
Abstract
Background
Excessive accumulation of lipids in the adipose tissue is a major problem in the present-day broiler industry. However, few studies have analyzed the expression of adipose tissue genes that are involved in pathways and mechanisms leading to adiposity in chickens. Gene expression profiling of chicken adipose tissue could provide key information about the ontogenesis of fatness and clarify the molecular mechanisms underlying obesity. In this study, Chicken Genome Arrays were used to construct an adipose tissue gene expression profile of 7-week-old broilers, and to screen adipose tissue genes that are differentially expressed in lean and fat lines divergently selected over eight generations for high and low abdominal fat weight.
Results
The gene expression profiles detected 13,234–16,858 probe sets in chicken adipose tissue at 7 weeks, and genes involved in lipid metabolism and immunity such as fatty acid binding protein (FABP), thyroid hormone-responsive protein (Spot14), lipoprotein lipase(LPL), insulin-like growth factor binding protein 7(IGFBP7) and major histocompatibility complex (MHC), were highly expressed. In contrast, some genes related to lipogenesis, such as leptin receptor, sterol regulatory element binding proteins1 (SREBP1), apolipoprotein B(ApoB) and insulin-like growth factor 2(IGF2), were not detected. Moreover, 230 genes that were differentially expressed between the two lines were screened out; these were mainly involved in lipid metabolism, signal transduction, energy metabolism, tumorigenesis and immunity. Subsequently, real-time RT-PCR was performed to validate fifteen differentially expressed genes screened out by the microarray approach and high consistency was observed between the two methods.
Conclusion
Our results establish the groundwork for further studies of the basic genetic control of growth and development of chicken adipose tissue, and will be beneficial in clarifying the molecular mechanism of obesity in chickens.
Similar content being viewed by others

Background
The chicken is an important model organism that bridges the evolutionary gap between mammals and other vertebrates [1]. Research on chickens (Gallus gallus) has had a significant impact on fundamental biology. The domestic chicken also provides a major protein source from meat and eggs for most human populations throughout the world. Its economic importance has made it the focus of numerous research projects, including a recent effort to sequence the entire chicken genome [2].
During the past 80 years, selective breeding has made spectacular progress in both egg and meat production traits. The modern commercial broiler is the product of intensive selection for rapid growth and enhanced muscle mass over many generations. Associated with these successes there have been a number of undesirable traits, such as ascites and lameness, reduced fertility, and reduced resistance to infectious diseases [3]. Selection for rapid growth has been also accompanied by increased fat deposition in these animals [4, 5]. Excessive fat is a major problem for the modern broiler industry, since it not only reduces carcass yield and feed efficiency, but also causes rejection of the meat by consumers [6] and difficulties in processing [7].
With the rapid development of molecular biotechnology, various studies have been performed to investigate the metabolic and genetic mechanisms involved in the regulation of fatness in chickens. Because de novo fatty acid synthesis in birds takes place mainly in the liver, most studies have been performed on hepatic tissue. There have been few analyses of the expression of adipose tissue genes involved in pathways and mechanisms leading to adiposity in chickens. In the present study, Chicken Genome Arrays were used to construct the gene expression profiles of 7-week-old broilers, and to screen genes that are differentially expressed in adipose tissue between lean and fat lines divergently selected over eight generations for high and low abdominal fat weight. Our study will be beneficial in clarifying the molecular mechanisms of obesity in chicken, and these data will contribute to related research on other species.
Results
Characterization of the two chicken lines
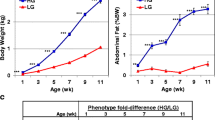
It is clear that the percentages of abdominal fat in the two lines have become very different after selecting for eight generations (Figure 1). In the eighth generation, the AFP is 2.95% in the fat chicken line and 1.55% in the lean line.

The separation of AFP between fat and lean lines. The lean and fat chicken lines derived from a commercial Arbor Acres (AA) grandsire line were created at the Northeast Agricultural University Animal Breeding Center in 1996. They have been selected for 8 generations up to 2004. The selection criteria were the proportion of abdominal fat and levels of very low density lipoproptein (VLDL) in males at 7 weeks of age. Significant differences in AFW and AFP between the two lines were apparent from the 4th generation. Selection was continued for 8 generations, with 15 sires and 4 hens per sire in each line for the G0 to G5 generations and 25 sires and 4 dams per sire in each line for generations G6 to G8. The number of fat line chickens from G1 to G8 was: 82, 88, 75, 81, 80, 78, 179, 165, respectively; and in lean line from G1 to G8: 124, 133, 127, 141, 139, 145, 258, 219, respectively. G represents generation. **p < 0.01, significant difference in AFP between the two lines.
The chickens used in this study differed significantly not in body weight but in abdominal fat weight and percentage of abdominal fat (Figure 2) and were chosen on that basis. AFP in the fat line was three times that in lean line (Table 1).

Comparison of BW, AFW and AFP in chickens used in the study. The birds were kept in similar environmental conditions and had access to feed and water ad libitum, and were fed with a commercial corn-soybean-based diet that met all NRC requirements for 7 weeks. BW was measured after 12 hours fasting, then the birds were slaughtered and the abdominal fat was isolated and weighed. The AFP was calculated as AFP (%) = AFW (g)/BW (g). BW, AFW and AFP data for the ten birds are shown in Table 1. BW is not significantly different between the two lines, but AFW and AFP differ significantly. Data are mean ± SD (n = 5). **p < 0.01, significant difference between fat line and lean line.
