
Abstract
Background
Members of the C-type lectin domain (CTLD) superfamily are metazoan proteins functionally important in glycoprotein metabolism, mechanisms of multicellular integration and immunity. Three genome-level studies on human, C. elegans and D. melanogaster reported previously demonstrated almost complete divergence among invertebrate and mammalian families of CTLD-containing proteins (CTLDcps).
Results
We have performed an analysis of CTLD family composition in Fugu rubripes using the draft genome sequence. The results show that all but two groups of CTLDcps identified in mammals are also found in fish, and that most of the groups have the same members as in mammals. We failed to detect representatives for CTLD groups V (NK cell receptors) and VII (lithostathine), while the DC-SIGN subgroup of group II is overrepresented in Fugu. Several new CTLD-containing genes, highly conserved between Fugu and human, were discovered using the Fugu genome sequence as a reference, including a CSPG family member and an SCP-domain-containing soluble protein. A distinct group of soluble dual-CTLD proteins has been identified, which may be the first reported CTLDcp group shared by invertebrates and vertebrates. We show that CTLDcp-encoding genes are selectively duplicated in Fugu, in a manner that suggests an ancient large-scale duplication event. We have verified 32 gene structures and predicted 63 new ones, and make our annotations available through a distributed annotation system (DAS) server http://anz.anu.edu.au:8080/Fugu_rubripes/ and their sequences as additional files with this paper.
Conclusions
The vertebrate CTLDcp family was essentially formed early in vertebrate evolution and is completely different from the invertebrate families. Comparison of fish and mammalian genomes revealed three groups of CTLDcps and several new members of the known groups, which are highly conserved between fish and mammals, but were not identified in the study using only mammalian genomes. Despite limitations of the draft sequence, the Fugu rubripes genome is a powerful instrument for gene discovery and vertebrate evolutionary analysis. The composition of the CTLDcp superfamily in fish and mammals suggests that large-scale duplication events played an important role in the evolution of vertebrates.
Similar content being viewed by others


Background
The superfamily of proteins containing the C-type (Ca-dependent) lectin-like domain (CTLD) is a large group of extracellular proteins characterized by evolutionary flexibility and functional versatility [1, 2]. Its members have been extensively studied because of their involvement in diverse physiological processes, and their ability to bind selectively a wide variety of ligands. As the superfamily name suggests, carbohydrates (in various contexts) are primary ligands for CTLDs and this binding is Ca-dependent [3]. However, the fold has been shown to specifically bind proteins [4], lipids [5] and inorganic compounds including CaCO3 and ice [6–9]. In several cases, the domain is multivalent and may bind both protein and sugar [10–12].
Three studies using the whole-genome approach have been published analyzing the distribution of the superfamily in C. elegans [13], D. melanogaster [14] and human [15]. An early study [2] attempted to generalize findings on vertebrate CTLD-containing proteins (CTLDcps), and to classify them into groups. This classification included 7 groups and, although not sufficient to describe later known CTLDcps even in mammals and other vertebrates, has been widely used by CTLD researchers. The recent work of Drickamer and Fadden [15] provided an updated classification of human and mouse CTLDcps, based on a comprehensive analysis of CTLDcps encoded by the human genome; this comprises 14 groups. These whole-genome studies and genome annotation projects demonstrated the relative abundance of CTLDcps and importance of the domain.
Known fish C-type lectins
A number of fish CTLDcp sequences have been reported separately in the literature and public sequence databases. The best-studied and most distinct set are serum antifreeze proteins (AFPs) from several cold-water-living species [7, 16, 17]. These sequences consist mostly of just a CTLD, and were classified as group VII members based on domain architecture. A three-dimensional structure of the sea raven antifreeze protein has been determined experimentally [18].
Apart from AFPs, several other soluble bony-fish CTLDcps have been described: 5 isoforms of Salmo salar serum lectin (SSL) [19], three collectins from different Cyprinidae carp family species [20], skin mucus protein AJL-2 [21] and two C-type lectins (eCL-1 and eCL-2) from gills of Japanese eel [22], two lectins from rainbow trout liver [23], a carp lectin [24], goldfish lectin OL-1 (GI: 26000685, unpublished), and a liver lectin from Gillichthys mirabilis (long-jawed mudsucker), annotated as "mannose receptor C" [25].
Known membrane-bound CTLDcps from bony fishes include a polycystic kidney disease protein 1 (PKD1) orthologue from Fugu [26], a rainbow trout Kupffer cell receptor homologue [27], and a set of putative killer cell receptors (KLR) identified recently [28]. Although predicted coding sequences for CTLDcps from winter flounder (GI:28394504, unpublished) and medaka fish [29] do not contain a recognizable transmembrane (TM) domain, based on CTLD sequence and, in the case of the medaka CTLDcp, domain structure, they should be assigned to group II, as the absence of TM regions may be a result of incomplete prediction.
The only known CTLDcp sequence from cartilaginous fishes is a tetranectin homologue from reef shark cartilage [30].
Fugu genome sequence
The Fugu rubripes genome, available since 2002 [31], is the second vertebrate genome sequenced. It is 8 times smaller than the human genome and is proving to be an effective instrument in analyzing the human genome because of its compactness, low content of repetitive elements and the relatively large evolutionary distance between fish and mammals, which is estimated to be about 430 Myr [32].
Currently three versions of the Fugu rubripes genome assembly are publicly available. The second version of the assembly (v.2), constructed from 4.1 million sequencing reads (5.4 X sequence coverage), was reported in the original publication announcing the completion of the Fugu rubripes genome sequencing [31]. The third version (v.3) was released in August 2002, has slightly better coverage (5.7X) and improved scaffold contiguity. Sequence data for all three assembly versions can be downloaded from the Joint Genome Institute web site [33]. The JGI site and the EnsEMBL web site [34] are the two main portals to the Fugu rubripes genome annotation. Although EnsEMBL and JGI annotations and genome browsers are different, they share the same gene and transcript structure predictions created by the EnsEMBL pipeline.
Several analyses of the draft Fugu genome sequence targeting different protein families have been published recently [35–39], which showed its usefulness for evolutionary and functional studies as well as gene discovery. Here we present an analysis of the presence of the CTLD superfamily in the draft assembly of the Fugu rubripes genome.
Results
Comparison of assembly versions 2 and 3
At the time this study was started, annotation of the v.3 assembly was not yet published; hence, most of our analysis was done with v.2 of the assembly and later mapped to the v.3 assembly. From our experience, there is no substantial difference between v.2 and v.3 assemblies in the amount of sequence information and its quality, although the v.3 assembly contains longer scaffolds due to more extensive linkage. Despite very high similarity at the sequence level, the v.3 assembly annotation contains no history information that would provide links between contigs, genes, and transcripts in the second and the third versions of the assembly. None of the stable identifiers for genes, transcripts or peptides from v.2 are present in v.3. This information cannot be generated by usual procedures used in EnsEMBL (e.g. ID Mapping Application, which is a part of EnsEMBL Java APIs) and has to be obtained by sequence comparisons. This lack of correspondence creates difficulties for the sequence analyzer and end point reader. To facilitate analysis and allow comparison, references to feature identifiers for both of the assemblies are given in Table 1.
Protein database searches
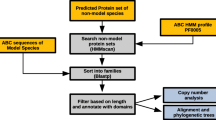
Due to almost complete lack of cDNA or EST sequences for Fugu rubripes, most of the EnsEMBL gene structure predictions are based on homology with known protein sequences from other organisms, mostly mammals. We expected a significant fraction of CTLDcps to be conserved between fish and human, and, therefore, to be predicted correctly by EnsEMBL in the Fugu genome. So our first approach to detecting Fugu CTLDcps was to search a sequence database of predicted Fugu proteins with a hidden Markov model (HMM) for the CTLD. This search returned 69 significant matches. Some of the identified genes had a description assigned to them, apparently derived from the description of the sequence they were found to be homologous to. These descriptions, however, could not be used as a reliable basis for assigning orthology and paralogy relationships. For example, a sequence, which we later identified as an Endo180 orthologue (SINFRUG00000058766 in v.2 assembly annotation) is described as "80 KDA SECRETORY PHOSPHOLIPASE A2 RECEPTOR PRECURSOR PLA2", while another gene, which we designated as an aggrecan orthologue (SINFRUG00000069597 in v.2 annotation) was annotated as "ADRENOLEUKODYSTROPHY PROTEIN (ALDP)". Therefore, we reviewed domain architecture and sequence similarity matches for each of the sequences found to verify phylogenetic relationships.
Homology detection
The results of Inparanoid [40] comparison (see Methods) of all human to all Fugu CTLDcps were used to initially cluster the set of Fugu proteins and detect approximate orthology/paralogy links. Inparanoid has an important advantage over phylogenetic tree reconstruction software, as it does not require a multiple alignment of sequences but creates a distance matrix of the local pairwise alignments. This method assigned putative human orthologues to 25 Fugu proteins. Orthology relationships for the other 44 sequences from the set were established by individual analyses.
Revision of CTLDcp gene structure predictions
While analyzing phylogenetic relationships predicted by Inparanoid, we discovered several systematic and sporadic mistakes in the EnsEMBL gene predictions. The most widespread mistake was a failure to include exons encoding TM domains into gene structure prediction. Consequently, almost all EnsEMBL-predicted Fugu CTLDcps were soluble proteins, whereas very few human CTLDcps are. Simple comparison with the GenScan [41] features overlapping the CTLD-encoding genes showed that absence of TM domains is a result of coding sequence (CDS) mis-prediction rather than a fundamental difference in Fugu CTLDcps. GenScan predictions, in turn, could not be used as a basis for our analysis because they sometimes contain regions that are absent from human or mouse orthologues, and often merge neighboring genes. Another general problem was observed with proteins that had a previously unknown domain architecture (see below). In such cases individual domains were split into separate gene models.
In addition to these systematic problems, there were multiple sporadic ones. For example, our analysis of the Fugu genome shows that, similarly to the human and mouse genomes, the selectin cluster is well conserved and contains all three selectin genes in tandem (SELE, SELL, SELP), located on scaffolds 1045 (32046–41921) and 166 (83937–93826) in the v.2 and v.3 Fugu genome annotations, respectively. However, the EnsEMBL annotation contains a prediction of two overlapping genes (v.2: SINFRUG00000085188 and SINFRUG00000085187; v.3: SINFRUG00000123102 and SINFRUG00000123101), one of which is located in the intron of the other (Figure 1).

