
Abstract
Background
Involvement of conservative molecular modules and cellular mechanisms in the widely diversified processes of eukaryotic cell morphogenesis leads to the intriguing question: how do similar proteins contribute to dissimilar morphogenetic outputs. Formins (FH2 proteins) play a central part in the control of actin organization and dynamics, providing a good example of evolutionarily versatile use of a conserved protein domain in the context of a variety of lineage-specific structural and signalling interactions.
Results
In order to identify possible plant-specific sequence features within the FH2 protein family, we performed a detailed analysis of angiosperm formin-related sequences available in public databases, with particular focus on the complete Arabidopsis genome and the nearly finished rice genome sequence. This has led to revision of the current annotation of half of the 22 Arabidopsis formin-related genes. Comparative analysis of the two plant genomes revealed a good conservation of the previously described two subfamilies of plant formins (Class I and Class II), as well as several subfamilies within them that appear to predate the separation of monocot and dicot plants. Moreover, a number of plant Class II formins share an additional conserved domain, related to the protein phosphatase/tensin/auxilin fold. However, considerable inter-species variability sets limits to generalization of any functional conclusions reached on a single species such as Arabidopsis.
Conclusions
The plant-specific domain context of the conserved FH2 domain, as well as plant-specific features of the domain itself, may reflect distinct functional requirements in plant cells. The variability of formin structures found in plants far exceeds that known from both fungi and metazoans, suggesting a possible contribution of FH2 proteins in the evolution of the plant type of multicellularity.
Similar content being viewed by others

Background
Proteins of the formin family (FH2 proteins) have an important role in the organization of the actin cytoskeleton in organisms as diverse as fungi, slime molds, metazoa and plants (reviewed in [1–3]). Formins have been implicated in processes such as budding of yeast cells, cytokinesis in Drosophila and Caenorhabditis, and formation of fruiting bodies in Dictyostelium (see e.g. [4–9]). Known mutations affecting formin function in vertebrates cause limb deformity and deafness [10, 11], again suggesting a role in morphogenetic processes.
Formins are defined by the presence of a hallmark domain, FH2, accompanied by a proline-rich FH1 domain and often also by other conserved sequence motifs shared only by a subset of FH2 proteins, such as the FH3 domain, a GTPase-binding domain (GBD), or coiled-coil regions [4, 12, 13]. The FH2 domain, whose structure has been recently determined [14, 15], acts as a dimer, nucleating new actin filaments by a novel Arp2/3 independent mechanism, which has been well documented in both yeast and metazoans [16–19]. This provides a mechanistic basis for the observed morphogenetic role of formins. The proline-rich FH1 motif binds profilin and contributes to the actin-nucleating activity and its regulation [20, 21]. Domains outside FH1 and FH2 provide a variety of "interfaces" for integration of the actin nucleating module into cellular regulatory networks (reviewed in [1, 2, 22]. For instance, a subfamily of Diaphanous-related formins may be mediating the effects of Rho class small GTPases on actin assembly and dynamics via a specific conserved domain [23, 24]. Other formins communicate with universal "adaptor" domains such as SH3 or WW (see e.g. [25–28], and at least indirectly even with the microtubule cytoskeleton [29–31].
Members of the formin family have been found also in higher plants, both experimentally [32] and by a bioinformatic approach [2, 33]. Formin-related sequences encoded by the complete Arabidopsis genome can be divided into two distinct subfamilies. One of them (Class I) contains mostly proteins with putative membrane insertion signals, and often with extensin-like proline-rich stretches in the predicted extracytoplasmic domain. This suggests a possible plant-specific mechanism of cytoskeleton-membrane connection, or even transmembrane anchorage of the cytoskeleton to the cell wall in case of plasmalemma-localized formins [33], which is now supported also by experimental data [32, 34]. No such motifs – and no conserved domains whatsoever besides FH1 and FH2 – were described in Class II formins so far. However, detailed analysis of the N-termini of Arabidopsis formins could not have been performed on the basis of a genome annotation where the majority of Class II formin genes appeared to be N-terminally truncated [2].
Here we report the results of a detailed structural and phylogenetic analysis of a collection of angiosperm formin-related sequences currently available in public databases, including the nearly complete rice genome. Using a comparative approach, we were able to refine the current annotation of the Arabidopsis formin-related genes, and to identify a novel N-terminal conserved domain shared by the majority of plant Class II formins. Moreover, the structure of this domain, which is related to the conserved and well-characterized protein phosphatase/tensin/auxilin fold, suggests a second possible plant-specific mechanism for integrating the formin-associated actin nucleation complexes into the cellular context.
Results and discussion
An inventory of Arabidopsis formins
An exhaustive in silicio search of the Arabidopsis genome has identified 22 occurrences of the FH2 domain in total of 21 annotated loci, described previously as AtFH1 to AtFH21 [2] see Table 1); the AtFH15 locus appears to encode two FH2 domains. Most of the formin-related genes reside at positions interspersed throughout the Arabidopsis genome. However, 5 loci (AtFH15, AtFH16, AtFH19, AtFH20 and AtFH21) form a tight cluster on chromosome 5.
At least partial cDNA sequences are available for 17 of the 21 loci. Expression data from the NASC microarray collection [35] suggest that the remaining genes are significantly expressed at least under some circumstances, although for AtFH12 and AtFH21 only very low transcript levels have been detected. Complete cDNAs have been sequenced only for AtFH1, AtFH5, AtFH9 and AtFH10, while the current genome annotation of the remaining genes is based mainly on automated splicing prediction. Such predictions are known to be error-prone, and inclusion of cDNA data can improve the annotation considerably [36, 37]. Homology among members of a large gene family can be used as an additional guide for identification of mispredicted intron-exon boundaries (see e.g. [38]). Taking into account both cDNA data and homology, we have found that the current annotation of 10 of the remaining 17 loci appears to be incorrect, and suggested modifications, although we still could not reliably identify the N-terminal exons of AtFH3 and AtFH12 (see Table 1 and Additional file 1).
Most of the suggested modifications represent extension of exons, or inclusion of extra exons that restore missing portions of the conserved FH2 domain, or of N-terminal regions of homology shared by multiple family members and revealed in TBLASTN searches (see Methods). This is the case of AtFH12, AtFH13, AtFH14, AtFH17, AtFH18 and AtFH20. In the case of AtFH20, the N-terminal portion of FH2, which was missing in the original prediction, was found in a neighboring gene (At5g07750), which therefore probably represents a part of the AtFH20 locus erroneously annotated as a separate gene.