Adipose tissue gene expression profile
The pattern of adipose tissue gene expression of chickens at 7 weeks was analyzed by oligonucleotide microarrays. Normalized data were used to analyze the total expressed genes. Depending on the individual bird, 13,234–16,858 probe sets were detected (bird 7-3-16 was excluded from subsequent analysis for the reason given below). Subsequently, the distribution of expression levels of genes in adipose tissue was calculated by JMP4.0 (Figure 3). The genes were ordered according to their mean expression levels; those with expression levels in the highest or lowest 1% were considered highly expressed or the converse. Some of the genes with especially high or low expression levels are named in Tables2 and 3.

The distribution of genes expressed in adipose tissue calculated by JMP. Raw data sets were normalized to total fluorescence, which represents the total amount of cRNA hybridized to a microarray using the software Affymetrix Microarray Suite 4.0. Data sets were excluded if the absolute call (Abs Call) was A (absent) or M (Marginal) according to the detection p-value in all arrays. Only expressed transcripts (the Abs Call was P, present) were used in the analysis. The y-axis is the signal intensity after normalization, which represents the relative expression level of genes. The signal intensity of most genes was under 10,000, that most genes are poorly expressed in chicken adipose tissue at 7 weeks.
We also believe that genes described as "not expressed" can provide useful information about the function of adipose tissue and lipid metabolism in chicken. Table 4 shows some of these genes, selected using the software Affymetrix Microarray Suite 4.0. Many genes allegedly involved in lipid metabolism and obesity were not expressed in adipose tissue in the 7-week-old chickens.
Analysis of consistency within the fat and lean lines
Although there is little difference among individuals in the AFW and AFP within each line, individuals in each line may differ in hereditary molecular characters. By comparing the consistency within each line, the selective effect can be evaluated properly, and chickens that deviate too much from the norm can be excluded from subsequent screening of differentially expressed genes. This makes the analysis of differentially expressed genes more reliable and credible. The cluster analysis results showed that the fat line individuals, except chicken 7-3-16, were more consistent that the lean line ones. Chicken 7-3-16 deviated too much from the other fat line chickens (R<0.6) so it was excluded from the screening of differentially expressed genes. The hierarchical clustering results are shown in Figure 4.

The hierarchical clustering results of five fat line and five lean line chickens. A: hierarchical clustering results for five fat line chickens. B: hierarchical clustering result for five lean line chickens. The data used for clustering were normalized data that excluded the "not expressed" genes. R represents the coefficient of correlation among individuals. The fat chicken line shared about 87% consistency, while the lean chicken line shared about 77% consistency. Sample 7-3-16 shared only 59% consistency with the other fat line chickens, so it was excluded from the subsequent screening of differentially expressed genes.
Identification of differentially expressed genes
In order to identify the differentially expressed genes, Significance Analysis of Microarrays (SAM) was performed on normalized data as described by Tusher et al. [8]. To avoid the low-variance problem in t-tests, SAM uses a statistical method similar to the t-test and estimates the false discovery rate by permutation of repeated measurements [8]. Subsequently, a two-class SAM analysis was performed on the log transformed data matrix (see Materials and Methods). A cutoff value, delta, depending on an arbitrary false positive rate, was chosen to identify genes that were significantly differentially expressed. For this analysis, a delta value of 0.8 was used (Figure 5). This led to the identification of a total of 230 differentially expressed genes: 153 were up-regulated and 77 were down-regulated in fat chickens compared to lean chickens (Figure 6). Highly differentially expressed genes were further selected by the fold change (fat/lean). These differentially expressed genes were mainly involved in lipid metabolism, energy metabolism, signal transduction, immunity and tumorigenesis. Table 5 is a summary of the most representative of these genes.

SAM Plot for Delta = 0.8. Differentially expressed genes were identified from normalized data using the Significance Analysis of Microarrays (SAM) algorithm implemented in a TIGR MultiExperiment Viewer. A cutoff value delta, depending on an arbitrary false positive rate, was chosen to identify significantly differentially expressed genes. For this analysis, a delta value of 0.8 was used, giving a reasonable cutoff of 3.13 and 4.73 in d-scores.

Cluster image of 230 significant differentially expressed genes. A total of 230 differentially expressed genes were identified by SAM algorithm: 153 were up-regulated and 77 were down-regulated in fat chickens compared to lean chickens. Colored bars indicate relative expression levels. Genes that are expressed at higher levels are assigned progressively brighter shades of red, while genes expressed at low levels are assigned shades of green.
Validation of gene expression data by quantitative real-time PCR
To validate the microarray results, we performed quantitative real-time PCR for: propionyl-coenzyme A-carboxylase (PCC), similar to 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma 2 (PBP2), tumor necrosis factor, alpha-induced protein 1 (TNFAIP1), fms-related tyrosine kinase 1 (FLT1), glycerol-3-phosphate dehydrogenase 2 (G3PD), low density lipoprotein-related protein 12 (LRP12), prostaglandin E receptor 3 (PER), suppression of tumorigenicity 7 (ST7), similar to endoplasmic reticulum oxidoreductin 1-Lbeta (ERO1), ataxin 3 (ATXN3), parvin, alpha (PARVA), CWF19-like 2 (CWF19), similar to Stxbp4 protein (Stxbp4), acyl-CoA oxidase (ACO), and suppressor of cytokine signaling 7(SOCS7) (Table 6). In all but one case (TNFAIP1), the real-time RT-PCR fold differences were in complete correspondence with the microarray data. Table 7 compares the microarray and real-time RT-PCR results.