Fugu genome sequence and annotation. A. Fugu selectin gene cluster annotation in the EnsEMBL database (v.2 annotation is shown, v.3 annotation is almost identical to v.2). Gene models predicted by us based on comparison with human selectins are shown in the grey box. As shown, the CTLD is encoded by the 5' exon in fSELP, fSELL and fSELE; the TM segment is encoded by the 3' exon. EnsEMBL predicted transcripts, GenScan predictions and similarity features are shown on the tracks below. Stable IDs for EnsEMBL transcripts are given. The TMHMM track shows ORFs encoding TransMembrane regions predicted by the TMHMM program (see Methods). B. Fragments of group VI genes found on various scaffolds. CTLD numbers indicate sequential number of CTLD in full-length MManR, while numbers for the CTLD in the partial sequences indicate the MManR CTLD sequence they are most closely homologous to.
To solve these problems, we had to manually revise the predicted structure for all genes encoding proteins detected by the protein-level searches, and correct them using supporting evidence available in the EnsEMBL database, as well as additional evidence generated by us. The latter included similarity features produced by genome-wide GeneWise and BLAST searches with CTLD profiles and sequences, transmembrane domain predictions, and similarity matches to the complete sequence of supposed human or mouse orthologues.
As the final stage of the CTLDcp identification process, we performed a set of DNA-level comparisons to ensure that the CTLD-containing loci that are not covered by EnsEMBL-predicted genes, or for which transcript predictions are wrong and, thus, not detectable by protein database searches, were not omitted from the analysis. This "quality control" step led to identification of an additional set of 25 well conserved CTLDcps, which had both new and known domain architectures, as well as additional individual CTLDs, which were merged with neighboring CTLDcp loci if appropriate.
Groups of Fugu CTLDcps
After all these searches, we had identified a set of 94 Fugu rubripes loci encoding CTLDcps (Table 1), which in total contain 173 individual CTLDs, including PTR/Link-type CTLDs [42]. Fugu CTLDcps were named according to their human orthologues, established on the basis of domain composition and sequence similarities. Where more than one homologue was present in Fugu, a name was produced by adding a suffix of the form "-FXX", where XX is a sequential number of the paralogue, to the name of the closest human homologue. Predicted CTLDcps that do not have homologues among the known CTLDcps have identifiers of the form ANZ000. A few of these novel genes were orthologous to loci in other vertebrate genomes supported by expression data, but otherwise are un-characterized, and were assigned descriptive names (CBCP, Bimlec, SEEC, CETM, NLSLH).
We have clustered Fugu CTLDcps using the classification scheme for human CTLDcps based on domain composition; this comprises 14 groups [15]. Link/PTR-domain-containing CTLDcps, apart from hyalectans, were placed into a separate group. Among the Fugu CTLDcps that did not have mammalian homologues we detected a distinct group of soluble dual-CTLD sequences, which we have called F1 (Figure 2). The remainder of the Fugu-specific CTLDcps were assigned to the U (Unclassified) group. Gene structure prediction for members of the U group is the lowest in quality, due to lack of supporting evidence apart from similarity to CTLD sequence profiles and GenScan predictions.

CTLDcps with novel domain architectures. Fugu CTLD-containing proteins, which do not fit into the existing CTLDcp classification are shown. Domain abbreviations are explained in the text. Roman numbers near names indicate suggested new group names for the new Fugu sequences, which also have new predicted human homologues. C-terminal CTLDs of DEC205-FUSE that are not present in the v.3 assembly are shown in light pink.
All but two groups of human CTLDcps have detectable representatives in the Fugu genome (Figure 3, Table 1). We did not detect any orthologues for groups V (NK cell receptors) and VII (lithostathine/Reg family). The member repertoire for most of the other groups is very well conserved between Fugu and human. However, groups II and III, which include some of the best-studied mammalian CTLDcps, have a significantly different member composition in Fugu. In summary:

Phylogenetic relationships between fish and human CTLDs. A phylogenetic tree built on a ClustalW alignment of a 95% non-redundant collection of predicted Fugu CTLDs and known human and fish CTLDs. Link domains and group VI CTLDs were excluded from the alignment. The tree was built by the neighbor-joining method with 100 bootstrap trials using the ClustalW program. PhyloDraw was used to draw the radial cladogram shown. Branches containing CTLDs from CTLDcps belonging to the same group are shaded; group numbers are marked. Lower case prefixes in the identifiers indicate taxonomic origin: h – Homo sapiens, f – Fugu rubripes, zbrfs – Danio rerio (zebrafish), g – Gillichthys mirabilis, gldhs – Carassius auratus (goldfish), carp – Cyprinus carpio (common carp), rsmlt – Osmerus mordax (rainbow smelt), slmn – Salmo salar (Atlantic salmon), wfldr – Pseudopleuronectes americanus (winter flounder), ahrng – Clupea harengus (Atlantic herring), servn – Hemitripterus americanus (sea raven), jpeel – Anguilla japonica (Japanese eel), medak – Oryzias latipes (Japanese medaka), c – Paralabidochromis chilotes (cichlid fish).
Group I
All four members of the lectican group that are present in human have orthologues in the Fugu genome. Each of the Fugu hyalectan genes is duplicated. One of the Fugu versican copies is split between two scaffolds in the v.2 assembly.
Group II
We found only one representative of the asialoglycoprotein receptor (ASGR) family in Fugu (HML2), while in human this family has 3 members encoded by a gene cluster on Ch 17 (ASGR1, ASGR2, HML2). The Fugu sequence was identified as an HML2 orthologue by phylogenetic analysis based on the alignment of CTLD sequences. Another clearly identifiable member of group II is the orthologue of scavenger cell receptor C-type lectin (SRCL), which is duplicated in Fugu and is 50% identical to the human SRCL. The rest of the group II Fugu CTLDcps (DC-SIGN-F1 – DC-SIGN-F8, XLCMCL) do not have clearly identifiable orthologues among known human CTLDcps, although phylogenetic analyses based on CTLD sequence alignment indicate that they are homologous to members of the group II subgroup containing DC-SIGN, Mincle and Dectin-2, which also appear as top hits in BLAST searches. However, this subset of group II Fugu sequences co-clusters in phylogenetic trees and is not similar enough to any tetrapod sequence to establish orthology. Four of the sequences (DC-SIGN-F2, DC-SIGN-F3, DC-SIGN-F4, DC-SIGN-F5) are located in a cluster on scaffold 75 in the v.3 assembly. Two members of the subgroup (DC-SIGN-F1 and DC-SIGN-F6) have unstable placements in phylogenetic trees, and may appear on a branch containing human/mouse group V sequences, if the latter are included in the alignment. This association is, however, unstable and may be due to mistakes in CDS prediction or phylogeny reconstruction. Alternatively, it is possible that these sequences are homologous to the common predecessor of group V and group II CTLDcps.
Group III
Although Fugu has two collectins, there are no orthologues for mannose binding proteins (MBPs) or pulmonary surfactant proteins (PSP), which are the best studied members of the group in human. Both of the Fugu collectins (COLEC10, MGC3279) are well conserved compared with their human orthologues and co-cluster with them in phylogenetic trees. No functional information is available for the novel collectin MGC3279, which was discovered in a large-scale cDNA sequencing project and maps to chromosome 2p25.3 in the v.31 NCBI assembly of the human genome, but the exceptionally high level of conservation between human and fish (~76% identity) strongly suggests that it is functional and important in both organisms. COLEC10 (collectin liver 1, CL-L1) was originally reported as limited to birds and mammals [43] based on the Zoo-blot analysis.
Group IV
As already mentioned, all three selectin genes found in other vertebrates are present in Fugu and have the same genome arrangement.
Group VI
We identified Fugu orthologues for all four human group VI members: macrophage mannose receptor (MManR), DEC-205 (CD205), phospholipase A2 receptor (PLA2R) and Endo180. In addition, there are 5 sequences (MManR-F1 – MManR-F5) showing high similarity to members of the group, four of which do not contain the minimal number of CTLDs (8) present in the known group VI sequences (Figure 1). The fragments belong to at least 3 group VI CTLDcps. Although the most parsimonious explanation of the presence of these fragments would be that each of the genes encoding an eight-CTLD molecule (MManR, Endo180 and PLA2R) was copied in a chromosome or genome duplication event, phylogenetic analysis indicates that all five sequences are paralogues of the MManR gene, which, thus, appears to have been duplicated several times.
There is one more potential group VI member in Fugu. A GenScan-predicted DEC-205-FUSE gene, which was assigned to the U group, encodes a large protein (~2000 residues) with multiple CTLDs clustered in two groups: 5 at the N terminus and 10 (7 in v.3 assembly) at the C terminus, with an LCCL domain [named after its presence in Limulus factor C, cochlear protein Coch-5b2, and late gestation lung protein Lgl1; [44]] and a coagulation factor 5/8 C-terminal domain (discoidin domain, FA58C) lying in the middle separating the two groups of CTLDs (see Figure 2). EnsEMBL predictions in the DEC-205-FUSE locus in both versions of the assembly contain a large (4 kb) intron in the region encoding LCCL, FA58C and 8 CTLDs at the center of the molecule. LCCL has been observed in a combination with a CTLD in an invertebrate protein [45], while FA58C has been found only in combination with LCCL, but not with a CTLD [46]. Although there is no supporting cDNA or EST evidence for our predicted gene structure, the small intron sizes (e.g. LCCL is separated by 135 bp from the downstream CTLD) and well-conserved CTLDs, suggest that the prediction may be correct if the corresponding region was correctly assembled. There is no orthologue for DEC-205-FUSE in the human genome.
Groups VIII and IX
We have identified Fugu orthologues for all known human members of groups VIII and IX. One member in each of these groups is duplicated in Fugu (Layilin and Tetranectin).
Group X
In addition to the PDK1 orthologue, which was identified previously [26], there is at least one more putative group X member, orthologous to a recently identified human and mouse PKD1 homologue PKD1L2 [47]. It is interesting to note that the GenScan-predicted Fugu PKD1L2 sequence is very similar to the sequences of human and mouse PKD1L2 cDNAs, even though the latter were deposited in GenBank at the beginning of June 2003 – after GenScan prediction. This example indicates that ab initio GenScan predictions on the Fugu genome can be very accurate.
