLAST hit for M. smegmatis GntR proteins with M. tuberculosis, M. avium, M. bovis, Mycobacterium ulcerans, Mycobacterium sp KMS, Mycobacterium sp. MCS, Mycobacterium vanbaalenii PYR-1 and B. subtilis.
Abbreviations
- M. tuberculosis :
-
Mycobacterium tuberculosis
- M. bovis :
-
Mycobacterium bovis
- M. avium para. :
-
Mycobacterium avium subsp. paratuberculosis
- M. smegmatis :
-
Mycobacterium smegmatis
- M. ulcerans :
-
Mycobacterium ulcerans
- M. sp KMS:
-
Mycobacterium sp. KMS
- M. sp. MCS:
-
Mycobacterium sp. MCS
- M. vanbaalenii PYR-1:
-
Mycobacterium vanbaalenii PYR-1.
References
Haydon DJ, Guest JR: A new family of bacterial regulatory proteins. FEMS. 1991, 63: 291-295. 10.1111/j.1574-6968.1991.tb04544.x.
Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Marchler GH, Mullokandov M, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Yamashita RA, Yin JJ, Zhang D, Bryant SH: CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res. 2005, 33: D192-196. 10.1093/nar/gki069.
Rigali S, Schlicht M, Hoskisson P, Nothaft H, Merzbacher M, Joris B, Titgemeyer F: Extending the classification of bacterial transcription factors beyond the helix-turn-helix motif as an alternative approach to discover new cis/trans relationships. Nucleic Acids Res. 2004, 32: 3418-3426. 10.1093/nar/gkh673.
Lee MH, Scherer M, Rigali S, Golden JW: PlmA, a new member of the GntR family, has plasmid maintenance functions in Anabaena sp. strain PCC 7120. J Bacteriol. 2003, 185: 4315-4325. 10.1128/JB.185.15.4315-4325.2003.
Rigali S, Derouaux A, Giannotta F, Dusart J: Subdivision of the helix-turn-helix GntR family of bacterial regulators in the FadR, HutC, MocR, and YtrA subfamilies. J Biol Chem. 2002, 277: 12507-12515. 10.1074/jbc.M110968200.
Casali N, White AM, Riley LW: Regulation of the Mycobacterium tuberculosis mce1 operon. J Bacteriol. 2006, 188: 441-449. 10.1128/JB.188.2.441-449.2006.
Vindal V, Ranjan S, Ranajan A: In silico analysis and characterization of GntR family of regulators from Mycobacterium tuberculosis. Tuberculosis. 2007, 87: 242-247. 10.1016/j.tube.2006.11.002.
Pellicer MT, Fernandez C, Badia J, Aguilar J, Lin EC, Baldom L: Cross-induction of glc and ace operons of Escherichia coli attributable to pathway intersection. Characterization of the glc promoter. J Biol Chem. 1999, 274: 1745-1752. 10.1074/jbc.274.3.1745.
Quail MA, Guest JR: Purification, characterization and mode of action of PdhR, the transcriptional repressor of the pdhR-aceEF-lpd operon of Escherichia coli. Mol Microbiol. 1995, 15: 519-529. 10.1111/j.1365-2958.1995.tb02265.x.
Nunez MF, Pellicer MT, Badia J, Aguilar J, Baldoma L: The gene yghK linked to the glc operon of Escherichia coli encodes a permease for glycolate that is structurally and functionally similar to L-lactate permease. Microbiology. 2001, 147: 1069-1077.
Lee HY, An JH, Kim YS: Identification and characterization of a novel transcriptional regulator, MatR, for malonate metabolism in Rhizobium leguminosarum bv. trifolii. Eur J Biochem. 2000, 267: 7224-7230. 10.1046/j.1432-1327.2000.01834.x.
Reizer A, Deutscher J, Saier MH, Reizer J: Analysis of the gluconate (gnt) operon of Bacillus subtilis. Mol Microbiol. 1991, 5: 1081-1089. 10.1111/j.1365-2958.1991.tb01880.x.
Poupin P, Ducrocq V, Hallier-Soulier S, Truffaut N: Cloning and characterization of the genes encoding a cytochrome P450 (PipA) involved in piperidine and pyrrolidine utilization and its regulatory protein (PipR) in Mycobacterium smegmatis mc2155. J Bacteriol. 1999, 181: 3419-3426.
Aravind L, Anantharaman V: HutC/FarR-like bacterial transcription factors of the GntR family contain a small molecule-binding domain of the chorismate lyase fold. FEMS Microbiol Lett. 2003, 222: 17-23. 10.1016/S0378-1097(03)00242-8.
Allison SL, Phillips AT: Nucleotide sequence of the gene encoding the repressor for the histidine utilization genes of Pseudomonas putida. J Bacteriol. 1990, 172: 5470-5476.
Quail MA, Dempsey CE, Guest JR: Identification of a fatty acyl responsive regulator (FarR) in Escherichia coli. FEBS Lett. 1994, 356: 183-187. 10.1016/0014-5793(94)01264-4.
Matthijs S, Koedam N, Cornelis P, De Greve H: The trehalose operon of Pseudomonas fluorescens ATCC 17400. Res Microbiol. 2000, 151: 845-851. 10.1016/S0923-2508(00)01151-7.
Chen CM, Ye QZ, Zhu ZM, Wanner BL, Walsh CT: Molecular biology of carbon-phosphorus bond cleavage. Cloning and sequencing of the phn (psiD) genes involved in alkylphosphonate uptake and C-P lyase activity in Escherichia coli B. J Biol Chem. 1990, 265: 4461-4471.