Discussion
Because de novo fatty acid synthesis in birds takes place mainly in the liver, most studies have been performed on hepatic tissues. The expression of some genes involved in lipid synthesis and secretion has been analyzed in the liver of lean and fat chickens. Bourneuf et al. [9] used a cDNA microarray to analyze the expression of genes in liver that are involved in pathways and mechanisms leading to adiposity, and found some genes differentially expressed between lean and fat chickens. Their research indicated that the mechanisms involved in the expression and regulation of lipogenic genes could play a key role in the ontogenesis of fatness in chickens from lean and fat lines. However, few studies have analyzed the functions of adipose tissue leading to adiposity in chickens.
Genome expression analysis aims to provide a broad and unbiased survey of the transcriptome, and requires true global coverage of a complex genome in a single microarray [10]. Significant progress has been made towards this goal and GeneChip microarrays have been improved significantly since they were invented [11].
The Chicken Genome Array, created by Affymetrix Inc. at the end of 2004 as part of the Ensembl annotation attempt at the complete chicken genome sequence (version 1, released May 2004), is a key research tool in chicken genomics. Our study provides, apparently for the first time, a comprehensive analysis of the chicken adipose tissue gene expression profile using this array. The gene expression profile results showed that 13,234–16,858 probe sets were detected in adipose tissue in 7-week-old chickens. Further study revealed that genes directly involved in lipid metabolism, including Spot14, LPL, adrenomedullin, pyruvate dehydrogenase kinase, isoenzyme 4 and the FABP family: FABP 3 (muscle and heart), FABP 4 (adipocyte) and FABP 5 (psoriasis-associated), were highly expressed in adipose tissue; FABP 4 is the most highly expressed gene in this tissue. These genes are mainly involved in fatty acid transport and degradation. Other genes that are closely related to lipid metabolism and obesity by signal transduction or regulation of transcription were also highly expressed: prosaposin (PSAP), retinol binding protein 7 (RBP7), retinoic acid receptor responder, IGFBP 7, thymosin beta 4, BMP7, superoxide dismutase 2 and annexin A5. In addition, many genes involved in the immune response and cytophylaxis, such as CD74 antigen, MHC class I glycoprotein, MHC class II beta 1 domain, MHC class II antigen alpha, lymphocyte antigen 6, beta-2 microglobulin, T-cell leukemia, Hsp70 and Hsp25, were also highly expressed in adipose tissue. The most highly expressed genes (A-FABP, LPL) were apparently not differentially expressed between fat and lean lines. This may not preclude a major involvement of these factors in regulating lipid metabolism in chick adipose tissue, and further work on protein expression is needed to understand the metabolism of this tissue better. Our results demonstrated primarily that the functions of adipose tissue in chicken are important in lipid metabolism by directly or indirectly regulating the synthesis, transport and degradation of lipids. Moreover, adipose tissue may play an important role in chicken immunity and cytophylaxis. These results will be help to clarify the adipose tissue gene expression profile and to choose appropriate methods for further study of these genes.
On the other hand, genes described as "not expressed", including obr,SREBP1, apoB,CCK,CCKR,IGF2 and lipoprotein (APOVLDLII), which have been studied extensively in other species (mainly Homo sapiens and Mus) and shown to affect obesity, were not detected in chicken adipose tissue in the present study. Possible reasons may be: (1) these genes are not expressed in chicken adipose tissue at all, or not at 7 weeks; (2) lipid metabolism in chicken is different from that in mammals. In birds, lipogenesis mainly occurs in the liver and is very limited in adipose tissue [12]. Although those genes were not detected in adipose tissue, they may regulate lipid metabolism indirectly. Another possible reason is gene interactions: the highly expressed genes may inhibit those with low expression levels and the "not expressed" ones when they participate the same biological responses.
Genetic variation in fatness was analyzed using two experimental lines (fat (FL) and lean (LL)) that were divergently selected for abdominal fat weight from a common genetic background. After selection for eight generations, the AFW and AFP differed significantly between the two lines (Figure 2). By consistency analysis within each line using hierarchical clustering of the total expressed genes, heterogeneity among individuals in each line was demonstrated (Figure 4). The hierarchical clustering results were barely satisfactory: the fat line showed higher consistency (about 87%) than the lean (about 77%). The consistency of each line is not sufficiently maintained. There may be two reasons for this. First, an insufficient number of generations have been used for selection, so the purity of each line will increase as selection continues. Second, our target trait is abdominal fat; after selection for eight generations the genes affecting abdominal fat may be better selected, but the genome array detects all genes expressed in adipose tissue, so genes affecting other traits, which have not been divergently selected, will reveal inter-individual differences within each chicken line. The higher consistency of the fat chicken line (about 87% compared to 77% for the lean line) shows that the fat line is purer than the lean line after being selected for eight generations. In addition, to make our study more reliable, the 7-3-16 individual was excluded from subsequent screening of differentially expressed genes because it deviated too much from the others.