Group XII
We found a single sequence resembling mammalian eosinophil major basic proteins (EMBPs) in Fugu (EMBPL). Although the similarity between the mammalian and the fish sequences is very low (~30% identity), several observations suggest that the Fugu EMBP-like sequence is an orthologue of one of the two mammalian genes. First, the overall domain architecture of the fish protein is similar to that of the EMBPs. Although the fish CTLD has a neutral pI (7.1), it is preceded by a 30-residue peptide with a predicted pI of 3.62, analogous to the longer acidic neck of the mammalian EMBPs. In the existing classification [15], the presence of the acidic neck is used as the defining feature of group XII distinguishing it from the other group of single-CTLD soluble proteins (VII). Second, in the phylogenetic trees EMBPL usually appears on the same branch as EMBPs (e.g. Figure 3), albeit with low bootstrap support. Third, the exon-intron structure of the CTLD region is identical in fish and mammalian genes. Finally, the fish sequence has the same rare substitution in the fourth position of the WIGL motif as the EMBP sequences (discussed in more detail below).
Group XIV
The thrombomodulin family is fully represented in Fugu, with one gene duplicated (C1qRP). In addition, a novel member of the family conserved between Fugu and mammals was identified, which we named CETM (for CTLD, EGF, TransMembrane domain) (see Figure 2). Multiple full-length cDNA and EST sequences from different tissues found in nucleotide databases indicate that mammalian CETM is ubiquitously expressed. The sequence of the CETM CTLD contains a putative carbohydrate-binding motif (EPN), which is normally associated with mannose specificity.
Antifreeze-protein-like sequences
We identified two putative CTLDcp-encoding loci with similarity to antifreeze proteins: AFPL-F1 and AFPL-F2 (antifreeze-protein-like), almost identical to each other and positioned in tandem on scaffold 1930 in the v.2 assembly. In v.3 of the assembly, the AFPL-encoding region was rearranged and one of the AFPL loci disappeared. The intron-exon structure of the CTLD-encoding region is identical to the structure of the sea raven antifreeze protein gene [48] with three intron insertions (upstream of C1, downstream of the WIGL motif, and between C2 and C3 [49]), and very similar to the structure of the Salmo salar serum lectins [19], where only the first two splice sites are present. The Fugu AFPL gene expression is confirmed by an EST sequence BU806418, which covers the whole predicted CDS.
Link domain containing CTLDcps
All link domain-containing proteins identified in mammals are represented in Fugu and often are highly conserved between fish and human (e.g. TSG-6, 72%; Stabilin-1, 45% identity); we will consider them as a single group despite their different domain architectures. Predicted members of the CD44 family (CD44 and lymphatic vessel endothelium-specific hyaluronan receptor (Lyve-1)), however, are much more divergent from their human homologues, and it is not clear whether the two loci found in Fugu are orthologues of the two human genes or paralogues which arose by duplication of an ancestral gene.
In a recently published comprehensive study of another family of the Link group, the hyaluronan and proteoglycan binding link proteins (HAPLN), four homologues were identified in vertebrates (mouse, human and partially zebrafish) each linked to one of the four lecticans [50]. As all lecticans (i.e. group I) are duplicated in Fugu, we were expecting to also find duplicate copies of all HAPLN members. However, orthologues of only three HAPLNs were found (CRTL1, BRAL1, HAPLN3), two of which are linked to hyalectans in the same way as in mammalian genomes (CRTL1 with Versican, BRAL1 with Brevican). The state of the assemblies does not allow to determine conclusively whether HAPLN3 is linked to Aggrecan or not. Only two of the Fugu lectican gene duplications are accompanied by corresponding HAPLN genes: Aggrecan-F1 is linked to HAPLN3-F1 and the CRTL1 paralogue is present downstream to Versican-F1 in two tandem copies (CRTL1-F1 and CRTL1-F2). In neither version of the assembly could the HAPLN4 homologue be identified downstream to Neurocan or Neurocan-F1. Sequence conservation levels within the HAPLN proteins compared with their human orthologues is quite high (e.g. 76% identity for CRTL1).
Fugu dual-CTLD CTLDcps
The members of this group are soluble proteins with two or three CTLDs, which we initially characterized as fragments of putative macrophage mannose receptor paralogues. However, phylogenetic analysis showed that these proteins constitute a separate group, with no mammalian orthologues detectable in sequenced genome and protein databases. The domain structure prediction is confirmed by three zebrafish cDNAs (CAE17649, CAE17650, CAE17651), which have the same domain organization, although conservation between zebrafish and Fugu sequences is only moderate (~30%). Another homologue with the same domain structure and similarity to the F1 group members, which was returned as the top-scoring hit by BLAST searches in the nrdb, is the SCARF2 protein from a planarian Girardia tigrina [51]. A hypothetical dual-CTLD protein from Drosophila (NP_609962), which presumably corresponds to the single member of group B in the Drosophila CTLDcp classification of Dodd and Drickamer [14], was also detected as a F1 homologue by BLAST.
Novel CTLDcps conserved between Fugu and mammals
Discovering novel superfamily members in existing database sequences is one of the most important and exciting outcomes of a systematic computer-based study such as this. We predicted putative Fugu orthologues for several uncharacterized mammalian CTLDcps (Bimlec, MGC3279, KIAA0534, CETM, SEEC, CBCP, NLSLH) that are well conserved between Fugu and mammals. Most of the predictions were supported by mammalian cDNA sequences from public databases, but for two of them (NLSLH and CBCP) no full-length cDNA from any organism was found in DBs. The high level of genomic sequence conservation over evolutionary time from fish to human, as in the case of NLSLH, and the presence of partial cDNA and EST sequences from rodents and human, as in the case of CBCP, were strongly suggestive that the predictions are correct. The novel CTLDcps that could be attributed to one of the 14 known groups have been discussed in the preceding sections for the corresponding groups; those that do not fit into the existing classification are described below.
A large (~2100 aa) proteoglycan (CBCP), containing a set of chondroitin sulphate proteoglycan (CSPG) repeats [52], which are homologous to the NG2 ectodomain [53], a calcium-binding Calx-β domain [54] and a CTLD, is a novel member of a protein family which had not been reported previously to have members containing CTLDs; examples of this family also include the human MCSP/CSPG4 [55] and mouse FRAS1 [56] genes. The prediction was supported by three overlapping but incomplete cDNA sequences from human and mouse, high levels of conservation between human and Fugu (~50% identity), and the compact structure of the predicted Fugu gene. CBCP has been placed in a new CTLD group, XVII; its domain structure is shown in Figure 2. We have cloned a full-length cDNA of mouse CBCP confirming the domain structure predicted in this study (A.N. Zelensky, in preparation). The CTLD of CBCP lacks Ca-binding residues, and its long loop region is short, resembling that of the group V CTLDs.
Another protein with a novel domain organization, whose prediction is strongly supported by available cDNAs, is SEEC (S CP, E GF, E GF, C TLD-containing protein) (see Figure 2), which is well conserved between human and Fugu. Although not described in a publication, a full-length human SEEC cDNA (AK074773) was sequenced in the NEDO high-throughput sequencing project. The predicted Fugu SEEC is 63% identical to the human sequence. The sperm-coating glycoprotein (SCP) domain, which is present in a broad set of organisms from yeast and plants to mammals, but whose function is unknown [57], is rarely observed in combination with other domains in proteins; in only one other known protein (from sea urchin) is it found together with an EGF domain [58], and SEEC is the first example of a CTLD-SCP combination. The potential Ca/carbohydrate-binding motif (QPD) characteristic of galactose specificity is present in the CTLD. SEEC has been placed in a new CTLD group XVI.
A predicted protein named "novel L-selectin homologue" (NLSLH) because its CTLD is most similar to selectin CTLDs is duplicated in Fugu (NLSLH and NLSLH-F1) but only moderately conserved (32% identity) between Fugu and human. The putative human orthologue is located on Ch1q25.1 about 18 Mb further from the centromere than the selectin cluster and is supported only by EST sequences (AA912157, AA889574), but not cDNAs. No conserved domains except for the CTLD could be detected in the human and Fugu NLSLH loci so, if the predictions are correct, NLSLH is a soluble single-CTLD-containing protein. Carbohydrate-binding motifs are not present in the NLSLH and NLSLH-F1 CTLDs.
Finally, a type I transmembrane protein Bimlec, whose prediction is supported by a full-length human cDNA, was placed in a new group XV.
Dating the CTLDcp duplications
We found 12 groups of unlinked Fugu-specific CTLDcp paralogues (Table 1), and attempted to determine the duplication dates using two approaches: (1) based on the estimation of the number of synonymous nucleotide substitutions (Ks) in the coding sequences and (2) based on the molecular clock hypothesis.
For all but two pairs of duplicated genes, Ks values estimated with four different methods (see Methods) were between 1.5–2.5, which indicates a complete saturation of the synonymous sites (Figure 4(A)). Ks values so high cannot provide an accurate estimation of the duplication age, but we can conclude with confidence that the CTLDcp gene duplications are at least 150 Myr old, which is the time required for complete saturation of silent sites assuming a mutation rate of 2.5 substitutions/silent site/billion years in fish [59]. If, however, Ks values presented in Figure 4(A) and the silent mutation rate are close to correct, the corresponding duplication timeframe is predicted to be 300–500 Myr.

CTLDcp duplication dates. A. Average number of synonymous substitutions per synonymous site (Ks) for CTLDcp paralogue pairs based on full-sequence (triangle) and CTLD-only (diamond) alignments, measured with four different methods (see Methods). Error bars show one standard deviation in the CTLD-only measurements. All possible pairwise alignments between the MManR fragments and between the three CRTL1 paralogues were analyzed. Only homologous regions were used for MManR fragment alignments. B. A linearized phylogenetic tree built by the neighbor-joining method from Poisson-corrected distances between ClustalW-aligned sequences of CTLDs 3–5 from Fugu, mouse and human MManRs. Sequence of the human PLA2R region containing CTLDs 3–5 was used as an outgroup. Thm – time of separation between human and mouse [96 Myr; 60], Tfish – time of separation between ray-finned and lobe-finned fishes [430 Myr; 32]. Time of duplication (Tdupl) was calculated using average between molecular clock calibrated with Thm and with Tfish.