Sung MH, Tanizawa K, Tanaka H, Kuramitsu S, Kagamiyama H, Hirotsu K, Okamoto A, Higuchi T, Soda K: Thermostable aspartate aminotransferase from a thermophilic Bacillus species. Gene cloning, sequence determination, and preliminary x-ray characterization. J Biol Chem. 1991, 266: 2567-2572.
Magarvey N, He J, Aidoo KA, Vining LC: The pdx genetic marker adjacent to the chloramphenicol biosynthesis gene cluster in Streptomyces venezuelae ISP5230: functional characterization. Microbiology. 2001, 147: 2103-2112.
Yoshida KI, Fujita Y, Ehrlich SD: An operon for a putative ATP-binding cassette transport system involved in acetoin utilization of Bacillus subtilis. J Bacteriol. 2000, 182: 5454-5461. 10.1128/JB.182.19.5454-5461.2000.
Yellaboina S, Seshadri J, Kumar MS, Ranjan A: PredictRegulon: a web server for the prediction of the regulatory protein binding sites and operons in prokaryote genomes. Nucleic Acids Res. 2004, 32: W318-320. 10.1093/nar/gkh364.
Yellaboina S, Ranjan S, Vindal V, Ranjan A: Comparative analysis of iron regulated genes in mycobacteria. FEBS Lett. 2006, 580: 2567-2576. 10.1016/j.febslet.2006.03.090.
Ranjan S, Seshadri J, Vindal V, Yellaboina S, Ranjan A: iCR: a web tool to identify conserved targets of a regulatory protein across the multiple related prokaryotic species. Nucleic Acids Res. 2006, 34: W584-587. 10.1093/nar/gkl202.
Ranjan S, Gundu RK, Ranjan A: MycoperonDB: a database of computationally identified operons and transcriptional units in Mycobacteria. BMC Bioinformatics. 2006, 7 (Suppl 5): S9-10.1186/1471-2105-7-S5-S9.
Hosoya S, Yamane K, Takeuchi M, Sato T: Identification and characterization of the Bacillus subtilis D-glucarate/galactarate utilization operon ycbCDEFGHJ. FEMS Microbiol Lett. 2002, 210: 193-199.
Belitsky BR, Sonenshein AL: GabR, a member of a novel protein family, regulates the utilization of gamma-aminobutyrate in Bacillus subtilis. Mol Microbiol. 2002, 45: 569-83. 10.1046/j.1365-2958.2002.03036.x.
The Institute for Genomic Research Comprehensive Microbial Resource. [http://cmr.tigr.org/tigr-scripts/CMR/CmrHomePage.cgi]
Eddy SR: Profile hidden Markov models. Bioinformatics. 1998, 14: 755-763. 10.1093/bioinformatics/14.9.755.
National Center for Biotechnology Information(NCBI) FTP site. [ftp://ftp.ncbi.nih.gov/genomes/Bacteria/]
Cuff JA, Clamp ME, Siddiqui AS, Finlay M, Barton GJ: JPred: a consensus secondary structure prediction server. Bioinformatics. 1998, 14: 892-893. 10.1093/bioinformatics/14.10.892.
Cheng J, Randall AZ, Sweredoski MJ, Baldi P: SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33: W72-76. 10.1093/nar/gki396.
Kelley LA, MacCallum RM, Sternberg MJ: Enhanced genome annotation using structural profiles in the program 3D-PSSM. J Mol Biol. 2000, 299: 499-520. 10.1006/jmbi.2000.3741.
Corpet F: Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16: 10881-10890. 10.1093/nar/16.22.10881.
Young CL, Barker WC, Tomaselli CM, Dayhoff MO: From Atlas of Protein Sequence and Structure. Edited by: Dayhoff MO. 1979, National Biochemical Foundation, Silver Spring, MD, 5 (Suppl 3): 73-93.
Fitch WM, Margoliash E: Construction of phylogenetic trees. Science. 1967, 155: 279-284. 10.1126/science.155.3760.279.
Page RD: TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996, 12: 357-358.
Felsenstein J: PHYLIP-Phylogeny Interface Package. Cladistics. 1989, 5: 164-166.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410.
Fulton DL, Li YY, Laird MR, Horsman BG, Roche FM, Brinkman FS: Improving the specificity of high-throughput ortholog prediction. BMC Bioinformatics. 2006, 28;7: 270-10.1186/1471-2105-7-270.
Acknowledgements
We thank TIGR for making M. smegmatis mc2-155 sequences available through their web portal. Research in AR's laboratory is supported by the grants from Department of Biotechnology, Department of Science and Technology, Indian Council of Medical Research (ICMR), Council of Scientific & Industrial Research (CSIR) NMITLI. VV is supported by the UGC fellowship. KS is supported by the CSIR NMITLI Grant. Authors acknowledge the assistance of Ms. P. Umadevi, SRF and Mr. Rohan Misra, JRF for proof reading the manuscript and making useful suggestions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
VV carried out the operator site prediction, subfamily data analysis, ortholog search and drafted the manuscript. KS participated in the multiple sequence alignment and structure based manual adjustment. AR participated in the study design and coordination. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Vindal, V., Suma, K. & Ranjan, A. GntR family of regulators in Mycobacterium smegmatis: a sequence and structure based characterization. BMC Genomics 8, 289 (2007). https://doi.org/10.1186/1471-2164-8-289
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-8-289