Significance Analysis of Microarrays (SAM) has been considered a canonical algorithm for identifying differentially expressed genes in microarray data analysis. A total of 230 differentially expressed genes were screened by SAM. Fifteen genes that were differentially expressed between the two chicken lines were validated by real-time RT-PCR. To validate the results further, in addition to the samples used in the array experiments, four RNA pools (two from the fat line and two from the lean) were used for real-time RT-PCR analysis to reveal the differentially expressed genes more exactly. The results of this validation were encouraging: in all but one case (TNFAIP1), the real-time RT-PCR fold differences correlated well with the microarray data. Rajeevan et al. [13] evaluated the efficiency of real-time RT-PCR for validating the differentially expressed genes identified by microarrays. Their results indicated that genes showing less than a 2-fold difference in expression were not likely to be validated by real-time RT-PCR. Bourneuf et al. [9] considered that confirmation of genes with fold changes <1.7 by microarray analysis and/or <2 by real-time RT-PCR was difficult and likely to produce false positives. According to this report, the fold change of TNFAIP1 in the microarray was -1.6, which is difficult to confirm by real-time RT-PCR, so it is not correct to affirm that the two methods gave contradictory results for this gene.
Our analysis results revealed that the expression levels of some important genes implicated in lipid metabolism were up-regulated in fat chickens: propionyl-coenzyme A-carboxylase, acyl-CoA oxidase, pyruvate dehydrogenase complex, glycerol-3-phosphate dehydrogenase, phosphatidylinositol, lysophospholipase 3, low density lipoprotein-related protein 12, etc. Pyruvate dehydrogenase catalyzes oxidative decarboxylation of pyruvate to form acetyl-CoA, which is a central metabolite [14–16]. Acetyl-CoA enters the Krebs cycle, and is the donor of acetate for synthesis of fatty acids, ketone bodies and cholesterol. The expression level of glycerol-3-phosphate dehydrogenase increased in diet-induced obese animals [17]. Propionyl-coenzyme A-carboxylase is the key enzyme in the catabolism of odd-chain fatty acids, isoleucine, threonine, methionine and valine [18]. Phosphatidylinositol is an important lipid, both as a key membrane constituent and as a participant in essential signaling processes in all plants and animals. These genes regulate lipid metabolism mainly by enhancing the synthesis of fatty acids. In addition, the expression of genes involved in energy metabolism, such as thioredoxin-like 4B, 5-oxoprolyl-peptidase, etc., were markedly down-regulated in fat line chickens, and the expression of genes participating in gluconeogenesis or glycolysis, including glycerol kinase (GK), ATPase, beta 1,3-galactosyltransferase etc. were up-regulated in this line. In fact, it is well recognized that both energy metabolism [19] and glycometabolism [20, 21] are highly related to obesity.
It is interesting that several genes in the protein tyrosine phosphatase pathway were more highly expressed in fat than in lean chickens: fms-related tyrosine kinase 1, receptor tyrosine kinase flk-1/VEGFR-2, a type III receptor tyrosine kinase, protein tyrosine phosphatase-like, member b, protein-tyrosine phosphatase MEG2, etc. This indicates that the protein tyrosine phosphatase pathway is very important in lipid metabolism, and further research is needed to elucidate the molecular mechanism.
Adipogenesis is promoted by the coordinated expression of many transcription factors [22, 23]. De-differentiation, or loss of the adipocyte phenotype, has been observed in response to tumor necrosis factor and transforming growth factor β [24, 25]. We also observed that several genes involved in tumorigenesis were differentially expressed between the two chicken lines. Expression of genes that inhibit tumorigenesis, such as tumor necrosis factor, alpha-induced protein 1, suppression of tumorigenicity 7 and thrombospondin 1, was down-regulated in fat line chickens, while the expression of genes promoting tumor formation, including ret proto-oncogene and turban tumor syndrome, was up-regulated. This indicates that tumorigenesis may be related to obesity. TNFAIP1 was first identified as a tumor necrosis factor a (TNFa) and interleukin-6(IL-6) inducible protein. It is induced rapidly and transiently by TNFa [26]. It is inferred that the in vivo effects of LPS on lipid metabolism are probably mediated by TNFa, which could be secreted by macrophages or by the adipose tissue itself [27, 28]. Albalat et al. [29] suggested that TNFa could be a key modulator of lipid metabolism in fish. Taking these reports and our data into consideration, we presume that TNFa may play a significant role in chicken lipid metabolism.
Conclusion
In conclusion, the chicken adipose tissue gene expression profile was investigated comprehensively in the present study, and specific genes that were differentially expressed in adipose tissue between a fat and a lean chicken line were identified with the aid of the Chicken Genome Array. Genes with high and low expression levels and "not-expressed" genes were identified, and 230 genes that were differentially expressed between the two chicken lines were screened out and confirmed by real-time RT-PCR. These genes were mainly involved in lipid metabolism, energy metabolism, signal transduction, tumorigenesis and immunity. Further analysis indicated that the pyruvate dehydrogenase complex, propionyl-coenzyme A-carboxylase, TNFa and the protein tyrosine phosphatase pathway may play key roles in lipid metabolism. If confirmed in future studies, these patterns of gene expression may contribute to understanding the molecular mechanism of obesity in chickens and provide potential targets for future therapy in humans.
Methods
Lean and fat chicken lines
The NEAU broiler lines divergently selected for abdominal fat content (NEAUHLF) have been selected since 1996 using percentage abdominal fat (%AFW) and plasma very low-density lipoprotein (VLDL) concentration as selection criteria. The G0 generation of the two lines came from the same grandsire line originating from the Arbor Acres breed, which was then divided into two lines according plasma VLDL concentration at 7 weeks. From G1 to G8, birds of each line were raised in two hatches. They were kept in similar environmental conditions and had access to feed and water ad libitum, and were fed with a commercial corn-soybean-based diet that met all NRC requirements (National Research Council, 1994). From hatching to 3 weeks the birds received a starter feed (3,100 kcal ME/kg and 210 g/kg CP), and from 4 weeks to slaughter they were fed a grower diet (3,000 kcal ME/kg and 190 g/kg CP). Plasma VLDL concentrations were measured for all birds at 7 weeks. Abdominal fat weight (AFW) of the male birds in the first hatch was measured and adjusted (%AFW) for body weight (BW) after slaughtering at 7 weeks. Birds with plasma VLDL concentration and AFP lower (lean line) or higher (fat line) than the population average value were selected as candidates for breeding, considering the body weights of male birds and egg production of female birds.