In order to date the duplications based on molecular clock measurements, we aligned duplicated Fugu CTLD sequences with their vertebrate orthologues present in GenBank, and built linearized phylogenetic trees based on the alignments. As human and mouse sequences were invariably available, the divergence time between these two species [96 Myr; 60] was used to calibrate the clock, together with the divergence time between Actinopterygii and Sarcopterygii [430 Myr; 32]. Symmetrical tree topology ((H, M) (F, F1)), expected for a Actinopterygian-specific duplication, was revealed by at least one phylogeny reconstruction method we used for the following six homologue groups (data not shown): brevican, neurocan, MManR, SRCL, tetranectin and HAPLN3, with duplications dated 369, 284, 397, 377, 360 and 312 Myr, respectively. A typical tree with symmetrical topology is shown for MManR in Figure 4(B). The other six alignments (aggrecan, versican, layilin, attractin, C1qRP, CRTL1) produced trees with topologies suggesting a duplication predating the split between Actinopterygian and Sarcopterygian. The portion of symmetrical topologies (50%) in the CTLD set is similar to the ratio reported by Taylor and coworkers in fish: 15 of 27 (55 %) [61], and 25 of 53 (45%) [62] for bigger and more heterogeneous gene collections.
Discussion
Draft assembly limitations
A systematic study based on draft-quality whole-genome data for an organism like Fugu rubripes has some limitations, as the genomic sequence is incomplete, fragmented and sometimes misassembled, and the expressed sequence information is scarce. On the other hand, many of the genomes that are currently being sequenced will be released and remain for sometime in the same state as the Fugu genome data are now. Indeed, more than a year after the initial release [31] very few improvements to the Fugu genomic data [v.3 assembly and EST sequencing project; [63]] have been published. Therefore, it is essential to extract useful biological information from draft-quality whole-genome sequences. Our study is such an attempt.
We have mentioned four limitations of the draft-state assembly – incompleteness, fragmentation, misassembly and lack of expression information. While the last might appear the biggest problem, we found that ab initio predictions combined with manual curation and interspecies comparison have proven to be very accurate (e.g. see PKD1L example), thanks to the compactness of the Fugu genome, smaller ratio between intron and intergenic region sizes compared with mammalian genes, wealth of data for comparative analysis etc. We do not expect that sequencing the remaining 5% of the Fugu genome, which is mostly heterochromatic regions, will lead to discovery of many new CTLDcps. From the comparison of the Fugu CTLDcp repertoire discovered by us and found in other fish species independently, the only surprising omission in our results is a MBP orthologue. MBP sequences have been found in several other fish species. Their absence in Fugu may represent a bona fide gene loss. As to the fragmentation, only a few of the CTLDcps are split between scaffolds, namely versican and some MManR paralogues (Figure 1). All of the fragmented genes are big, and in most cases the fragments can be combined easily to reveal the full sequence. Finally, misassembly signs were observed in several CTLDcp loci while comparing two versions of the assembly. These showed as presence of repeated regions in the v.2 assembly, which disappeared in the v.3 assembly.
Two groups identified in higher vertebrates are not detectable in Fugu
We could not detect CTLDcp representatives for groups V (NK cell receptors) and VII (lithostathine) in the Fugu genome. CTLDs in the members of these groups have lost their carbohydrate-binding activities, and perform functions that have, apparently, evolved after evolutionary separation of tetrapods from fish, or which are mediated by other proteins in fish. For example, group VII members are secreted into the digestive tract – a system that is very flexible evolutionally. Group V is probably one the youngest and most rapidly evolving sets of CTLDcps; its component members vary significantly even between rodents and human, a phenomenon connected to the co-evolution with the acquired immune system proteins that group V CTLDcps interact with.
Our conclusion on the absence of group V CTLDcps in the Fugu genome is at odds with the conclusions of two studies describing group V CTLDcp evolution in chordates. A recent paper describes possible CD94 homologues (cichlid killer cell lectin receptor, cKLR) in bony fishes Paralabidochromis chilotes and Oreochromis niloticus, which are encoded by a large multi-gene family with at least 10 members [28]. Another recent work described sequencing of a CD94 homologue in a tunicate [64].
The decision by Sato et al. [28] to assign putative fish killer cell receptors to group V rather than to group II was based on several considerations, including gene structure, absence of canonical Ca2+/carbohydrate-binding residues, and phylogenetic analysis based on the CTLD alignment. The latter consideration is mentioned as the most important one. However, as the authors themselves note, bootstrap values for placing cKLR on the group V branch, are "low to moderate". Indeed, we found that in phylogenetic trees built using different methods (maximum parsimony, distance estimation method with PAM matrix followed by neighbor-joining tree reconstruction, maximum likelihood) from the ClustalW alignments of cKLR sequences with group V and group II CTLD sequences from Fugu, mouse and human, cKLR placement is unstable. As shown in Figure 5, on a tree built by the neighbor-joining method we found cKLR on the branch containing the Fugu-specific subset of group II CTLDcps (DC-SIGN-F1 – DC-SIGN-F8), most of which do contain residues required for Ca2+/carbohydrate binding. On a tree built by the maximum parsimony method, we found cKLR on a separate branch equally related to group II and group V sequences (not shown). Also, a BLAST search with the complete cKLR sequence (GI 31789959) in the non-redundant NCBI protein database returns members of the ASGR subgroup of group II as top matches. Therefore, we judge that sufficient support for assignment of cKLR to group V is lacking and the question of the presence of the NK-cell receptor family in fishes is still open.

Relationships between fish, mouse and human group V and II CTLDs. Non-redundant set of CTLD sequences from known human and mouse CTLDcps classified as groups II and V, Fugu CTLDcps classified as group II, and putative killer cell receptor from Paralabidochromis chilotes (cKLR) were aligned with ClustalW. A consensus phylogenetic tree was built from 100 bootstrap trials using the protdist (with PAM distance matrix) and neighbor programs from the PHYLIP package. Black triangle shows position of cKLR. Bootstrap values higher than 40 are indicated.
As to the putative CD94 homologue from tunicates, it is indeed more similar to CD94 than to any other CTLDcp. However, the low level of sequence homology and the lack of evidence for existence of group V CTLDcps in more advanced taxa does not allow a confident statement that the sequence from tunicates is a CD94 orthologue, rather than a result of convergent evolution.
Expansion of the innate immunity CTLDcp groups in Fugu
Unlike pairwise unlinked duplications (see below), tandem duplications and other gene family expansions are limited to two groups, namely the DC-SIGN subgroup of group II and MManR. In mammals, members of these subgroups play an important role in innate immune responses. In particular, DC-SIGN is actively studied due to its ability to bind and internalize a broad range of bacterial and viral pathogens, including HIV-1 and Mycobacterium tuberculosis (reviewed in [65]), while MManR is also implicated in binding and phagocytosis of a wide range of microorganisms [66]. Expansion of these groups, most notably the DC-SIGN subgroup, in Fugu may reflect a larger role for innate immunity in host defense in lower vertebrates. Interestingly, multi-copy clusters comprising at least 10 genes encoding close cKLR homologues were identified in another cichlid fish species Oreochromis niloticus [28], which suggests another parallel between the expanded DC-SIGN subgroup in puffer fish and cKLRs of cichlids.
There are no extra members, however, in the Fugu collectin group – another CTLD group directly involved in innate immunity in mammals. Moreover, the mannose binding protein (MBP), which is the best-studied mammalian collectin involved in lectin complement activation pathway, was not detected by us. The absence of MBP orthologues in Fugu is rather puzzling, as MBP sequences have been found in several other fish species (Danio rerio, Cyprinus carpio and Carassus auratus; [20]), and are well conserved within the Cyprinidae carp family. The collectin family is also present and expanded in the Urochordate Ciona intestinalis with nine collectin genes identified in the draft genome sequence [67], although it is not clear whether one of these nine genes is an MBP orthologue. Given the role of MBPs in complement activation in mammals, and their presence and level of conservation in the carp family, it is possible that the Fugu MBP orthologue does exist but is not covered by the draft genome sequence. Complement-activating C-type lectins from lower organisms have been identified but not completely sequenced [68]; they have multiple CTLDs as in CPL-III from the protochordate Clavelina picta [69] or lack the collagen domain and show more similarity to other CTLDcps such as the glucose-binding lectin (GBL) from another tunicate, Halocynthia roretzi [70].
Fugu dual CTLD molecules – a missing link between vertebrate and invertebrate CTLDs?
Previous whole-genome studies of the CTLD superfamily in two invertebrates [13, 14] failed to identify any groups of CTLDcps common to both invertebrates and vertebrates. A group of predicted dual CTLD-containing proteins in Fugu (F1) may be the first vertebrate group that has detectable homologues in invertebrates. Alternatively, it is possible that none of the Fugu F1 group members are in fact orthologous to the invertebrate sequences, as sequence similarities are only moderate (~30% ID) and the domain architecture is simple and could have evolved independently in different lineages. However, several observations suggest that at least F1 members from Fugu and zebrafish and SCARF proteins from Girardia tigrina evolved from the same predecessor. First, similarity levels between fish sequences and between fish and planarian sequences are about the same. It is unlikely that the fish sequences are unrelated, which implies that F1 members are evolving quickly, and only major structural features of these molecules are under selective pressure [49]. Second, the CTLDs of planarian and, in all cases at least one CTLD of the fish dual-CTLDcps, contain residues characteristic of Ca/carbohydrate binding. In vertebrates, ability to bind carbohydrates is associated with the oldest CTLDcps groups, and is considered to be an ancestral feature of the CTLD. Indeed, both vertebrate CTLDcp groups that we failed to find in Fugu (V and VII) have lost sugar-binding properties. This is also the case for the antifreeze proteins from fish and snake venom CTLDcps, which have only been found in the corresponding clades. Third, similar domain organization (two CTLDs, no transmembrane domain) is also observed in two other known groups of invertebrate CTLDcps: immulectins from various insect species [71, 72] and nine proteins from C. elegans, classified as group D1 by Drickamer and Dodd [13]. Despite identical domain structure, none of these proteins shows statistically significant homology to the fish F1 group members or their putative homologues from planarian or Drosophila. Altogether, this indicates that domain structure alone cannot establish an evolutionary link between the fish and invertebrate sequences. Hence, the suggestive link between the F1 group fish members and the planarian and Drosophila proteins is even more interesting.
CTLDcp classification update
The existing classification of CTLDcps is generally accepted and popularly used in studies of the superfamily and recently has been updated [15]. The classification divides CTLDcps into monophyletic groups of proteins with identical overall domain architecture based on a combination of structural and phylogenetic information. Although two previous large-scale studies [13, 14] showed it to be inapplicable for description of invertebrate CTLDcps, our analysis of the puffer fish genome indicates that it is sufficient to describe the superfamily in all vertebrates, with only minor modifications and some extensions.