In three loci (AtFH3, AtFH4 and AtFH16) attempts to restore a missing exon within FH2 revealed probable frameshift errors in the genome sequence. For AtFH3, we have included an internal exon exhibiting homology to the very closely related sequenced AtFH5 cDNA and a 3' extension of the ORF that was suggested by an alternative GenScan prediction (see Methods) as a new exon preceded by an unusually short (11 bp) intron. Since the smallest (protozoan) introns reported so far are 13 bp short and the majority of short introns in plants exceed the length of 30 bp [39], we suspect that the presumed "intron", which would contain a stop codon, may in fact be a part of a contiguous exon disrupted by omission of 1 base. Also in the internal exon, an extra base must be introduced in order to maintain the reading frame. Since both suspect areas are extremely GC-rich (and therefore notoriously difficult to sequence), we believe that an error in the genomic data is a likely explanation in both cases. For AtFH4, the original annotation predicts an intron disrupted by an in-frame stop codon at the position of the conserved G-N-X-M-N motif. However, this intron is poorly supported by WebGene and GenScan predictions and apparently not spliced in a sequenced cDNA, which contains an extra base in this area and restores the reading frame within a highly conserved portion of FH2. For AtFH16, the splicing pattern could have been inferred with a reasonable confidence, since most of the locus is covered by cDNAs. However, a contiguous reading frame throughout the conserved FH2 domain can be maintained only by inserting an extra base in a GC-rich area not covered by cDNA, again suggesting a sequencing error.
In case of AtFH15, the locus with two FH2 domains, a sequenced cDNA ends by a stretch corresponding to a presumed intron, which contains multiple stop-codons. We believe that the locus either represents two related neighboring genes (further referred to as AtFH15a and AtFH15b), or produces multiple gene products by alternative splicing. Evidence for cDNA-supported alternative splicing, documented in metazoan formins [40], has been found also for AtFH13 and AtFH20, as well as for two rice formin homologues (see below and Additional file 2). In the following structural analysis only the longest predicted proteins have been taken into account.
Nine of the eleven mispredicted loci code for formins previously classified as Class II, while most Class I formins appear to be predicted correctly. The complex structure of Class II formin genes, which possess substantially more intron-exon boundaries than their Class I relatives, may be sufficient to explain the difference [36]. Moreover, both mispredicted Class I loci appear to contain sequencing errors, and one of them, AtFH3, is expressed almost exclusively in pollen according to the results of a recent microarray analysis [35, 41]. This narrow tissue specificity might be associated with a modification of the "housekeeping" splicing apparatus, whose function has been so far characterized mainly on the basis of data from vegetative tissues. We therefore believe that the difficulties in predicting AtFH3 structure (including the lack of a reliable N-terminus) may partly reflect the particular expression pattern of this gene.
Phylogeny of the plant FH2 proteins
Previous phylogenetic analyses of plant formins [2, 33] included only Arabidopsis data. We have used the re-annotated Arabidopsis formin sequences as a query for identifying genes encoding FH2 proteins from available angiosperm sequences in the public databases, including the nearly complete rice genome and a recently published large collection of rice cDNAs [42]. At least partial cDNA or genomic sequences corresponding to 79 putative formin-related genes from cotton, soybean, barley, tomato, trefoil, alfalfa, tobacco, rice, pea, sorghum, potato, wheat, grapevine and maize have been found (Additional files 2 to 4).
Complete FH2 domain sequence could have been reconstructed for 29 of the non-arabidopsis sequences. This subset, together with 22 Arabidopsis FH2 domains and a selection of fungal, slime mold and metazoan formins, has been used to construct an unrooted phylogenetic tree (Fig. 1) using the NJ method [43]. For the remaining sequences, closest neighbors have been determined using the BLAST algorithm (Table 2). All of the plant FH2 domains studied so far, including the incomplete ones, can be unequivocally assigned to one of the two previously proposed classes [2]; Table 2). Also the overall domain composition and domain order of available complete plant formins – i.e. sequences outside FH2 – reflects rather well the dichotomy between Class I and Class II formins (see below).

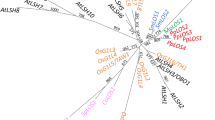
An unrooted phylogenetic tree of the plant FH2 domains. For description of the plant genes see Table 1 and Additional file 2. Selected fungal and metazoan sequences are included: fission yeast Cdc12 (Sp CDC12, CAA92232.1), Fus1 (Sp FUS1, T43296) and For3 (Sp FOR3, CAA22841.1), budding yeast Bni1 (Sc BNI1, P41832) and Bnr1 (Sc BNR1, P40450), Dictyostelium ForA (Dd ForA, BAC16796.1), ForB (Dd ForB, BAC16797.1) and ForC (Dd ForC, BAC16798.1), Caenorhabditis Cyk-1 (Ce CYK1, AAM15566.1), Drosophila Diaphanous (Dm Dia, P48608) and Cappucino (Dm Capp, 2123320A), mouse Formin (Mm FOR, Q05860) and Diaphanous (Mm Dia, AAC53280.1), fugu Formin (Fr FH, AAC34395.1), human FHOS (Hs FHOS, AAD39906.1). Symbols at nodes denote percentual bootstrap values (out of 500 replicates); no symbol means less than 50 % node stability, the sequence used as forced root for tree construction is marked by an arrow. For complete or nearly complete plant genes, sequences are color-coded according to their overall domain structure (see Fig. 3). Proteins encoded by the Arabidopsis chromosome V cluster are denoted by an asterisk.
The presence of two classes of formins appears to be a general feature of plants. However, although several subclasses containing representatives of more than one species can be distinguished within the two classes (see Fig. 1 – branches Ia to Ie, IIa and IIb, Table 1, Table 2), only occasionally true orthology between genes from different species was established. A similar pattern has been previously observed for another large plant gene family encoding the plethora of phospholipase D isoforms [38]. Very closely related proteins that might represent true orthologues (Table 2, sequences in bold) were found mostly within limited taxonomical groups such as the grasses (barley, rice, sorghum, wheat), the legumes (soybean, alfalfa), or Solanaceae (tobacco, potato and tomato). The observed pattern of paralog distribution may suggest that a number of gene duplications or polyploidization events occurred relatively recently compared to the separation of the angiosperm lineages included in the analysis. An extreme example of such a recent gene multiplication is presented by the well-defined subgroup of Class II genes corresponding to the clustered loci on the Arabidopsis chromosome V (marked by asterisk in Fig. 1).
Structural diversity of the plant FH2 domains
Surprisingly, major differences between Class I and Class II formins have been found within the relatively well-conserved FH2 domain. Hallmark features for both classes of plant formins can be identified already at the level of amino acid sequence in the C-terminal portion of the FH2 domain, which is less conserved than the central area around the G-N-X-M-N motif (see Additional file 5). While Class I formins contain a consensus V/I-R-D-F-L motif about 170–190 aa from the conserved core, Class II proteins possess a signature M-H-Y-L/Y-C-K, located usually 31 aa downstream of the central motif.