Selection was continued for 8 generations, with 15 sires and 4 hens per sire in the fat line and 10 sires and 4 hens per sire in the lean line for the G0 to G5 generation, then 25 sires and 4 dams per sire in each line for generations G6 through G8. The phenotypic correlation coefficient between body weight and AFW is 0.3789 (P < 0.01), and AFP is 0.1765(P < 0.01) at 7 wk of age based on data of eighth generation population, and the heritability of AFW and AFP were calculated based on the pedigree of eighth generation using the software MTDFREML. The heritability of AFW and AFP at G8 was considered high: AFW is 0.55, and AFP is 0.57.
Sample preparation
Birds were slaughtered at 7 weeks and abdominal fat was isolated, immediately frozen in liquid nitrogen and stored at -80°C. The ten birds used in the present study were chosen by the percentage of abdominal fat (AFP): five had the highest AFP and the other five had the lowest (Table 1). Total RNA was extracted from 300 to 800 mg bulk abdominal adipose tissue using an RNeasy Lipid Tissue Mini Kit (QIAGEN, Valencia, CA) according to the manufacturer's recommendations, and quantified by spectrophotometry. mRNA was isolated using an Oligotex mRNA Mini Kit (QIAGEN, Valencia, CA). cDNA was prepared by oligo-dT-primed reverse transcription using Superscript II (Life Technologies, Inc.). Labeled cRNA probes were prepared using an IVT Labeling Kit (Affymetrix, Inc.) according to the manufacturer's protocol.
Microarrays, hybridization and scanning
The GeneChip Chicken Genome Array used in present study was created by Affymetrix Inc. at the end of 2004, with comprehensive coverage of over 38,000 probe sets representing 32,773 transcripts corresponding to over 28,000 chicken genes. The Chicken Genome Array also contains 689 probe sets for detecting 684 transcripts from 17 avian viruses. Sequence information for this array was selected from the following public data sources: GenBank, UniGene (Build 18; May, 2004) and Ensembl (version 1, released on May, 2004).
Twenty micrograms of cRNA were fragmented at 94°C for 35 min in a 5 × fragmentation buffer containing 200 mM Tris-acetate (pH 8.1), 500 mM KOAc, 150 mM MgOAc. Prior to hybridization, the fragmented cRNA was heated at 95°C for 5 min in 1 × MES hybridization buffer (100 mM MES, 1 M NaCl, 20 mM EDTA, 0.01% Tween20) and 0.1 mg/ml herring sperm DNA, then at 45°C for 5 min, before loading on to the Affymetrix probe array cartridge (Affymetrix, Inc.). After prehybridization in 300 μl 1 × Hybridization Buffer at 45°C for 10 min, the Chicken Genome Arrays were incubated for 16 h at 45°C at constant rotation (60 rpm) using the manufacturer's hybridization buffer. Following hybridization, the arrays were washed with 6 × SSPE-T (0.9 M NaCl/60 mM NaH2PO4/6 mM EDTA/0.01% Tween20) at 25°C on a fluidics station (Affymetrix) for 10 × 2 cycles, then washed with 0.1 M MES/0.1 M NaCl/0.01% Tween20 at 50°C for 4 × 15 cycles. The arrays were stained with a streptavidin-phycoerythrin conjugate (Molecular Probes, Eugene, OR), followed by 10 × 4 wash cycles. To enhance the signals, the arrays were further stained with anti-streptavidin antibody for 10 min followed by a 10-minute staining with the streptavidin-phycoerythrin conjugate. After 15 × 4 additional wash cycles, the arrays were scanned at 560 nm using a confocal scanner (Affymetrix Gene array Scanner3000).
Microarray data normalization
Raw data sets were normalized to total fluorescence, which represents the total amount of cRNA hybridized to a microarray, using the software Affymetrix Microarray Suite 4.0. Data sets were excluded if the absolute call (Abs Call) was A (absent) or M (Marginal) according to the detection p-value in all arrays. Only expressed transcripts (the Abs Call was P, present) were used in further analysis.
The raw data have been deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus and have been assigned the following GEO accession numbers: GSM197816, GSM197817, GSM197818, GSM197819, GSM197820, GSM197821, GSM197822, GSM197823, GSE197824 and GSE197825
Analysis of gene expression profile in 7-week broilers
The gene expression profile was investigated by three methods. First, according to the data normalization and Affymetrix Microarray Suite 4.0 software results, a gene was considered "not detected" by the scanner if its absolute call was A; we call these genes "not expressed" in this paper. The distribution of the expressed transcript (the Abs Call was P) was characterized by JMP4.0 according to its expression level, and genes with high and low expression levels were defined on the basis of this result.
Hierarchical clustering analysis of the variation within each line
The fat and lean chicken lines have been selected for eight generations, so it is expected that the differences among individuals within each line are slight. Hierarchical clustering analysis of the genes expressed in five birds from each line was conducted to measure the consistency within each line. The value of R (coefficient correlation) was calculated and used to evaluate inter-line variation. Any individual that deviated too much from the others was excluded from the subsequent screening for differentially expressed genes.