Our newly discovered CTLDcps, with a few exceptions, do not fit into the existing classification because of their unique domain architecture. We propose several new groups to accommodate the novel CTLDcps which have been found in both higher and lower vertebrates and are supported by cDNA sequences:
• XV – Bimlec (type I transmembrane protein), which in phylogenetic trees is not placed on the same branch as group VIII sequences, has a distinct exon-intron structure of the CTLD region and a neck not similar to the neck region of the group VIII sequences;
• XVI – SEEC, based on unique domain architecture;
• XVII – CBCP, based on unique domain architecture;
Additional groups may be required for the sequences not supported by sufficient expression data (NLSLH) and other sequences from the "unclassified" group whose presence in higher vertebrates is not clear. Also, clade-specific groups, such as fish antifreeze proteins (AFP), dual-CTLD sequences (group F1) predicted by us and so far identified only in fish, or snake venom CTLDcps which lack orthologues in other vertebrates, are required.
It has been suggested previously [19, 48] that AFPs belong to group VII based on their domain architecture and exon-intron structure. However, our phylogenetic analysis of an alignment of CTLD sequences from all known human and mouse CTLDcps and 26 different fish CTLD-containing protein sequences identified by searching the NCBI protein database with BLAST, indicates that they constitute a phylogenetically distinct group including all known soluble fish CTLD-containing proteins, except Cyprinidae collectins. As to the exon-intron structure, introns in the group XII (EMBP) CTLDs are at exactly the same positions as in group VII and AFP-like CTLDs, which suggests that all three groups are closely related but does not allow classification of the fish AFP-like sequences to either of the mammalian groups. Interestingly, just like most of the AFPs, mammalian EMBPs contain an atypical WIGL motif with a glycine in the fourth position, a substitution not observed in any other mammalian CTLD we analyzed. Taken together, these observations indicate that in a broader evolutionary perspective the differences between some of the groups including CTLDcps with a very similar domain architecture (VII, XII and AFP; II and V) become less distinct, which makes classification of the "intermediate" or "ancestral" sequences, equally related to more than one group, problematic.
Selective duplication of the Fugu CTLDcp-encoding genes and the whole-genome duplication hypothesis
The hypothesis that whole-genome duplications were one of the main driving forces in vertebrate evolution, providing genetic material for increased diversity and progressive development [73], and that there were two rounds of whole-genome duplication in vertebrate phylogeny (the 2R hypothesis) [73, 74], is actively debated [75, 76]. A more recent whole-genome duplication is suggested for the Actinopterygian branch [61]. Ray-finned fish are the most diverse group of vertebrates, and based on the initial observation that each of the four human HOX gene clusters has two homologues in zebrafish [77] it was suggested that they have undergone an additional round of a whole-genome duplication after the divergence from Sarcopterygian about 430 Myr ago [61]. Analysis of the genome duplication in fish can give a picture of a duplicated genome after 300–400 Myr of evolution and fill the gap between the now generally accepted recent tetraploidizations in plants [78] and yeast [79] and the alleged more ancient duplication(s) of the ancestral vertebrate genome.
Although many fish genes are indeed duplicated [61, 77, 80–82], it is not clear whether the copies were created by a complete genome duplication (autopolyploidy), merge of different genomes (allopolyploidy), regional duplication, or simply a series of tandem duplications. Attempts to show that ancient tetraploidization (has not) occurred usually involve: (i) searching for an excess of paralogue groups where the number of members is double the number of alleged duplications (i.e. 2 in case of Actinopterygian duplication, and 4 in case of vertebrate duplication, the "one to four rule") [74, 76]; (ii) showing that a statistically significant number of duplications took place at approximately the same time by molecular clock estimation or synonymous substitution counting [83, 84]; (iii) using phylogenetic methods to assess the relation between duplication and speciation events [61]; and (iv) showing that duplicated genes are arranged in paralogous blocks on chromosomes (paralogons) [62, 85, 86]. We used these approaches to analyze the nature of the observed CTLDcp duplications in Fugu.
Our results clearly show that tandem gene copying is a mechanism of CTLD family evolution and led to generation of three gene clusters: DC-SIGN-F2 – DC-SIGN-F5 (4 genes), CRTL1-F1 and CRTL1-F2, and AFPL-F1 and AFPL-F2. Members of the two latter clusters are nearly identical and may be an assembly artifact. Twelve other duplicated genes are not linked in the current assembly and have sequences much more diverged than tandem duplicates. Of the 12 genes only MManR, which has 3 paralogues, is present in more than two copies. We consider this is important evidence in favor of a whole-genome duplication, as sporadic duplications cannot explain such a strong bias towards two-member paralogue groups. Unfortunately, the results of duplication time estimations are less conclusive as they give only a broad timeframe for the possible duplication events of about 300–400 Myr. As in the case of some other fish gene families reported previously [61, 62, 87, 88], molecular phylogeny reconstruction performed by us often indicates that duplications occurred before the divergence between fish and tetrapods. However, this could be an artifact of the method caused by different selection pressures on duplicates. Unfortunately, there is practically no overlap between vertebrate and invertebrate CTLD families, so we could not use invertebrate sequences to refine phylogenetic analysis. To conclude: phylogenetic relationships between CTLD paralogues and estimated duplication time distribution indicate that there was a burst in duplication activity in the Fugu genome 300–400 Myr ago. While we cannot determine definitively the nature of the duplications (tandem, regional or whole-genome), a pronounced bias in the number of two-member paralogue groups strongly suggests that there was a single large-scale or whole-genome duplication event in fish.
Another interesting observation is that CTLDcp genes were either duplicated, or retained after a large-scale duplication, in a pronounced selective manner. One group (I) is duplicated completely, while in other groups only partial duplications are found. Interestingly, group I (lecticans), which in tetrapods contain four large (>2000 amino acids) proteins, very similar to each other in sequence and domain structure, is a good candidate for demonstrating the 2 R hypothesis. If the four lecticans arose as a result of the alleged two rounds of the whole-genome duplication early in vertebrate history, the fact that the family was also completely duplicated in fish and retained after the duplication appears very non-random and implies some functional explanation. In the human genome, all four genes encoding lecticans are located on different chromosomes (1, 5, 15 and 19), but it is not clear whether they are linked in Fugu.
Another group that conforms to the 2 R hypothesis is group VI, which in tetrapods has four members with almost identical domain structure in mammals (Pla2R, MManR, DEC-205 and Endo180). Though in the Fugu genome we identified 7 group VI sequences, some of which are fragmented (Figure 1), phylogenetic analysis shows that only one member of the family (MManR) was quadruplicated, while others are present in a single copy. Both molecular clock and Ks-based methods date the MManR duplications at approximately the same time as other CTLDcp gene duplications. Phylogenetic trees, built on alignment of the overlapping portions (Figure 1) of the complete sequences and three largest fragments (fMManR-F1, fMManR-F2, fMManR-F3) have symmetrical structure, with fMManR-F1, fMManR-F2 and fMManR-F3 forming a separate branch (Figure 4(B)). A whole-genome duplication, generating fMManR and fMManR-F1, followed by tandem duplications of fMManR-F1, producing fMManR-F2 and fMManR-F3, can explain this topology.
Conclusions
We have performed an analysis of the CTLD superfamily composition in Fugu rubripes. Although the sequence assembly is in the draft state and lacks physical mapping information and native cDNA sequences that could be used to make and verify gene predictions, the quality of the data is good enough despite these limitations to answer many important questions. Our study demonstrates that all but two groups of CTLDcps present in mammals are also found in fish, that most of the groups have the same composition as in mammals, and that the missing groups are the evolutionarily most dynamic ones involved in physiological processes that may be specific to higher vertebrates. We also identified at least one distinct fish-specific CTLD group, which could be the first known vertebrate CTLD group also found in invertebrates.
The compactness of the Fugu genome makes it an extremely convenient reference sequence for identification of new genes based on supporting similarity features, and we were able to identify and predict the structure of several new CTLD-containing genes highly conserved between Fugu and human. The new sequences are supported by cDNA and EST sequences from databases and have previously unknown domain architectures. We are now characterizing some of these sequences experimentally. We also show that CTLDcp-encoding genes are selectively duplicated in Fugu, in a manner that suggests an ancient large-scale duplication event in fish.
Methods
Corrected gene predictions are made available as a distributed annotation system (DAS) [89] resource [90], which can be viewed in the EnsEMBL genome browser. The data source names for predictions based on assemblies v.2 and v.3 are fugu_ctld_1 and fugu_ctld_2, respectively. Transcript sequences (in FASTA format) for the CTLDcp-encoding genes created or modified by us (stable IDs starting with ANU) and their translations are also provided in the additional file 1 and additional file 2, respectively.
Searches and gene annotations were done on version 2 of the Fugu rubripes genome assembly [31] downloaded from the EnsEMBL web site [91, 92]. When the third version of the assembly was released, we mapped gene annotations onto it. Mapping was done on the basis of SSAHA [93] matches in the v.3 assembly for exons predicted on the v.2 assembly. The v.2 assembly is currently accessible at the Singapore IMCB site [94] and on our server [95], which is pre-configured to display the DAS track with our annotations and contains a reference table with hyperlinks for all of the Fugu CTLDcp genes discussed. Version 3 of the assembly can be found on the main EnsEMBL web site [34]. The EnsEMBL genome browser can be easily configured to display our gene models as a DAS track.
We used a multi-step approach to find genes encoding CTLDs. First, a hidden Markov model (HMM) profile of the CTLD was used to scan a FASTA database of EnsEMBL-predicted genes with the hmmsearch program from the HMMER package [96]. To detect orthologues and paralogues, the set of Fugu sequences found was compared with the 95% non-redundant set of sequences of human CTLDcps that could be found in the Entrez proteins database, using the Inparanoid program [40]. All of the 25 orthology links detected by Inparanoid were checked manually.
Because of systematic and sporadic errors in EnsEMBL gene predictions, we had to manually revise the structure of each of the 69 genes encoding proteins detected by the HMM-based search. This was done using the Apollo genome annotation software [97] connected to a local installation of the EnsEMBL database. To facilitate annotation, several additional feature tracks were added to the EnsEMBL database:
a) Similarity features detected by GeneWise [98] search of Fugu scaffold sequences with a CTLD HMM built in a global alignment mode. This was done to detect well conserved CTLDs while avoiding many false positives.
b) Same as a), but with an HMM built in the local alignment mode; this was done to detect highly conserved fragmented CTLDs;
c) Similarity features detected by a TBLASTN search of Fugu scaffold sequences using all known human CTLD sequences; this was done to detect CTLDs that are less conserved;
d) ORFs encoding putative transmembrane (TM) domains. To create this track a database of all possible ORFs longer than 45 bp was produced and translated into protein sequence using the EMBOSS programs. This was then scanned with the TMHMM program [99] to detect ORFs that encode putative TM domains.