Data on 3-dimensional structure of the FH2 domain have been published recently for the yeast Bni1p formin [14], see also Fig. 2). Its FH2 domain folds into a structure consisting from N-terminal "lasso", connected by a predominantly helical linker to the globular "knob" region, which is followed by a coiled-coil assembly of three α-helices and a "post" domain, again predominantly α-helical. The protein can dimerize through interaction of the lasso and post domains, producing flexible ring-like "head to tail" dimers with putative actin-binding sites on the part of the inner surface of the ring, provided by the knob. Residues directly participating in the actin-binding site have been identified by site-directed mutagenesis of selected positions conserved among Bni1p and metazoan formins of the Diaphanous type. Mutants unable to form dimers do not nucleate actin in vitro, and proteolytically cleaved "hemidimers" have been shown to block barbed end elongation, acting as capping proteins rather than nucleators [14, 15]. Apparently, dimerization can add another dimension to the diversity of plant formins, since, in theory, the 22 Arabidopsis FH2 domains could produce up to 484 different homo- and heterodimers. However, the actual number will be lower, since formins do exhibit tissue-specific expression patterns, as documented by analysis of available expression data [35]. Moreover, structural differences may prevent heterodimerization of some protein pairs.

Summary of structural variation in plant FH2 domains. Structure of the yeast Bni1p FH2 domain (PDB 1UX5), with marked positions of major insertions (arrows), deletions (flags pointing towards the missing portion of sequence) and conserved site mutations (colored balls) found in Arabidopsis formins. Grey balls denote positions of insertions found in multiple proteins, numbers correspond to conserved amino acid positions in Bni1p.
Comparison of Arabidopsis FH2 domain sequences with the sequence of Bni1p (see Additional file 5) revealed surprising plant-specific features in Class I formins (Fig. 2). All Class I formins except AtFH9 and AtFH10 have a small or non-polar amino acid at the position corresponding to K1639 of Bni1p, a conserved Lys residue contributing to the actin binding site and required for efficient nucleation [14]. However, all Class I proteins have a relatively large insertion (12–54 aa) in the vicinity (position 1620 of Bni1p), suggesting an alternative construction of the actin-binding site. It is worth noting that the shortest insertions were found in AtFH9 and AtFH10, i. e. the only Class I formins that have kept – or, more likely, restored – the consensus Lys residue. On the other hand, AtFH9 and AtFH10, which are mutually closely related (see Fig. 1), exhibit deviations from the Bni1p/Diaphanous consensus in portions of the molecule that are involved in dimerization (a deletion in the lasso of AtFH9, altered structure of the post in AtFH10). It is tempting to speculate that these alterations might result in a restriction of (hetero)dimerizing capability of AtFH9 and AtFH10, although we cannot, at present, predict which dimers will be preferred or excluded.
On the other hand, the overall structure of most Class II formins basically corresponds to the Bni1p/Diaphanous consensus, with several notable exceptions. AtFH20 contains two insertions in different strands of the coiled-coil part of the molecule. Insertions of 15–43 aa have been found also in the post region of AtFH13, AtFH15a and AtFH16, close to the site of the common insertion in Class I formins. AtFH13 has also an insertion in the lasso region, with possible effect on dimerization.
The case of AtFH15a and its neighbour or splicing variant AtFH15b is rather enigmatic, since both proteins miss substantial portions of the FH2 domain (part of post and coiled-coil in AtFH15a, lasso in AtFH15b) and, moreover, each of them lacks one of two residues important for actin nucleation – an essential isoleucine (I1431 of Bni1) in AtFH15a and a lysine (K1601 of Bni1) in AtFH15b. We suspect that AtFH15a and AtFH15b may present two out of many possible splicing alternatives of the complex AtFH15 locus, which may be encoding multiple proteins, including perhaps both a "complete" active formin and regulatory variants without nucleation and/or dimerization activity.
Two more proteins have mutations in the conserved positions required for actin nucleation. In AtFH21, the K1601 mutation is accompanied by a large insertion in the flexible linker. This appears to be a result of a partial gene duplication involving also the lasso-linker area of FH2, which has been subsequently lost from the posterior copy. Most dramatic deviation from the conserved structure has been found in AtFH12, which lacks both I1431 and K1601 and the whole lasso-linker assembly, while the presumed dimerization region of the post is altered. Such a protein may perhaps present a naturally occurring "hemidimer" variant, acting as a barbed end cap rather than a nucleating centre.
Plant formins exhibit variable domain composition
Following the phylogenetic analysis, we have examined the overall domain composition of plant FH2-containing proteins, searching for known sequence or structure motifs. Several patterns of conserved domain order can be distinguished in the complete plant formin sequences (Fig. 3). We further refer to these patterns as structural types A through F. Besides of the conserved domains, some formins contain long stretches of sequence (55–870 amino acids) lacking any conserved motifs, located either between FH1 and FH2 (AtFH6, AtFH21) or C-terminally (AtFH16, OsFH14).

Domain composition of plant FH2 proteins. Schematic representation of the domain composition and order encountered in plant FH2 proteins (domains of variable size, such as FH1, and unique sequences not to scale). Note that only structures E and F correspond to those found outside the plant kingdom.
Most plant formins contain proline-rich sequences, often called FH1 in the formin context. However, neither the FH3 domain nor additional motifs common in FH proteins, such as the DBD motif shared by diaphanous-related formins, or the coiled coil, were found in plant FH proteins (a single coiled-coil domain, located between FH1and FH2, has been found in AtFH21).
It has been noted previously [33] that most Arabidopsis Class I formins contain putative secretion or membrane insertion signals and transmembrane segments, indicating that they may be integral membrane proteins. A second proline-rich domain, reminiscent of some cell wall proteins such as the extensins, is often located in the presumed extracytoplasmic portion of the protein (Fig. 3, structure A). Indeed, association with insoluble cellular fractions has been reported for one of the presumed transmembrane formins [32], providing support for the hypothesis that this type of formins may mediate anchorage of actin nucleation sites to the cell wall across the plasmalemma. We found similar sequences also in the majority of complete non-arabidopsis Class I sequences (see color coding in Fig. 1), although transmembrane segments appeared to be on the edge of significance for the rice sequences OsFH10 and OsFH11. We believe that also AtFH3 may be a type A (transmembrane) formin, since its current predicted sequence appears to be 5'-truncated, and its closest relative, AtFH5, exhibits type A structure. The only non-membrane Class I formin in Arabidopsis is AtFH7, which resembles "standard" animal formins with FH1 and FH2 motifs but possesses a unique repetitive structure in its N-terminal half. No other Class I proteins of this structure have been found so far, and it remains to be clarified whether this is a representative of a common, though less abundant, Class I formin type, or a relatively recent modification, present perhaps only in one or a handful of species. The same can be said about the remaining non-membrane Class I formin from rice, OsFH14, which has an extremely long C-terminal extension unparalleled elsewhere. However, until complete cDNA sequence becomes available, we cannot exclude the possibility of artifacts resulting from wrong splicing prediction in the weakly conserved parts of these loci.