Identification of differentially expressed genes
Differentially expressed genes were identified from normalized data using the Significance Analysis of Microarrays (SAM) algorithm [8] implemented in a TIGR MultiExperiment Viewer. According to the SAM algorithm, genes are identified as differentially expressed on the basis of expression differences among the sample groups and the consistency of these differences; a score is assigned to each gene on the basis of a change in its expression relative to the standard deviation of repeated measurements for that gene. A gene is deemed 'significant' if its score surpasses a certain threshold. SAM calculates a false discovery rate (FDR), which is the median percentage of genes that are likely to be identified as significantly changed by chance. The threshold can be adjusted to identify different sets of putatively significant genes and the FDR is changed accordingly. The number of significantly changed genes in each experiment depends on a threshold with an acceptable FDR, selected by the investigator [8, 30, 31]. Clustering was achieved using uncentered Pearson correlations and average linkage clustering, and was displayed in TreeView. In the present study, a ?-value of 0.8 was chosen, giving a reasonable cutoff of 3.13 and 4.73 in d-scores.
Real-time quantitative RT-PCR
Oligonucleotide primers were designed to amplify a fragment containing sequences from two adjacent exons in order to avoid contamination with genomic DNA. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was considered to be a stably expressed housekeeping gene and was used as an internal reference gene. The primer pairs used to analyze gene expression and the size of product are shown in Table 6.
cDNA was prepared using an RNA PCR Kit (AMV) Ver3.0 (Takara) starting with 500 ng of total RNA from the following samples: (1) fat line: 7-3-3, 7-3-8, 7-3-15, 7-3-16, 7-3-24; (2) lean line: 7-1-1, 7-1-2, 7-1-4, 7-1-11, 7-1-17; (3) four RNA pools (two from each line), each containing three samples that were not used for the array experiments. First-strand cDNA synthesis was performed at 42°C for 30 min. The 10 μl reaction mixture also contained 1 × AMV reaction buffer, 1 mM each dNTP, 0.125 pmol oligo-dT-adaptor primer, 5 mM MgCl2, 10 U RNase inhibitor, and 2.5 U avian myeloblastosis virus (AMV) reverse transcriptase XL. After incubation, the mixture was heated at 99°C for 5 min to extinguish reverse transcriptase activity. Relative quantification of the expression of selected genes was performed using SYBR® Premix Ex Taq™ (Takara). The reaction mixtures were incubated in an ABI Prism 7300 Sequence Detection System (Applied Biosystems) programmed to conduct one cycle at 95°C for 10 s and 40 cycles at 95°C for 5 s and 60°C for 31 s. The dissociation curves were analyzed using Dissociation Curve 1.0 software (ABI) for each PCR reaction to detect and eliminate possible primer-dimer artifacts. Results (fold changes) were expressed as 2ΔΔCt with ΔΔCt = (Ctij - CtGAPDHj) - (Cti1 - CtGAPDH1), where Ctij and CtGAPDHj are the Ct for gene i and for GAPDH in a pool or a sample (named j), and Cti1 and CtGAPDH1 are the Ct in pool 1 or sample 1, expressed as the standard [9].
Abbreviations
- ACO:
-
acyl-CoA oxidase
- ACX3:
-
acyl-CoA oxidase
- AFW:
-
abdominal fat weight
- AFP:
-
percentage of abdominal fat
- ApoB:
-
apolipoprotein B
- ATXN3:
-
ataxin 3
- BW:
-
body weight
- CCK:
-
cholecystekinin
- CCKR:
-
cholecystekinin receptor
- CWF19L2:
-
CWF19-like 2, cell cycle control (S. pombe)
- ERO1:
-
similar to endoplasmic reticulum oxidoreductin 1-Lbeta
- FABP:
-
fatty acid binding protein
- FLT1:
-
fms-related tyrosine kinase 1
- G3PD:
-
glycerol-3-phosphate dehydrogenase 2
- IGF2:
-
insulin-like growth factor 2
- IGFBP7:
-
insulin-like growth factor binding protein 7
- LPL:
-
lipoprotein lipase
- LRP12:
-
low density lipoprotein-related protein 12
- NEAU:
-
Northeast Agricultural University
- PARVA:
-
parvin, alpha
- PBP2:
-
similar to 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma 2
- PCC:
-
propionyl-coenzyme A-carboxylase
- PER:
-
prostaglandin E receptor 3
- SAM:
-
Significance Analysis of Microarrays
- SOCS7:
-
Suppressor of cytokine signaling 7
- Spot14:
-
thyroid hormone-responsive protein
- SREBP:
-
sterol regulatory element binding proteins
- ST7:
-
suppression of tumorigenicity 7
- Stxbp4:
-
similar to Stxbp4 protein
- TNFAIP1:
-
Tumor necrosis factor, alpha-induced protein 1.