To verify whether there are CTLDs that were not covered by EnsEMBL gene predictions, we searched for all significant CTLD similarity features detected by GeneWise which do not overlap with any of the genes analyzed in the first stage. This step led to detection of 25 new CTLD-coding genes, including most of the ones that have previously uncharacterized domain organization. At the next stage we analyzed the loci with different CTLD similarity features detected by genewisedb search with a local alignment HMM. Finally, the features identified by BLAST and not overlapping with already detected genes were analyzed. This set of features mostly contained only partial CTLD matches.
We translated both the new and already predicted gene CDSs into protein sequences and performed another Inparanoid comparison. Phylogenetic relationships were analyzed with the programs from the Phylip package [100]. ClustalW [101] guiding trees were used for quick phylogeny estimation and in cases where a proper multiple alignment could not be made.
BioPerl [102] and EnsEMBL Perl modules were used to automate all stages of the analysis. Domain architectures were analyzed with the SMART web service [103].
To estimate the proportion of substitutions in synonymous sites, we aligned translated sequences of the duplicated CTLDcp-encoding genes with ClustalW, using either whole sequence or sequence for the CTLD-encoding region only, and built nucleotide sequence alignments based on the protein alignments. Ks estimations were performed with four methods: Lynch and Connery [104] and Li [105], both implemented in the ntdiffs package [104]; and Nei and Gojobori [106] and Yang and Nielsen [107], both implemented in the yn00 program from the PAML package.
Duplication dating using the calibrated molecular clock approach was performed as in [83]. Alignments of CTLD-containing regions of Fugu paralogues and their mammalian orthologues were made with ClustalW. The MEGA2 program [108] was used to build linearized trees from Poisson-corrected distances, p-distances and Gamma-corrected distances by the neighbor-joining method with 1000 bootstrap samplings. The global clock was calibrated using divergence times 96 Myr and 430 Myr for human-mouse and fish-mammal splits, respectively [32, 60, 83].
Abbreviations
- AFP:
-
antifreeze protein
- AFPL:
-
AFP-like
- CBCP:
-
C alx-β and C TLD-containing P rotein
- CDS:
-
coding sequence
- CETM:
-
C TLD, E GF, T ransM embrane domain
- CTLD:
-
C-type-lectin-like domain
- CTLDcp:
-
CTLD-containing protein
- DAS:
-
distributed annotation system
- EMBP:
-
eosinophil major basic protein
- EST:
-
expressed sequence tag
- HMM:
-
hidden Markov model
- MBP:
-
mannose-binding protein
- Myr:
-
million years
- NLSLH:
-
N ovel L-S eLectin H omologue
- PKD1:
-
polycystic kidney disease protein 1
- SEEC:
-
S CP, E GF, E GF, C TLD
- TM:
-
transmembrane.
References
Drickamer K: C-type lectin-like domains. Curr Opin Struct Biol. 1999, 9: 585-590. 10.1016/S0959-440X(99)00009-3.
Drickamer K: Evolution of Ca(2+)-dependent animal lectins. Prog Nucleic Acid Res Mol Biol. 1993, 45: 207-232.
Weis WI, Drickamer K: Structural basis of lectin-carbohydrate recognition. Annu Rev Biochem. 1996, 65: 441-473. 10.1146/annurev.bi.65.070196.002301.
Natarajan K, Dimasi N, Wang J, Mariuzza RA, Margulies DH: Structure and Function of Natural Killer Cell Receptors: Multiple Molecular Solutions to Self, Nonself Discrimination. Annu Rev Immunol. 2002, 20: 853-885. 10.1146/annurev.immunol.20.100301.064812.
Sano H, Kuroki Y, Honma T, Ogasawara Y, Sohma H, Voelker DR, Akino T: Analysis of chimeric proteins identifies the regions in the carbohydrate recognition domains of rat lung collectins that are essential for interactions with phospholipids, glycolipids, and alveolar type II cells. J Biol Chem. 1998, 273: 4783-4789. 10.1074/jbc.273.8.4783.
Geider S, Baronnet A, Cerini C, Nitsche S, Astier JP, Michel R, Boistelle R, Berland Y, Dagorn JC, Verdier JM: Pancreatic lithostathine as a calcite habit modifier. J Biol Chem. 1996, 271: 26302-26306. 10.1074/jbc.271.42.26302.
Ewart KV, Li Z, Yang DS, Fletcher GL, Hew CL: The ice-binding site of Atlantic herring antifreeze protein corresponds to the carbohydrate-binding site of C-type lectins. Biochemistry. 1998, 37: 4080-4085. 10.1021/bi972503w.
Mann K, Siedler F: The amino acid sequence of ovocleidin 17, a major protein of the avian eggshell calcified layer. Biochem Mol Biol Int. 1999, 47: 997-1007.
Weiss IM, Kaufmann S, Mann K, Fritz M: Purification and characterization of perlucin and perlustrin, two new proteins from the shell of the mollusc Haliotis laevigata. Biochem Biophys Res Commun. 2000, 267: 17-21. 10.1006/bbrc.1999.1907.
Yokoyama WM: Natural killer cell receptors. Curr Opin Immunol. 1998, 10: 298-305. 10.1016/S0952-7915(98)80168-4.
Matsumoto N, Ribaudo RK, Abastado JP, Margulies DH, Yokoyama WM: The lectin-like NK cell receptor Ly-49A recognizes a carbohydrate- independent epitope on its MHC class I ligand. Immunity. 1998, 8: 245-254. 10.1016/S1074-7613(00)80476-8.
Kijimoto-Ochiai S: CD23 (the low-affinity IgE receptor) as a C-type lectin: a multidomain and multifunctional molecule. Cell Mol Life Sci. 2002, 59: 648-664. 10.1007/s00018-002-8455-1.
Drickamer K, Dodd RB: C-Type lectin-like domains in Caenorhabditis elegans: predictions from the complete genome sequence. Glycobiology. 1999, 9: 1357-1369. 10.1093/glycob/9.12.1357.
Dodd RB, Drickamer K: Lectin-like proteins in model organisms: implications for evolution of carbohydrate-binding activity. Glycobiology. 2001, 11: 71R-79R. 10.1093/glycob/11.5.71R.
Drickamer K, Fadden AJ: Genomic analysis of C-type lectins. Biochem Soc Symp. 2002, 59-72.
Ng NF, Trinh KY, Hew CL: Structure of an antifreeze polypeptide precursor from the sea raven, Hemitripterus americanus. J Biol Chem. 1986, 261: 15690-15695.
Achenbach JC, Ewart KV: Structural and functional characterization of a C-type lectin-like antifreeze protein from rainbow smelt (Osmerus mordax). Eur J Biochem. 2002, 269: 1219-1226. 10.1046/j.1432-1033.2002.02761.x.
Gronwald W, Loewen MC, Lix B, Daugulis AJ, Sonnichsen FD, Davies PL, Sykes BD: The solution structure of type II antifreeze protein reveals a new member of the lectin family. Biochemistry. 1998, 37: 4712-4721. 10.1021/bi972788c.
Richards RC, Hudson DM, Thibault P, Ewart KV: Cloning and characterization of the Atlantic salmon serum lectin, a long-form C-type lectin expressed in kidney. Biochim Biophys Acta. 2003, 1621: 110-115. 10.1016/S0304-4165(03)00045-X.
Vitved L, Holmskov U, Koch C, Teisner B, Hansen S, Skjodt K: The homologue of mannose-binding lectin in the carp family Cyprinidae is expressed at high level in spleen, and the deduced primary structure predicts affinity for galactose. Immunogenetics. 2000, 51: 955-964. 10.1007/s002510000232.
Tasumi S, Ohira T, Kawazoe I, Suetake H, Suzuki Y, Aida K: Primary structure and characteristics of a lectin from skin mucus of the Japanese eel Anguilla japonica. J Biol Chem. 2002, 277: 27305-27311. 10.1074/jbc.M202648200.
Mistry AC, Honda S, Hirose S: Structure, properties and enhanced expression of galactose-binding C-type lectins in mucous cells of gills from freshwater Japanese eels (Anguilla japonica). Biochem J. 2001, 360: 107-115. 10.1042/0264-6021:3600107.
Bayne CJ, Gerwick L, Fujiki K, Nakao M, Yano T: Immune-relevant (including acute phase) genes identified in the livers of rainbow trout, Oncorhynchus mykiss, by means of suppression subtractive hybridization. Dev Comp Immunol. 2001, 25: 205-217. 10.1016/S0145-305X(00)00057-4.
Fujiki K, Bayne CJ, Shin DH, Nakao M, Yano T: Molecular cloning of carp (Cyprinus carpio) C-type lectin and pentraxin by use of suppression subtractive hybridisation. Fish Shellfish Immunol. 2001, 11: 275-279. 10.1006/fsim.2000.0331.
Gracey AY, Troll JV, Somero GN: Hypoxia-induced gene expression profiling in the euryoxic fish Gillichthys mirabilis. Proc Natl Acad Sci U S A. 2001, 98: 1993-1998. 10.1073/pnas.98.4.1993.
Sandford R, Sgotto B, Aparicio S, Brenner S, Vaudin M, Wilson RK, Chissoe S, Pepin K, Bateman A, Chothia C, Hughes J, Harris P: Comparative analysis of the polycystic kidney disease 1 (PKD1) gene reveals an integral membrane glycoprotein with multiple evolutionary conserved domains. Hum Mol Genet. 1997, 6: 1483-1489. 10.1093/hmg/6.9.1483.
Zhang H, Robison B, Thorgaard GH, Ristow SS: Cloning, mapping and genomic organization of a fish C-type lectin gene from homozygous clones of rainbow trout (Oncorhynchus mykiss). Biochim Biophys Acta. 2000, 1494: 14-22. 10.1016/S0167-4781(00)00198-6.
Sato A, Mayer WE, Overath P, Klein J: Genes encoding putative natural killer cell C-type lectin receptors in teleostean fishes. Proc Natl Acad Sci U S A. 2003, 100: 7779-7784. 10.1073/pnas.1235938100.
Matsuo MY, Asakawa S, Shimizu N, Kimura H, Nonaka M: Nucleotide sequence of the MHC class I genomic region of a teleost, the medaka (Oryzias latipes). Immunogenetics. 2002, 53: 930-940. 10.1007/s00251-001-0427-3.