While examining the predicted protein structures of Class II formins, we have noticed an area of mutual similarity in the N-terminal half of a subset of these proteins. This area exhibits also considerable similarity to the structurally well-characterized PTEN domain known from metazoans (see below; Fig. 4). Subsequently, we have found this motif, located N-terminally from the conventional FH1 and FH2 domains, in a majority of Class II formins (type D). The only exceptions are the rather diverged AtFH12 protein, AtFH17 and a group of mutually related formins encoded by genes of the cluster on the Arabidopsis chromosome V that apparently arose by a relatively recent series of gene duplication events. A clear distinction between Class I and Class II formins is therefore not restricted to the structural features of the FH2 domain itself, but extends also to features outside FH1 and FH2; presence of the PTEN-related domain can be considered a hallmark feature of a subset of Class II plant formins.

The PTEN domain of selected plant formins. For terminology of the plant proteins see Tables 1 and 2; the remaining sequence in the alignment is human PTEN (HsPTEN, AAD13528.1). Amino acids conserved between at least one of the plant sequences and PTEN are shown in yellow for the protein phosphatase-related domain and in light blue for the C2 domain; residues conserved between at least six plant formins are inverted, and marked by asterisks if found also in PTEN. The lipid/protein phosphatase signature is in red, the putative regulatory phosphorylation site (T383) in dark blue. Note that only OsFH3 can be phosphorylated at the corresponding position. Secondary structure prediction for AtFH13 is shown above the alignment (a – α-helix, b – β-sheet); results for other Arabidopsis formins were analogous.
Aberrations from the characteristic domain composition of Class I and Class II formins (types A and D, respectively) appear to be mostly Arabidopsis-specific, and often associated with relatively recent gene duplications (a partial internal duplication involving a segment of the FH2 domain has been identified in AtFH21). Although an Arabidopsis-specific tendency to duplicate formin-related sequences cannot be excluded a priori, we believe that a more likely explanation is that duplicated sequences are notoriously difficult to analyse, and therefore tend to be the last ones to make their way into genome databases.
A phosphatase/tensin/PTEN-related domain in most Class II formins
While screening for known domains in plant formins using the SMART package [44], we found a significant match to the undefined specifity protein phosphatase domain (PTPc_DSPc, SM0012, BLAST E = 9. 10-6) in the N-terminal part of AtFH18. Position-specific iterated BLAST [45] revealed a conserved domain (KOG2283 – clathrin coat dissociation kinase GAK/PTEN/auxilin) in the same region. The conserved domain falls into an area exhibiting high degree of conservation between AtFH18 and three other Arabidopsis family members (AtFH13, 14 and 20), and a related motif has been found also in several non-Arabidopsis Class II plant formins (OsFH3, 5, 6, 7 and 12, MtFH6; see Fig. 4).
Search of the 3D-PSSM protein fold library [46] with the multiple alignment of AtFH13, 14, 18 and 20 as a probe predicted significant structural similarity to a well-defined three-dimensional structure element, the c1d5ra_ fold. The prototype of this fold, the PTEN tumor suppressor (PDB 1d5r), is a member of a wider superfamily that includes, among others, also protein phosphatases, tensin and the tensin/auxilin domain of the cyclin G associated protein kinase (GAK) – i.e. sequences that have defined the KOG2283 domain. The presence of a PTEN-related structural motif has been independently confirmed by another sequence-structure threading method – FUGUE [47], while a third algorithm – SAM-T99 [48] – failed to recognize any conserved elements in this area. However, a secondary structure similar to that established for PTEN has been independently predicted from the sequences of AtFH13, 14 and 18 by PSIPRED [49]. Taken together, these results indicate the presence of a conserved sequence and structure motif in multiple Class II plant formins. We will further refer to this sequence/structure element as the PTEN domain.
The prototype of the PTEN domain is the human PTEN (phosphatase and tensin-related) antioncogene, whose mutation results in rapid development of multi-organ tumors in humans [50, 51]. The conserved part of human PTEN protein consists of two structural units. First unit (corresponding to positions 7–185 of the human protein) has the structural similarity to dual specificity protein phosphates, contains the active site signature of protein phosphatases, HCXXGXXR, and possesses both a lipid phosphatase activity with a strong affinity to PtdIns(3,4,5)P3 and a weak protein tyrosine phosphatase activity [52, 53]. The second structural unit, related to a class of domains collectively referred to as C2, will be discussed in more detail below.
A portion of the PTEN molecule, including the phosphatase-like domain, exhibits significant similarity to the N-terminal domain of tensin, a multifunctional component of integrin-mediated focal adhesions known to participate in cell motility and cell adhesion [50, 54]. A related domain was found also in some metazoan auxilins (proteins involved in uncoating of clathrin-coated vesicles), and in the cyclin G-associated protein kinase (GAK), which has an auxilin-like domain [50, 55].
Some of the metazoan proteins sharing the PTEN domain are involved in the structural organization of cell regions exhibiting a complex cytoskeletal pattern. Tensin is associated with actin in focal adhesions, acting both as a barbed-end cap and a cross-linking protein [56]. Overexpression of PTEN results in alteration of the structure of the actin cytoskeleton [57]. Moreover, although PTEN does not exhibit a specific intracellular localization, it binds to proteins localized to tight junctions in epithelia, and appears to be essential for embryonic development in mice. Together with its apparent participation in the control of cell growth, adhesion, migration, invasion and apoptosis, this suggests a possible role in the building of the cell surface and in cellular processes that involve a substantial contribution of the actin cytoskeleton [53]. In plants, homologous proteins may be therefore expected to participate in the construction of the cytoplasmic portion of the cell wall – membrane – cytoskeleton continuum.
The PTEN-related domain may have a structural rather than catalytic role
To our surprise, PTEN domains in plant formin sequences bear mutations that make both protein and lipid phosphatase activity very unlikely (Fig. 4). The last arginine residue in the phosphatase active site, which has been shown to be crucial for catalysis [58, 59], is replaced by hydrophobic or small polar residues in the plant proteins. Another residue essential for catalysis, Asp 92, that act as a general acid to faciliate protonation of the phenolic oxygen atom of the tyrosyl group [60] was in the plant formins substituted by glycine. We therefore believe that the function of plant PTEN domains is rather structural than catalytic.
Such a structural role could perhaps be attributed mainly to the second portion of the conserved PTEN motif. This second conserved unit (corresponding to positions 186–351 of human PTEN) is similar to the C2 domain that is known from a variety of proteins with multiple functions including membrane fusion, vesicular transport, GTPase regulation, protein phosphorylation, and protein degradation. The C2 domain mediates protein-membrane or protein-protein interactions, often dependent on the presence of Ca2+ ions. However, not all C2 domains exhibit the same membrane-association mechanism. Some of them, such as those of synaptotagmin or phospholipase Cβ, bind to membrane surface, while others, such as the C2 domain of phospholipase A2, invade the membrane by insertion of variable C2 domain loops [61]. The C2 domain of PTEN cannot bind calcium ions. Instead, two stretches of basic residues mediate peripheral binding to the membrane by electrostatic interaction [62]. The PTEN C2 domain therefore defines a specific class of calcium-independent C2 domains [61].