References
Hillier LW, Miller W, Birney E, Warren W, Hardison RC, Ponting CP, Bork P, Burt DW, Groenen MA, Delany ME, Dodgson JB, Chinwalla AT, Cliften PF, Clifton SW, Delehaunty KD, Fronick C, Fulton RS, Graves TA, Kremitzki C, Layman D, Magrini V, McPherson JD, Miner TL, Minx P, Nash WE, Nhan MN, Nelson JO, Oddy LG, Pohl CS, Randall-Maher J, Smith SM, Wallis JW, Yang SP, Romanov MN, Rondelli CM, Paton B, Smith J, Morrice D, Daniels L, Tempest HG, Robertson L, Masabanda JS, Griffin DK, Vignal A, Fillon V, Jacobbson L, Kerje S, Andersson L, Crooijmans RP, Aerts J, van der Poel JJ, Ellegren H, Caldwell RB, Hubbard SJ, Grafham DV, Kierzek AM, McLaren SR, Overton IM, Arakawa H, Beattie KJ, Bezzubov Y, Boardman PE, Bonfield JK, Croning MD, Davies RM, Francis MD, Humphray SJ, Scott CE, Taylor RG, Tickle C, Brown WR, Rogers J, Buerstedde JM, Wilson SA, Stubbs L, Ovcharenko I, Gordon L, Lucas S, Miller MM, Inoko H, Shiina T, Kaufman J, Salomonsen J, Skjoedt K, Wong GK, Wang J, Liu B, Wang J, Yu J, Yang H, Nefedov M, Koriabine M, Dejong PJ, Goodstadt L, Webber C, Dickens NJ, Letunic I, Suyama M, Torrents D, von Mering C, Zdobnov EM, Makova K, Nekrutenko A, Elnitski L, Eswara P, King DC, Yang S, Tyekucheva S, Radakrishnan A, Harris RS, Chiaromonte F, Taylor J, He J, Rijnkels M, Griffiths-Jones S, Ureta-Vidal A, Hoffman MM, Severin J, Searle SM, Law AS, Speed D, Waddington D, Cheng Z, Tuzun E, Eichler E, Bao Z, Flicek P, Shteynberg DD, Brent MR, Bye JM, Huckle EJ, Chatterji S, Dewey C, Pachter L, Kouranov A, Mourelatos Z, Hatzigeorgiou AG, Paterson AH, Ivarie R, Brandstrom M, Axelsson E, Backstrom N, Berlin S, Webster MT, Pourquie O, Reymond A, Ucla C, Antonarakis SE, Long M, Emerson JJ, Betran E, Dupanloup I, Kaessmann H, Hinrichs AS, Bejerano G, Furey TS, Harte RA, Raney B, Siepel A, Kent WJ, Haussler D, Eyras E, Castelo R, Abril JF, Castellano S, Camara F, Parra G, Guigo R, Bourque G, Tesler G, Pevzner PA, Smit A, Fulton LA, Mardis ER, Wilson RK: Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004, 432: 695-716. 10.1038/nature03154.
Wicker T, Robertson JS, Schulze SR, Feltus FA, Magrini V, Morrison JA, Mardis ER, Wilson RK, Peterson DG, Paterson AH, Ivarie R: The repetitive landscape of the chicken genome. Genome Res. 2005, 15: 126-136. 10.1101/gr.2438005.
Burt DW: Applications of biotechnology in the poultry industry. Worlds Poult Sci J. 2002, 58: 5-13. 10.1079/WPS20020002.
Nones K, Ledur MC, Ruy DC, Baron EE, Melo CM, Moura AS, Zanella EL, Burt DW, Coutinho LL: Mapping QTLs on chicken chromosome 1 for performance and carcass traits in a broiler x layer cross. Anim Genet. 2006, 37: 95-100. 10.1111/j.1365-2052.2005.01387.x.
Havenstein GB, Ferket PR, Scheideler SE, Rives DV: Carcass composition and yield of 1991 vs 1957 broilers when fed "typical" 1957 and 1991 broiler diets. Poult Sci. 1994, 73: 1795-1804.
Kessler AM, Snizek PN, Brugalli I: Manipulação da quantidade de gordura na carcaça de frangos In: Anais da Conferência APINCO de Ciência e Tecnologia Avícolas. 2000, APINCO, Campinas, SP, Brazil Press
Chambers JR: Genetics of growth and meat production in chickens. Poultry breeding and genetics. Edited by: Crawford RD. 1990, Amsterdam: Elsevier Press
Tusher VG, Tibshirani R, Chu G: Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001, 98: 5116-5121. 10.1073/pnas.091062498.
Bourneuf E, Hérault F, Chicault C, Carré W, Assaf S, Monnier A, Mottier S, Lagarrigue S, Douaire M, Mosser J, Diot C: Microarray analysis of differential gene expression in the liver of lean and fat chickens. Gene. 2006, 372: 162-170. 10.1016/j.gene.2005.12.028.
Zhu T: Global analysis of gene expression using GeneChip microarrays. Curr Opin Plant Biol. 2003, 6: 418-425. 10.1016/S1369-5266(03)00083-9.
Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittman M, Wang C, Kobayashi M, Horton H, Brown EL: Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol. 1996, 14: 1675-1680. 10.1038/nbt1296-1675.
Saadoun A, Leclercq B: In vivo lipogenesis of genetically lean and fat chickens: effects of nutritional state and dietary fat. J Nutr. 1987, 117: 428-435.
Rajeevan MS, Vernon SD, Taysavang N, Unger ER: Validation of array-based gene expression profiles by real-time (kinetic) RT-PCR. J Mol Diagn. 2001, 3: 26-31.
Reed LJ: Regulation of mammalian pyruvate dehydrogenase complex by a phosphorylation-dephosphorylation cycle. Curr Top Cell Regul. 1981, 18: 95-106.
Reed LJ: A trail of research from lipoic acid to alpha-keto acid dehydrogenase complexes. J Biol Chem. 2001, 276: 38329-38336. 10.1074/jbc.R100026200.