Neame PJ, Young CN, Treep JT: Primary structure of a protein isolated from reef shark (Carcharhinus springeri) cartilage that is similar to the mammalian C-type lectin homolog, tetranectin. Protein Sci. 1992, 1: 161-168.
Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P, Christoffels A, Rash S, Hoon S, Smit A, Gelpke MD, Roach J, Oh T, Ho IY, Wong M, Detter C, Verhoef F, Predki P, Tay A, Lucas S, Richardson P, Smith SF, Clark MS, Edwards YJ, Doggett N, Zharkikh A, Tavtigian SV, Pruss D, Barnstead M, Evans C, Baden H, Powell J, Glusman G, Rowen L, Hood L, Tan YH, Elgar G, Hawkins T, Venkatesh B, Rokhsar D, Brenner S: Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science. 2002, 297: 1301-1310. 10.1126/science.1072104.
Ahlberg PE, Milner AR: The origin and early diversification of tetrapods. Nature. 1994, 368: 507-514. 10.1038/368507a0.
Joint Genome Institute web portal to the Fugu rubripes genome assembly and annotation. [http://genome.jgi-psf.org/fugu6/fugu6.info.html]
EnsEMBL portal to the Fugu rubripes genome annotation. [http://www.ensembl.org/Fugu_rubripes/]
Maglich JM, Caravella JA, Lambert MH, Willson TM, Moore JT, Ramamurthy L: The first completed genome sequence from a teleost fish (Fugu rubripes) adds significant diversity to the nuclear receptor superfamily. Nucleic Acids Res. 2003, 31: 4051-4058. 10.1093/nar/gkg444.
Oshiumi H, Tsujita T, Shida K, Matsumoto M, Ikeo K, Seya T: Prediction of the prototype of the human Toll-like receptor gene family from the pufferfish, Fugu rubripes, genome. Immunogenetics. 2003, 54: 791-800.
Jones AK, Elgar G, Sattelle DB: The nicotinic acetylcholine receptor gene family of the pufferfish, Fugu rubripes. Genomics. 2003, 82: 441-451. 10.1016/S0888-7543(03)00153-8.
Jiang Y, Doolittle RF: The evolution of vertebrate blood coagulation as viewed from a comparison of puffer fish and sea squirt genomes. Proc Natl Acad Sci U S A. 2003, 100: 7527-7532. 10.1073/pnas.0932632100.
Zimek A, Stick R, Weber K: Genes coding for intermediate filament proteins: common features and unexpected differences in the genomes of humans and the teleost fish Fugu rubripes. J Cell Sci. 2003, 116: 2295-2302. 10.1242/jcs.00444.
Remm M, Storm CE, Sonnhammer EL: Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol. 2001, 314: 1041-1052. 10.1006/jmbi.2000.5197.
Burge C, Karlin S: Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997, 268: 78-94. 10.1006/jmbi.1997.0951.
Brissett NC, Perkins SJ: The protein fold of the hyaluronate-binding proteoglycan tandem repeat domain of link protein, aggrecan and CD44 is similar to that of the C- type lectin superfamily. FEBS Lett. 1996, 388: 211-216. 10.1016/0014-5793(96)00576-5.
Ohtani K, Suzuki Y, Eda S, Kawai T, Kase T, Yamazaki H, Shimada T, Keshi H, Sakai Y, Fukuoh A, Sakamoto T, Wakamiya N: Molecular cloning of a novel human collectin from liver (CL-L1). J Biol Chem. 1999, 274: 13681-13689. 10.1074/jbc.274.19.13681.
Trexler M, Banyai L, Patthy L: The LCCL module. Eur J Biochem. 2000, 267: 5751-5757. 10.1046/j.1432-1327.2000.01641.x.
Muta T, Miyata T, Misumi Y, Tokunaga F, Nakamura T, Toh Y, Ikehara Y, Iwanaga S: Limulus factor C. An endotoxin-sensitive serine protease zymogen with a mosaic structure of complement-like, epidermal growth factor-like, and lectin-like domains. J Biol Chem. 1991, 266: 6554-6561.
Kobuke K, Furukawa Y, Sugai M, Tanigaki K, Ohashi N, Matsumori A, Sasayama S, Honjo T, Tashiro K: ESDN, a novel neuropilin-like membrane protein cloned from vascular cells with the longest secretory signal sequence among eukaryotes, is up-regulated after vascular injury. J Biol Chem. 2001, 276: 34105-34114. 10.1074/jbc.M105293200.
Li A, Tian X, Sung SW, Somlo S: Identification of two novel polycystic kidney disease-1-like genes in human and mouse genomes. Genomics. 2003, 81: 596-608. 10.1016/S0888-7543(03)00048-X.
Loewen MC, Gronwald W, Sonnichsen FD, Sykes BD, Davies PL: The ice-binding site of sea raven antifreeze protein is distinct from the carbohydrate-binding site of the homologous C-type lectin. Biochemistry. 1998, 37: 17745-17753. 10.1021/bi9820513.
Zelensky AN, Gready JE: Comparative analysis of structural properties of the C-type-lectin-like domain (CTLD). Proteins. 2003, 52: 466-477. 10.1002/prot.10626.
Spicer AP, Joo A, Bowling R. A., Jr.: A Hyaluronan Binding Link Protein Gene Family Whose Members Are Physically Linked Adjacent to Chrondroitin Sulfate Proteoglycan Core Protein Genes: THE MISSING LINKS. J Biol Chem. 2003, 278: 21083-21091. 10.1074/jbc.M213100200.
Shagin DA, Barsova EV, Bogdanova E, Britanova OV, Gurskaya N, Lukyanov KA, Matz MV, Punkova NI, Usman NY, Kopantzev EP, Salo E, Lukyanov SA: Identification and characterization of a new family of C-type lectin-like genes from planaria Girardia tigrina. Glycobiology. 2002, 12: 463-472. 10.1093/glycob/cwf056.
Staub E, Hinzmann B, Rosenthal A: A novel repeat in the melanoma-associated chondroitin sulfate proteoglycan defines a new protein family. FEBS Lett. 2002, 527: 114-118. 10.1016/S0014-5793(02)03195-2.
Nishiyama A, Dahlin KJ, Prince JT, Johnstone SR, Stallcup WB: The primary structure of NG2, a novel membrane-spanning proteoglycan. J Cell Biol. 1991, 114: 359-371. 10.1083/jcb.114.2.359.
Schwarz EM, Benzer S: Calx, a Na-Ca exchanger gene of Drosophila melanogaster. Proc Natl Acad Sci U S A. 1997, 94: 10249-10254. 10.1073/pnas.94.19.10249.
Pluschke G, Vanek M, Evans A, Dittmar T, Schmid P, Itin P, Filardo EJ, Reisfeld RA: Molecular cloning of a human melanoma-associated chondroitin sulfate proteoglycan. Proc Natl Acad Sci U S A. 1996, 93: 9710-9715. 10.1073/pnas.93.18.9710.
McGregor L, Makela V, Darling SM, Vrontou S, Chalepakis G, Roberts C, Smart N, Rutland P, Prescott N, Hopkins J, Bentley E, Shaw A, Roberts E, Mueller R, Jadeja S, Philip N, Nelson J, Francannet C, Perez-Aytes A, Megarbane A, Kerr B, Wainwright B, Woolf AS, Winter RM, Scambler PJ: Fraser syndrome and mouse blebbed phenotype caused by mutations in FRAS1/Fras1 encoding a putative extracellular matrix protein. Nat Genet. 2003, 34: 203-208. 10.1038/ng1142.
Szyperski T, Fernandez C, Mumenthaler C, Wuthrich K: Structure comparison of human glioma pathogenesis-related protein GliPR and the plant pathogenesis-related protein P14a indicates a functional link between the human immune system and a plant defense system. Proc Natl Acad Sci U S A. 1998, 95: 2262-2266. 10.1073/pnas.95.5.2262.
Ohbayashi H, Mantoku T, Yamamoto T, Nomura K, Suzuki N: Primary structure of a 120 kDa protein associated with the fucose sulfate glycoconjugate constituting the acrosome reaction-inducing substance of the sea urchin, Hemicentrotus pulcherrimus. Dev Growth Differ. 1998, 40: 641-650.
Li Wen-Hsiung: Molecular evolution. 1997, Sunderland, Mass., Sinauer Associates
Nei M, Xu P, Glazko G: Estimation of divergence times from multiprotein sequences for a few mammalian species and several distantly related organisms. Proc Natl Acad Sci U S A. 2001, 98: 2497-2502. 10.1073/pnas.051611498.
Taylor JS, Van de Peer Y, Braasch I, Meyer A: Comparative genomics provides evidence for an ancient genome duplication event in fish. Philos Trans R Soc Lond B Biol Sci. 2001, 356: 1661-1679. 10.1098/rstb.2001.0975.
Taylor JS, Braasch I, Frickey T, Meyer A, Van de Peer Y: Genome duplication, a trait shared by 22000 species of ray-finned fish. Genome Res. 2003, 13: 382-390. 10.1101/gr.640303.
Clark MS, Edwards YJ, Peterson D, Clifton SW, Thompson AJ, Sasaki M, Suzuki Y, Kikuchi K, Watabe S, Kawakami K, Sugano S, Elgar G, Johnson SL: Fugu ESTs: new resources for transcription analysis and genome annotation. Genome Res. 2003, 13: 2747-2753. 10.1101/gr.1691503.
Khalturin K, Becker M, Rinkevich B, Bosch TC: Urochordates and the origin of natural killer cells: identification of a CD94/NKR-P1-related receptor in blood cells of Botryllus. Proc Natl Acad Sci U S A. 2003, 100: 622-627. 10.1073/pnas.0234104100.
Van Kooyk Y, Geijtenbeek TB: DC-SIGN: escape mechanism for pathogens. Nat Rev Immunol. 2003, 3: 697-709. 10.1038/nri1182.
Linehan SA, Martinez-Pomares L, Gordon S: Macrophage lectins in host defence. Microbes Infect. 2000, 2: 279-288. 10.1016/S1286-4579(00)00300-2.
Azumi K, De Santis R, De Tomaso A, Rigoutsos I, Yoshizaki F, Pinto MR, Marino R, Shida K, Ikeda M, Arai M, Inoue Y, Shimizu T, Satoh N, Rokhsar DS, Du Pasquier L, Kasahara M, Satake M, Nonaka M: Genomic analysis of immunity in a Urochordate and the emergence of the vertebrate immune system: "waiting for Godot". Immunogenetics. 2003, 55: 570-581. 10.1007/s00251-003-0606-5.