We were unable to detect C2 domains in plant formins by any of the software used to find PTEN similarity (see above and Methods). This can be due to a huge sequence divergence within the C2 domains family, reflecting the diversity of functions they participate in. However, upon visual inspection of sequence alignment, we found obvious sequence similarity to the C2 domain of human PTEN protein, including the regions that make PTEN Ca2+ independent (Fig. 4).
Using the human PTEN C2 domain as a template, we were able to produce 3D models of three representative plant formin C2 domains, AtFH13, AtFH14 and AtFH15, further strengthening the notion that plant Class II formins do possess a C2 domain. Surface charge distributions of all three formin C2 domains resemble those of phospholipase A2 (PLA2) rather than PTEN (Fig. 5). Membrane association of the PLA2 C2 domain is Ca2+-dependent and occurs via hydrophobic interactions with zwitterionic phospholipids and insertion of C2 loops into the membrane. However, since the formin C2 domains appear to be unable to bind Ca2+ (at least not in the same manner as PLA2), although they perhaps could be neutralised by other means, it remains to be decided experimentally whether plant Class II formins interact with membranes via their C2 domains.

Model of the C2 domain of AtFH13. 3D model of the AtFH13 C2 domain and its predicted surface potential compared to that of human PTEN (PDB 1d5r) and calcium-free human PLA2 (PDB 1bci) C2 domains (red – negative, blue – positive). All models are oriented membrane side upwards. Analogous results have been obtained also for AtFH14 and AtFH18.
Phosphorylation of a threonine residue downstream from the C2 domain (at position 383 of human PTEN) was recently found to modulate the ability of PTEN to modify cell migration in culure [63]. However, the phosphorylated motif, including the crucial threonine, is not conserved in plant sequences, suggesting that this regulation may be specific for the metazoan lineage (Fig. 4).
It is therefore tempting to speculate that the widespread occurrence of the PTEN domain among plant Class II formins may suggest a function analogous to that proposed for the transmembrane segments in their Class I counterparts. A variant PTEN domain that has lost its catalytic activity by mutation but retained its intracellular localisation may act as an anchor positioning the actin-nucleating sites (FH2) to some intracellular (and possibly cell surface/plasmalemma-associated) structures.
Conclusions
One of the most intriguing questions of contemporary molecular biology is how can vastly dissimilar organisms develop utilizing a limited repertoire of basically similar molecules derived from a relatively small set of conserved protein domains? Processes of eukaryotic cell morphogenesis, such as shaping of the actin cytoskeleton, provide a good example of such a versatile usage of conserved molecular mechanisms. Well-conserved protein domains, such as the FH1/FH2 motifs shared by formins and participating in actin nucleation, are being used for a broad range of cellular tasks in various eukaryotic lineages. It is plausible to assume that the diversity of cellular functions may to a large extent stem from the context those domains assume within the framework of larger, multidomain protein molecules.
We have examined the phylogeny and molecular context of the FH2 domain in available angiosperm formin homologues. Besides of confirming the existence of two classes of plant formins, suggested previously solely on the basis of Arabidopsis data, we could identify a novel domain shared by a significant portion of Class II formins and possibly involved in the association between formin-based actin nucleation modules and other cellular structures.
Another interesting aspect is the extremely dynamic evolution of the angiosperm formin family, with ample evidence for multiple gene duplication events (sometimes accompanied by major domain rearrangements) not seen in other species studied so far. At the moment, we can only speculate whether this is a feature specific for a small selection of plant taxa, or a general characteristic of plant formins. However, we hope that publication of more complete plant genomes can help to resolve this question in a not so distant future.
The enormous variety of formins within a single plant organism far exceeds that found in fungi and metazoans, where only a handful of formin genes exist, although their diversity can be enhanced by alternative splicing [13] and heterodimerization [14]. However, even the relatively small Arabidopsis genome contains no less than 21 FH2 domain-encoding loci, while possibilities for alternative splicing and dimerization remain open, resulting in literally hundreds of possible species of the functional formin dimer. It is tempting to speculate that this diversity may be related to the demands of actin-nucleation site positioning with respect to precisely defined cellular surfaces in the context of a multicellular body consisting of cells endowed with rather rigid surface structures. Consequently, a major increase in formin diversity could be expected at the transition between unicellular green algae and multicellular plants. However, testing of this hypothesis would have to wait until more complete data from algal and gymnosperm genome projects become available.
Methods
Identification of FH2-containing plant genes
Arabidopsis loci encoding putative formin homologues have been identified by BLASTP and TBLASTN searches [64] of both predicted protein and complete genome sequence databases (at TAIR and NCBI) using previously characterized members of the family [33] as the query. The same loci have been found by both approaches. Analogous searches have been performed with the most diverged sequences from the first round as the query, until no new significant matches appeared. Presence of FH2 domains in predicted candidate open reading frames has been confirmed by a SMART search [44] for all genes. Sequences from other plant species coding for related proteins have been identified in analogous searches of Entrez nr, est and htgs databases.
Detection of gene expression
For Arabidopsis genes with no available cDNA sequence, microarray slides with highest transcript levels within the NASCArrays data set [35] have been detected using the Spot History tool. Visual inspection of the Detection parameter in full slide data has been used to confirm that detected expression levels were significantly above zero.
Gene structure predictions
For every Arabidopsis formin-related gene, exhaustive TBLASTN searches of the Entrez nr and est databases have been performed in order to identify all cDNAs derived from the chromosomal locus. Resulting cDNA sequences were aligned to the genomic and predicted ORF sequences with the aid of the MACAW program [65]. Conflicts between the genomic and cDNA sequence have been resolved in favor of the genomic version unless strong evidence suggested genomic sequence errors (see Results and Discussion). In cases of either a complex splicing pattern without experimental support or apparent deletions in the FH2 portion of the sequence, an alternative splicing prediction has been obtained using GenScan [66, 67]. GenScan usually agreed reasonably well with the original genome annotation, although it did miss exons occasionally. WebGene [68] or MZEF [69] have been used as well in some cases, however these programs appeared to be inferior with respect to both performance (agreement with cDNA or the FH2 consensus) and output readability. The original splicing predictions have been modified for some genes, taking into account alternative predictions and the structure of closest homologues (see Results, Table 1 and Additional file 1). Programs of the Sequence manipulation suite [70] version 2 [71] have been used for general sequence manipulation, assembly and translation tasks.
For non-arabidopsis genomic sequences, gene models have been produced in an analogous manner, based on combination of existing genome annotation (if available), GenScan and WebGene predictions and alignment to the closest Arabidopsis relatives.
Protein sequence alignments and phylogeny reconstruction
To produce an alignment of FH2 domain sequences, selected representative members of divergent branches of the family have been aligned using ClustalW [72] configured to run as a helper application under the BioEdit package [73], using the BLOSUM matrix series. The resulting core alignment has been modified manually using BioEdit, taking into account independently produced alignments of selected subsets of sequences, made with the aid of the MACAW software [65] as described previously [33]. Sequences closely related to those already present in the core alignment have been merged to the alignment manually in the BioEdit environment. Alignments of the PTEN domain have been produced in an analogous manner, taking into account 3D structure data (see below).