Martin E, Rosenthal RE, Fiskum G: Pyruvate dehydrogenase complex: metabolic link to ischemic brain injury and target of oxidative stress. J Neurosci Res. 2005, 79: 240-247. 10.1002/jnr.20293.
Lopez IP, Marti A, Milagro FI, Zulet Md Mde L, Moreno-Aliaga MJ, Martinez JA, De Miguel C: DNA microarray analysis of genes differentially expressed in diet-induced (cafeteria) obese rats. Obes Res. 2003, 11: 188-194.
Van Greevenbroek MM, Vermeulen VM, De Bruin TW: Identification of novel molecular candidates for fatty liver in the hyperlipidemic mouse model, HcB19. J Lipid Res. 2004, 45: 1148-1154. 10.1194/jlr.M400062-JLR200.
Labib M: The investigation and management of obesity. J Clin Pathol. 2003, 56: 17-25. 10.1136/jcp.56.1.17.
Sun Y, Liu S, Ferguson S, Wang L, Klepcyk P, Yun JS, Friedman JE: Phosphoenolpyruvate carboxykinase overexpression selectively attenuates insulin signaling and hepatic insulin sensitivity in transgenic mice. J Biol Chem. 2002, 277: 23301-23307. 10.1074/jbc.M200964200.
Boden G, Cheung P, Stein TP, Kresge K, Mozzoli M: FFA cause hepatic insulin resistance by inhibiting insulin suppression of glycogenolysis. Am J Physiol Endocrinol Metab. 2002, 283: E12-19.
MacDougald O, Lane M: Transcriptional regulation of gene expression during adipocyte differentiation. Annu Rev Biochem. 1995, 64: 345-373. 10.1146/annurev.bi.64.070195.002021.
Gregoire F, Smas CM, Sul HS: Understanding adipocyte differentiation. Physiol Rev. 1998, 78: 783-809.
Ron D, Brasier AR, McGehee RE, Habener JF: Tumor necrosis factor-induced reversal of adipocyte phenotype of 3T3-L1 cells is preceded by a loss of nuclear CCAAT/Enhancer binding protein (C/EBP). J Clin Invest. 1992, 89: 223-233.
Nadler ST, Stoehr JP, Schueler KL, Tanimoto G, Yandell BS, Attie AD: The expression of adipogenic genes is decreased in obesity and diabetes mellitus. Proc Natl Acad Sci USA. 2000, 97: 11371-11376. 10.1073/pnas.97.21.11371.
Wolf FW, Marks RM, Sarma V, Byers MG, Katz RW, Shows TB, Dixit VM: Characterization of a novel tumor necrosis factor-alpha-induced endothelial primary response gene. J Biol Chem. 1992, 267 (2): 1317-1326.
Sewter CP, Digby JE, Prins J, O'Rahilly S: Regulation of tumor necrosis factor-alpha release from human adipose tissue in vitro. J Endocrinol. 1999, 163: 33-38. 10.1677/joe.0.1630033.
MacKenzie S, Planas JV, Goetz FW: LPS-stimulated expression of a tumor necrosis factor-alpha mRNA in primary trout monocytes and in vitro differentiated macrophages. Dev Comp Immunol. 2003, 27: 393-400. 10.1016/S0145-305X(02)00135-0.
Albalat A, Liarte C, MacKenzie S, Tort L, Planas JV, Navarro I: Control of adipose tissue lipid metabolism by tumor necrosis factor-alpha in rainbow trout (Oncorhynchus mykiss). J Endocrinol. 2005, 184: 527-534. 10.1677/joe.1.05940.
Chowers I, Gunatilaka TL, Farkas RH, Qian J, Hackam AS, Duh E, Kageyama M, Wang C, Vora A, Campochiaro PA, Zack DJ: Identification of novel genes preferentially expressed in the retina using a custom human retina cDNA microarray. Invest Ophthalmol Vis Sci. 2003, 44: 3732-3741. 10.1167/iovs.02-1080.
Hedtjarn M, Mallard C, Eklind S, Gustafson-Brywe K, Hagberg H: Global gene expression in the immature brain after hypoxia-ischemia. J Cereb Blood Flow Metab. 2004, 24: 1317-1332. 10.1097/00004647-200412000-00002.
Acknowledgements
The authors would like to acknowledge the members of the Poultry Breeding Group of the College of Animal Science and Technology in Northeast Agricultural University for managing the birds and collecting data. This research was supported by a National Natural Science Foundation Key Project (No.30430510), the Program for New Century Excellent Talents in University (No. NCET- 04-0343), and the National Basic Research Program (No.2006CB102105).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
HBW contributed to the design of the microarray, participated in the interpretation of data, performed the verification of microarray data, drafted and wrote the manuscript. HL co-led the conception and design of the study, contributed to the design of the microarray, participated in the interpretation of data, and contributed to writing the manuscript. QGW participated in the design of the study and interpretation of data, and contributed to writing the manuscript. XYZ carried out analysis and interpretation of microarray data and assisted in production of the microarray. SZW participated in the design of the study and contributed to writing the manuscript. YXW participated in the design of the study and contributed to writing the manuscript. XPW performed microarray hybridizations, generated microarray data, and assisted in production of the microarray. All authors submitted comments on drafts, and read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wang, HB., Li, H., Wang, QG. et al. Profiling of chicken adipose tissue gene expression by genome array. BMC Genomics 8, 193 (2007). https://doi.org/10.1186/1471-2164-8-193
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-8-193