Nair SV, Pearce S, Green PL, Mahajan D, Newton RA, Raftos DA: A collectin-like protein from tunicates. Comp Biochem Physiol B Biochem Mol Biol. 2000, 125: 279-289. 10.1016/S0305-0491(99)00180-7.
Vasta GR, Quesenberry M, Ahmed H, O'Leary N: C-type lectins and galectins mediate innate and adaptive immune functions: their roles in the complement activation pathway. Dev Comp Immunol. 1999, 23: 401-420. 10.1016/S0145-305X(99)00020-8.
Sekine H, Kenjo A, Azumi K, Ohi G, Takahashi M, Kasukawa R, Ichikawa N, Nakata M, Mizuochi T, Matsushita M, Endo Y, Fujita T: An ancient lectin-dependent complement system in an ascidian: novel lectin isolated from the plasma of the solitary ascidian, Halocynthia roretzi. J Immunol. 2001, 167: 4504-4510.
Yu XQ, Gan H, Kanost MR: Immulectin, an inducible C-type lectin from an insect, Manduca sexta, stimulates activation of plasma prophenol oxidase. Insect Biochem Mol Biol. 1999, 29: 585-597. 10.1016/S0965-1748(99)00036-3.
Kim SR, Lee KS, Kim I, Kang SW, Nho SK, Sohn HD, Jin BR: cDNA sequence of a novel immulectin homologue from the silkworm, Bombyx mori. Int J Indust Entomol. 2003, 6: 99-102.
Ohno Susumu: Evolution by gene duplication. 1970, Berlin, New York, Springer-Verlag
Ohno S: Gene duplication and the uniqueness of vertebrate genomes circa 1970-1999. Semin Cell Dev Biol. 1999, 10: 517-522. 10.1006/scdb.1999.0332.
Wolfe KH: Yesterday's polyploids and the mystery of diploidization. Nat Rev Genet. 2001, 2: 333-341. 10.1038/35072009.
Hughes AL, Friedman R: 2R or not 2R: testing hypotheses of genome duplication in early vertebrates. J Struct Funct Genomics. 2003, 3: 85-93. 10.1023/A:1022681600462.
Amores A, Force A, Yan YL, Joly L, Amemiya C, Fritz A, Ho RK, Langeland J, Prince V, Wang YL, Westerfield M, Ekker M, Postlethwait JH: Zebrafish hox clusters and vertebrate genome evolution. Science. 1998, 282: 1711-1714. 10.1126/science.282.5394.1711.
Blanc G, Hokamp K, Wolfe KH: A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res. 2003, 13: 137-144. 10.1101/gr.751803.
Wong S, Butler G, Wolfe KH: Gene order evolution and paleopolyploidy in hemiascomycete yeasts. Proc Natl Acad Sci U S A. 2002, 99: 9272-9277. 10.1073/pnas.142101099.
Williams H, Brenner S, Venkatesh B: Identification and analysis of additional copies of the platelet-derived growth factor receptor and colony stimulating factor 1 receptor genes in fugu. Gene. 2002, 295: 255-264. 10.1016/S0378-1119(02)00736-9.
Robinson-Rechavi M, Marchand O, Escriva H, Bardet PL, Zelus D, Hughes S, Laudet V: Euteleost fish genomes are characterized by expansion of gene families. Genome Res. 2001, 11: 781-788. 10.1101/gr.165601.
Wittbrodt J, Meyer A, Schartl M: More genes in fish?. BioEssays. 1998, 20: 511-515. 10.1002/(SICI)1521-1878(199806)20:6<511::AID-BIES10>3.0.CO;2-3.
Gu X, Wang Y, Gu J: Age distribution of human gene families shows significant roles of both large- and small-scale duplications in vertebrate evolution. Nat Genet. 2002, 31: 205-209. 10.1038/ng902.
Lynch M, Conery JS: The evolutionary fate and consequences of duplicate genes. Science. 2000, 290: 1151-1155. 10.1126/science.290.5494.1151.
Wolfe KH, Shields DC: Molecular evidence for an ancient duplication of the entire yeast genome. Nature. 1997, 387: 708-713. 10.1038/42711.
McLysaght A, Hokamp K, Wolfe KH: Extensive genomic duplication during early chordate evolution. Nat Genet. 2002, 31: 200-204. 10.1038/ng884.
Van de Peer Y, Taylor JS, Meyer A: Are all fishes ancient polyploids?. J Struct Funct Genomics. 2003, 3: 65-73. 10.1023/A:1022652814749.
Robinson-Rechavi M, Marchand O, Escriva H, Laudet V: An ancestral whole-genome duplication may not have been responsible for the abundance of duplicated fish genes. Curr Biol. 2001, 11: R458-9. 10.1016/S0960-9822(01)00280-9.
Dowell RD, Jokerst RM, Day A, Eddy SR, Stein L: The Distributed Annotation System. BMC Bioinformatics. 2001, 2: 7-10.1186/1471-2105-2-7.
Fugu rubripes CTLDcp annotation DAS resource. [http://anz.anu.edu.au/das/]
Clamp M, Andrews D, Barker D, Bevan P, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, Durbin R, Eyras E, Gilbert J, Hammond M, Hubbard T, Kasprzyk A, Keefe D, Lehvaslaiho H, Iyer V, Melsopp C, Mongin E, Pettett R, Potter S, Rust A, Schmidt E, Searle S, Slater G, Smith J, Spooner W, Stabenau A, Stalker J, Stupka E, Ureta-Vidal A, Vastrik I, Birney E: Ensembl 2002: accommodating comparative genomics. Nucleic Acids Res. 2003, 31: 38-42. 10.1093/nar/gkg083.
Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, Durbin R, Eyras E, Gilbert J, Hammond M, Huminiecki L, Kasprzyk A, Lehvaslaiho H, Lijnzaad P, Melsopp C, Mongin E, Pettett R, Pocock M, Potter S, Rust A, Schmidt E, Searle S, Slater G, Smith J, Spooner W, Stabenau A, Stalker J, Stupka E, Ureta-Vidal A, Vastrik I, Clamp M: The EnsEMBL genome database project. Nucleic Acids Res. 2002, 30: 38-41. 10.1093/nar/30.1.38.
Ning Z, Cox AJ, Mullikin JC: SSAHA: a fast search method for large DNA databases. Genome Res. 2001, 11: 1725-1729. 10.1101/gr.194201.
EnsEMBL portal to v.2 of the Fugu rubripes genome annotation. [http://scrappy.fugu-sg.org/Fugu_rubripes/]
Reference table for Fugu rubripes CTLDcp genes. [http://anz.anu.edu.au:8080/Fugu_rubripes/]
Eddy SR: Profile hidden Markov models. Bioinformatics. 1998, 14: 755-763. 10.1093/bioinformatics/14.9.755.
Lewis SE, Searle SM, Harris N, Gibson M, Lyer V, Richter J, Wiel C, Bayraktaroglir L, Birney E, Crosby MA, Kaminker JS, Matthews BB, Prochnik SE, Smithy CD, Tupy JL, Rubin GM, Misra S, Mungall CJ, Clamp ME: Apollo: a sequence annotation editor. Genome Biol. 2002, 3: 1-14. 10.1186/gb-2002-3-12-research0082.
Birney E, Durbin R: Using GeneWise in the Drosophila annotation experiment. Genome Res. 2000, 10: 547-548. 10.1101/gr.10.4.547.
Krogh A, Larsson B, von Heijne G, Sonnhammer EL: Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001, 305: 567-580. 10.1006/jmbi.2000.4315.
Felsenstein J: PHYLIP (Phylogeny Inference Package) version 3.5c. 1993, Distributed by the author. Department of Genetics, University of Washington, Seattle
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22: 4673-4680.
Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, Fuellen G, Gilbert JG, Korf I, Lapp H, Lehvaslaiho H, Matsalla C, Mungall CJ, Osborne BI, Pocock MR, Schattner P, Senger M, Stein LD, Stupka E, Wilkinson MD, Birney E: The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002, 12: 1611-1618. 10.1101/gr.361602.
Letunic I, Goodstadt L, Dickens NJ, Doerks T, Schultz J, Mott R, Ciccarelli F, Copley RR, Ponting CP, Bork P: Recent improvements to the SMART domain-based sequence annotation resource. Nucleic Acids Res. 2002, 30: 242-244. 10.1093/nar/30.1.242.
Conery JS, Lynch M: Nucleotide substitutions and the evolution of duplicate genes. Pac Symp Biocomput. 2001, 167-178.
Li WH: Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J Mol Evol. 1993, 36: 96-99.
Nei M, Gojobori T: Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986, 3: 418-426.
Yang Z, Nielsen R: Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 2000, 17: 32-43.
Kumar S, Tamura K, Jakobsen IB, Nei M: MEGA2: molecular evolutionary genetics analysis software. Bioinformatics. 2001, 17: 1244-1245. 10.1093/bioinformatics/17.12.1244.
Acknowledgements
JEG is supported by the ANU IAS block grant, and ANZ is supported by a PhD scholarship from the ANU. We would like to thank Elia Stupka and the Fugu bioinformatics team at the Singapore Institute of Molecular and Cell Biology for help and allowing us access to the pre-release versions of the v.3 assembly annotation. We also thank Anders Krogh for providing us with a copy of the TMHMM program for local use.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
ANZ carried out the bioinformatics studies and participated in the interpretation of its results. JEG conceived the study and participated in the interpretation of its results. Both authors participated in writing the manuscript and approved its final form.
Electronic supplementary material
12864_2004_153_MOESM1_ESM.FASTA
Additional File 1: Transcript sequences for re-annotated Fugu CTLD genes. The file contains cDNA sequences (in FastA format) for all CTLDcp-encoding genes that were re-annotated by us (sequence identifiers starting with ANU). (FASTA 121 KB)
12864_2004_153_MOESM2_ESM.FASTA
Additional File 2: Protein sequences for re-annotated Fugu CTLD genes. The file contains protein product sequences (in FastA format) for all CTLDcp-encoding genes that were re-annotated by us (sequence identifiers starting with ANU). (FASTA 43 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Zelensky, A.N., Gready, J.E. C-type lectin-like domains in Fugu rubripes . BMC Genomics 5, 51 (2004). https://doi.org/10.1186/1471-2164-5-51
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-5-51