All positions containing gaps in at least one sequence have been removed prior to the construction of the phylogenetic tree. An unrooted phylogenetic tree based on the resulting alignment has been produced with the aid of the Treecon package [74] using the neighbor-joining (NJ) algorithm [43] with Poisson correction for distance estimation.
Protein domain recognition
The SMART program package [44, 75] version 4 has been used for identification of known domains, secretory signals (by the SignalP method – [76]), transmembrane segments (by the TMHMM algorithm – [77]) and repetitive sequence motifs within predicted formin sequences. Unless stated otherwise, only signals above the default significance threshold have been taken into account.
Additional series of domain searches has been performed using reverse position-specific BLAST [45] against the CDD database (version 1.65), which contains a larger selection of domains than the SMART collection (including FH3 and DBD), with the same results.
3D structure searches and alignments
The 3D-PSSM threading algorithm [46] has been used to find possible known protein folds in the shared N-terminal domain of AtFH13, 14, 18 and 20, using a multiple alignment of the four sequences as a probe. The result appeared to be highly significant (over 95 % confidence, PSSM E = 0.00179 for the 1d5ra_ fold). For independent confirmation, a FUGUE version v2.s.07 [47, 78] search against the HOMSTRAD fold library has been performed for two selected sequences (AtFH13 and AtFH14), resulting in identification of the same fold in both cases as "certain". A third algorithm, SAM-T99 [48, 79] produced no significant results. However, this program is known to have a very powerful filter against false negatives that loses some positives, especially those with predominantly helical structures (see manuals at the program website).
Secondary structure predictions and homology modelling
Secondary structure of type II formins was predicted using a PSIPRED server [49, 80].
The structure of the human Phosphoinositide phosphotase PTEN (1d5r) was used as a template for modeling the C2 domains of AtFH13, AtFH14 and AtFH18. The template and the target structures were aligned with ClustalW [72] and the resulting alignment was manually edited with help of the secondary structure prediction outputs.
The WHAT IF program [81] was used for modeling as described [82]. The sequence alignments and coordinates of the models are available as Additional files 6 to 9.
SwissProt Deep View [83, 84] has been used to calculate electrostatic potentials of the 3-D models of formins and to generate their figures, which have been graphically visualized using the PovRay 3.5 raytracing software [85].
References
Wallar BJ, Alberts AS: The formins: active scaffolds that remodel the cytoskeleton. Trends Cell Biol. 2003, 13: 435-446. 10.1016/S0962-8924(03)00153-3.
Deeks MJ, Hussey P, Davies B: Formins: intermediates in signal transduction cascades that affect cytoskeletal reorganization. Trends Plant Sci. 2002, 7: 492-498. 10.1016/S1360-1385(02)02341-5.
Zigmond SH: Formin-induced nucleation of actin filaments. Curr Opin Cell Biol. 2004, 16: 99-105. 10.1016/j.ceb.2003.10.019.
Castrillon DH, Wasserman SA: Diaphanous is required for cytokinesis in Drosophila and shares domains of similarity with the limb deformity gene. Development. 1994, 120: 3367-3377.
Evangelista M, Blundell K, Longtine MS, Chow CJ, Adames N, Pringle JR, Peter M, Boone C: Bni1p, a yeast formin linking Cdc42p and the actin cytoskeleton during polarized morphogenesis. Science. 1997, 276: 118-122. 10.1126/science.276.5309.118.
Fujiwara T, Tanaka K, Mino A, Kikyo M, Takahashi K, Shimizu K, Takai Y: Rho1p-Bni1p-Spa2p interactions: implication in localization of bni1p at the bud site and regulation of the actin cytoskeleton in saccharomyces cerevisiae. Mol Biol Cell. 1998, 9: 1221-1233.
Magie CR, Meyer MR, Gorsuch MS, Parkhurst SM: Mutations in the Rho1 small GTPase disrupt morphogenesis and segmentation during early Drosophila development. Development. 1999, 126: 5353-5364.
Ozaki-Kuroda K, Yamamoto Y, Nohara H, Kinoshita M, Fujiwara T, Irie K, Takai Y: Dynamic localization and function of Bni1p at the sites of directed growth in Saccharomyces cerevisiae. Mol Cell Biol. 2001, 21: 827-839. 10.1128/MCB.21.3.827-839.2001.
Huckaba TM, Pon LA: Cytokinesis: Rho and Formins Are the Ringleaders. Curr Biol. 2002, 12: R813-R814. 10.1016/S0960-9822(02)01316-7.
Trumpp A, Blundell PA, de la Pompa JL, Zeller R: The chicken limb deformity gene encodes nuclear proteins expressed in specific cell types during morphogenesis. Genes Dev. 1992, 6: 14-28.
de la Pompa JL, James D, Zeller R: The limb deformity proteins during avian neurulation and sense organ development. Dev Dyn. 1995, 204: 156-167.
Petersen J, Nielsen O, Egel R, Hagan IM: FH3, a domain found in formins, targets the fission yeast formin FUS1 to the projection tip during conjugation. J Cell Biol. 1998, 141: 1217-1228. 10.1083/jcb.141.5.1217.
Zeller R, Haramis AG, Zuniga A, McGuigan C, Dono R, Davidson G, Chabanis S, Gibson T: Formin defines a large family of morphoregulatory genes and functions in establishment of the polarising region. Cell Tissue Res. 1999, 296: 85-93. 10.1007/s004410051269.
Xu Y, Moseley JB, Sagot I, Poy F, Pellman D, Goode BL, Eck MJ: Crystal structures of a formin homology-2 domain reveal a tethered dimer architecture. Cell. 2004, 116: 711-723. 10.1016/S0092-8674(04)00210-7.
Shimada A, Nyitrai M, Vetter IR, Kuhlmann D, Bugyi B, Narumiya S, Geeves MA, Wittinghofer A: The core FH2 domain of diaphanous-related formins is an elongated actin binding protein that inhibits polymerization. Mol Cell. 2004, 13: 511-522. 10.1016/S1097-2765(04)00059-0.
Evangelista M, Pruyne D, Amberg DC, Boone C, Bretscher A: Formins direct Arp2/3-independent actin filament assembly to polarize cell growth in yeast. Nat Cell Biol. 2002, 4: 32-41. 10.1038/ncb718.
Pruyne D, Evangelista M, Yang C, Bi E, Zigmond SH, Bretscher A, Boone C: Role of formins in actin assembly: nucleation and barbed-end association. Science. 2002, 297: 612-615. 10.1126/science.1072309.
Severson AF, Baillie DL, Bowerman B: A Formin Homology Protein and a Profilin Are Required for Cytokinesis and Arp2/3-Independent Assembly of Cortical Microfilaments in C. elegans. Curr Biol. 2002, 12: 2066-2075. 10.1016/S0960-9822(02)01355-6.
Li F, Higgs HN: The mouse formin mDia1 is a potent actin nucleation factor regulated by autoinhibition. Curr Biol. 2003, 13: 1335-1340. 10.1016/S0960-9822(03)00540-2.
Pring M, Evangelista M, Boone C, Yang C, Zigmond SH: Mechanism of formin-induced nucleation of actin filaments. Biochemistry. 2003, 42: 486-496. 10.1021/bi026520j.
Kovar DR, Kuhn JR, Tichy AL, Pollard TD: The fission yeast cytokinesis formin Cdc12p is a barbed end actin filament capping protein gated by profilin. J Cell Biol. 2003, 161: 875-887. 10.1083/jcb.200211078.
Cvrčková F, Bavlnka B, Rivero F: Evolutionarily conserved modules in actin nucleation: lessons from Dictyostelium and plants. Protoplasma. 2004,
Alberts AS: Diaphanous-related Formin homology proteins. Curr Biol. 2002, 12: R796-R796. 10.1016/S0960-9822(02)01309-X.
Olson MF: GTPase Signalling: New Functions for Diaphanous-Related Formins. Curr Biol. 2003, 13: R360-R362. 10.1016/S0960-9822(03)00277-X.
Fujiwara T, Mammoto A, Kim Y, Takai Y: Rho small G-protein-dependent binding of mDia to an Src homology 3 domain-containing IRSp53/BAIAP2. Biochem Biophys Res Commun. 2000, 271: 626-629. 10.1006/bbrc.2000.2671.
Kamei T, Tanaka K, Hihara T, Umikawa M, Imamura H, Kikyo M, Ozaki K, Takai Y: Interaction of Bnr1p with a novel Src homology 3 domain-containing Hof1p. Implication in cytokinesis in Saccharomyces cerevisiae. J Biol Chem. 1998, 273: 28341-28345. 10.1074/jbc.273.43.28341.
Tominaga T, Sahai E, Chardin P, McCormick F, Courtneidge SA, Alberts A: Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol Cell. 2000, 5: 13-25. 10.1016/S1097-2765(00)80399-8.
Yayoshi-Yamamoto S, Taniuchi I, Watanabe T: FRL, a novel formin-related protein, binds to Rac and regulates cell motility and survival of macrophages. Mol Cell Biol. 2000, 20: 6872-6881. 10.1128/MCB.20.18.6872-6881.2000.
Chang F: Movement of a cytokinesis factor cdc12p to the site of cell division. Curr Biol. 1999, 9: 849-852. 10.1016/S0960-9822(99)80372-8.
Ishizaki T, Morishima Y, Okamoto M, Furuyashiki T, Kato T, Narumiya S: Coordination of microtubules and the actin cytoskeleton by the effector mDia1. Nat Cell Biol. 2001, 3: 8-14. 10.1038/35050598.
Kato T, Watanabe T, Morishima Y, Fujita A, Ishizaki T, Narumiya S: Localization of a mammalian homolog of Diaphanous, mDia1, to the mitotic spindle in HeLa cells. J Cell Sci. 2001, 114: 775-784.
Banno H, Chua NH: Characterization of the arabidopsis formin-like protein AFH1 and its interacting protein. Plant Cell Physiol. 2000, 41: 617-626.
Cvrčková F: Are plant formins integral membrane proteins?. Genome Biology. 2000, 1: research001-
Cheung AY, Wu H.-m.: Overexpression of an Arabidopsis formin stimulates supernumerary actin cable formation from pollen tube cell membrane. Plant Cell. 2004, 16: 257-269. 10.1105/tpc.016550.
Craigon DJ, James N, Okyere J, Higgins J, Jotham J, May S: NASCArrays: a repository for microarray data generated by NASC's transcriptomics service. Nucleic Acids Res. 2004, 32 Database issue: D575-D577. 10.1093/nar/gkh133. Database issue
Haas BJ, Volfovsky N, Town CD, Troukhan M, Alexandrov N, Feldmann KA, Flavell RB, White O, Salzberg SL: Full-length messenger RNA sequences greatly improve genome annotation. Genome Biol. 2002, 3: research0029-10.1186/gb-2002-3-6-research0029.
Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith R.K.Jr., Hannick LI, Maiti R, Ronning CM, Rusch DB, Town CD, Salzberg SL, White O: Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31: 5654-5666. 10.1093/nar/gkg770.
Eliáš M, Potocký M, Cvrčková F, Zárský V: Molecular diversity of phospholipase D in angiosperms. BMC Genomics. 2002, 3: 2-10.1186/1471-2164-3-2.
Deutsch M, Long M: Intron-exon structures of eukaryotic model organisms. Nucleic Acids Res. 1999, 27: 3219-3228. 10.1093/nar/27.15.3219.
Wang CC, Chan DC, Leder P: The mouse formin (Fmn) gene: genomic structure, novel exons, and genetic mapping. Genomics. 1997, 39: 303-311. 10.1006/geno.1996.4519.
Honys D, Twell D: Comparative analysis of the Arabidopsis pollen transcriptome. Plant Physiol. 2003, 132: 640-652. 10.1104/pp.103.020925.
Kikuchi S, Satoh K, Nagata T, Kawagashira N, Doi K, Kishimoto N, Yazaki J, Ishikawa M, Yamada H, Ooka H, Hotta I, Kojima K, Namiki T, Ohneda E, Yahagi W, Suzuki K, Ohtsuki K, Shishiki T, Otomo Y, Murakami K: Collection, mapping, and annotation of over 28,000 cDNA clones from japonica rice. Science. 2003, 301: 376-379. 10.1126/science.1081288.
Saitou N, Nei M: The neighbor-joining method: a new method for reconstructing phylogenetictrees. Mol Biol Evol. 1987, 4: 406-425.
Schultz J, Milpetz F, Bork P, Ponting C: SMART, a simple modular architecture research tool: Identification of signalling domains. Proc Natl Acad Sci U S A. 1998, 95: 5857-5864. 10.1073/pnas.95.11.5857.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database searchprograms. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
Kelley LA, MacCallum RM, Sternberg MJE: Enhanced Genome Annotation using Structural Profiles in the Program 3D-PSSM. J Mol Biol. 2000, 299: 499-520. 10.1006/jmbi.2000.3741.
Shi J, Blundell T, Mizuguchi K: FUGUE: Sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J Mol Biol. 2001, 310: 243-257. 10.1006/jmbi.2001.4762.
Karplus K, Hu B: Evaluation of protein multiple alignments by SAM-T99 using the BAliBASE multiple alignment test set. Bioinformatics. 2001, 17: 713-720. 10.1093/bioinformatics/17.8.713.
Jones DT: Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999, 292: 195-202. 10.1006/jmbi.1999.3091.
Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R: PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997, 275: 1943-1946. 10.1126/science.275.5308.1943.
Steck PA, Pershouse MA, Jasser SA, Yung WKA, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DHF, Tavtigian SV: Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nature Genetics. 1997, 15: 356-362.
Li L, Ernsting BR, Wishart MJ, Lohse DL, Dixon JE: A family of putative tumor suppressors is structurally and functionally conserved in humans and yeast. J Biol Chem. 1997, 272: 29403-29406. 10.1074/jbc.272.47.29403.
Yamada KM, Araki M: Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis. J Cell Sci. 2001, 114: 2375-2382.
Lo SH: Molecules in focus: tensin. Int J Biochem Cell Biol. 2004, 36: 31-34. 10.1016/S1357-2725(03)00171-7.
Lemmon SK: Clathrin uncoating: auxilin comes to life. Curr Biol. 2001, 11: R49-R52. 10.1016/S0960-9822(01)00010-0.
Lo SH, Janmey PA, Hartwig JH, Chen LB: Interactions of tensin with actin and identification of its three distinct actin-binding domains. J Cell Biol. 1994, 125: 1067-1075. 10.1083/jcb.125.5.1067.
Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM: Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998, 280: 1614-1617. 10.1126/science.280.5369.1614.
Barford D, Flint AJ, Tonks NK: Crystal structure of human protein tyrosine phosphatase 1B. Science. 1994, 263: 1397-1404.
Stuckey JA, Schubert HL, Fauman EB, Zhang ZY, Dixon JE, Saper MA: Crystal structure of Yersinia protein tyrosine phosphatase at 2.5 A and the complex with tungstate. Nature. 1994, 370: 571-575. 10.1038/370571a0.
Jia Z, Barford D, Flint AJ, Tonks NK: Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science. 1995, 268: 1754-1758.
Murray D, Honig B: Electrostatic control of the membrane targeting of C2 domains. Mol Cell. 2002, 9: 145-154. 10.1016/S1097-2765(01)00426-9.
Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP: Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999, 99: 323-334. 10.1016/S0092-8674(00)81663-3.
Raftopoulou M, Etienne-Manneville S, Self A, Nicholls S, Hall A: Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science. 2004, 303: 1179-1181. 10.1126/science.1092089.
Gish W, States DJ: Identification of protein coding regions by database similarity search. Nature Genetics. 1993, 3: 266-272.
Schuler GD, Altschul SF, Lipman DJ: A workbench for multiple alignment construction analysis. Proteins. 1991, 9: 180-190.
Burge C, Karlin S: Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997, 268: 78-94. 10.1006/jmbi.1997.0951.
Burge C: Modeling dependencies in pre-mRNA splicing signals. Computational Methods in Molecular Biology. Edited by: SalzbergS, SearlsD and KasifS. 1998, Amsterdam, Elsevier Science, 127-163.
Milanesi L, D'Angelo D, Rogozin IB: GeneBuilder: interactive in silico prediction of genes structure. Bioinformatics. 1999, 15: 612-621. 10.1093/bioinformatics/15.7.612.
Zhang MQ: Identification of protein coding regions in the human genome by quadratic discriminant analysis. Proc Natl Acad Sci U S A. 1997, 94: 565-568. 10.1073/pnas.94.2.565.
Stothard P: The Sequence Manipulation Suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques. 2000, 28: 1102-1104.
The Sequence Manipulation Suite 2. 2004, [http://bionformatics.org/sms2/]
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22: 4673-4680.
Hall TA: BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999, 41: 95-98.
Van de Peer Y, De Wachter R: TREECON for Windows: a software package for the construction and drawing of evolutionary trees for the Microsoft Windows environment. Comput Appl Biosci. 1994, 10: 569-570.
Letunic I, Goodstadt L, Dickens NJ, Doerks T, Schultz J, Mott R, Ciccarelli F, Copley RR, Ponting C, Bork P: Recent improvements to the SMART domain-based sequence annotation resource. Nucleic Acids Res. 2002, 30: 242-244. 10.1093/nar/30.1.242.
Nielsen H, Krogh A: Prediction of signal peptides and signal anchors by a hidden Markov model. Proceedings of the Sixth International Conference on Intelligent Systems for Molecular Biology (ISMB 6). 1998, Menlo Park, California, AAAI Press, 122-130.
Krogh A, Larsson B, von Heijne G, Sonnhammer EL: Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001, 305: 567-580. 10.1006/jmbi.2000.4315.
FUGUE:Sequence-structure homology recognition and alignment engine. 2004, [http://www-cryst.bioc.cam.ac.uk/~fugue/]
UCSC HMM Applications. 2004, [http://www.cse.ucsc.edu/research/compbio/HMM-apps/]
McGuffin LJ, Jones DT, Bryson K: The PSIPRED protein structure prediction server. Bioinformatics. 2000, 16: 404-405. 10.1093/bioinformatics/16.4.404.
Vriend G: WHAT IF: a molecular modeling and drug design program. J Mol Graph. 1990, 8: 52-56. 10.1016/0263-7855(90)80070-V.
Chinea G, Padron G, Hooft RW, Sander C, Vriend G: The use of position-specific rotamers in model building by homology. Proteins. 1995, 23: 415-421.
Guex N, Peitsch MC: SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis. 1997, 18: 2714-2723.
Deep View Swiss-PdbViewer. 2004, [http://www.expasy.org/spdbv/]
POV-Ray - the Persistence of Vision Raytracer. 2004, [http://www.povray.org]
Acknowledgements
We thank Brendan Davies for sharing data prior to publication and Mike Deeks for helpful discussion. This work has been supported by the Grant Agency of the Czech Republic Grant 204/02/1461 to FC, VŽ and DP and by Uppsala University and the Linnaeus Centre for Bioinformatics funds to MN. Part of the salaries of FC and VŽ has been provided by the MŠMT ČR Project J13/98:113100003.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
FC conceived of the study, participated in database searches and sequence analyses, carried out the phylogenetic analysis and drafted the manuscript. MN carried out the 3D structure analyses and model building. DP participated in database searching and re-annotation of Arabidopsis genes. VŽ participated in the microarray data analysis and substantially contributed to the design of the study and writing of the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2004_146_MOESM1_ESM.txt
Additional File 1: Nucleotide sequence alignments used to generate revised ORF predictions for selected Arabidopsis formins (TXT 479 KB)
12864_2004_146_MOESM2_ESM.xls
Additional File 2: Non-arabidopsis plant FH2 proteins included in the analysis GenBank/EMBL/DDBJ accession numbers are shown where available; AF3, AF4 – see Additional files 3 or 4; NA – not available. (XLS 26 KB)
12864_2004_146_MOESM4_ESM.txt
Additional File 4: Translations of non-arabidopis, non-rice ESTs or EST assemblies encoding (partial) FH2 proteins. (TXT 14 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Cvrčková, F., Novotný, M., Pícková, D. et al. Formin homology 2 domains occur in multiple contexts in angiosperms. BMC Genomics 5, 44 (2004). https://doi.org/10.1186/1471-2164-5-44
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-5-44