
Abstract
Background
The identification of genes and uncovering the role they play in diseases is an important and complex challenge. Genome-wide linkage and association studies have made advancements in identifying genetic variants that underpin human disease. An important challenge now is to identify meaningful disease-associated genes from a long list of candidate genes implicated by these analyses. The application of gene prioritization can enhance our understanding of disease mechanisms and aid in the discovery of drug targets. The integration of protein-protein interaction networks along with disease datasets and contextual information is an important tool in unraveling the molecular basis of diseases.
Results
In this paper we propose a computational pipeline for the prioritization of disease-gene candidates. Diverse heterogeneous data including: gene-expression, protein-protein interaction network, ontology-based similarity and topological measures and tissue-specific are integrated. The pipeline was applied to prioritize Alzheimer's Disease (AD) genes, whereby a list of 32 prioritized genes was generated. This approach correctly identified key AD susceptible genes: PSEN1 and TRAF1. Biological process enrichment analysis revealed the prioritized genes are modulated in AD pathogenesis including: regulation of neurogenesis and generation of neurons. Relatively high predictive performance (AUC: 0.70) was observed when classifying AD and normal gene expression profiles from individuals using leave-one-out cross validation.
Conclusions
This work provides a foundation for future investigation of diverse heterogeneous data integration for disease-gene prioritization.
Similar content being viewed by others


Background
The rapid accumulation of high-throughput data along with advances in network biology have been fundamental in improving our knowledge of biological systems and complex disease. The emergence of network medicine [1] has explored disease complexity through the systematic identification of disease pathways and modules. Via the analysis of network topology and dynamics, key discoveries have been made including identification of novel disease genes and pathways, biomarkers and drug targets for disease [2]. Network theory is making important contributions to the topological study of biological networks, such as Protein-Protein Interaction Networks (PPIN) [3]. The study by Xu et al. [4] analyzed topological features of a PPIN and observed that hereditary disease-genes from the Online Mendelian Inheritance in Man (OMIM) database [5] have a larger degree and tendency to interact with other disease-genes in literature curated networks. Both Chuang et al. [6] and Taylor et al. [7] have indicated that the alterations in the physical interaction network may be a indicator of breast cancer prognosis. Goh et al. [8] demonstrated that the majority of disease genes are nonessential and located in the periphery of functional networks. Research by [9] discovered that genes connected to diseases with similar phenotypes are more likely to interact directly with each other.
Identification of candidate genes associated with physiological disorders are a fundamental task in the analysis of complex diseases [10]. Genome-wide association studies and linkage analysis have been pivotal in the identification of candidate genes, however, the large list of resultant genes returned are time-consuming and expensive to analyze [11]. The availability of high-throughput molecular interaction network provides and the application of network analysis tools such as clustering or graph partitioning have proved valuable in disease gene prioritization [12]. For instance, PPIN data integrated with genome-wide expression profiles using DNA arrays and/or next generation sequencing enabling the modeling of networks have aided our understanding of how biological networks operate. A number of computational approaches to prioritize candidate genes have been proposed including: ToppGene [13] and GeneWanderer [14] which rank candidate genes based on known associations with disease genes using diverse data sources and methodology. The study by Vanunu et al. [15] applied a diffusion- based method named PRINCE to prioritize genes in prostate cancer, AD and type 2 diabetes. Wu et al. proposed the resource AlignPI [16] which applied a network alignment approach predict disease genes. The algorithm VAVIEN [17] was also developed to prioritize disease genes based on topological features of PPINs.
These diverse studies confirm the importance and need of improving methods to integrate diverse 'omic' sources to uncover candidate disease genes in biological systems. To address this need, we have developed a prioritization pipeline, which integrates diverse heterogeneous information. We illustrate the implementation of this framework using Alzheimer's Disease (AD) as a Case Study. AD is the most common neurodegenerative disease which is both genetically complex and heterogeneous. Pathological characteristics of AD include presence of amyloid peptide plaques, mature senile plaques and neurofibrillary tangles and loss of neurons in conjunction with the presence of oxidative stress [18]. AD can be divided into two categories early onset AD (EOAD) (patients < 65) and late onset AD (LOAD) (patients > 65). A set of gene mutations including APP, PSEN1 and PSEN2 involved in the amyloid beta and tau pathways have been associated with hereditary AD. Using genome-wide association studies, Lambert et al. [19] identified the gene encoding APOE in LOAD as a risk factor along with 11 new loci. Furthermore, studies have suggested that AD is a multifactorial disease in which many pathways are involved. This highlights the progress, which has been made in determining the genetic underpinnings of AD. However, there is further need for an understanding of AD mechanisms to develop more specific diagnostic tests and novel drug therapies to target this disease.
Our proposed pipeline integrates AD gene-expression and network data along with ontology-based semantic similarity, topological information and tissue information. The integration of biological data such as semantic similarity which is independent of the gene expression profiles and PPIN used to obtain the significant hubs is advantageous in providing objective prioritization criteria. To evaluate the perfromance of our proposed method we (1) train an Random Forest classifier using features generated using the prioritized gene candidates to predict AD sample outcome using leave-one out cross-validation (LOOCV); (2) perform enrichment analysis and (3) compare the candidate gene list to a manually curated reference dataset of verified known and susceptible AD disease genes. Furthermore, we investigate the tissues in which AD candidate disease-genes are expressed through incorporation of tissue-specific expression data.
The remainder of the paper is organized as follows, in Section 2 the integrative framework is described along with details on datasets and PPINs used in the analysis. Section 3 provides a summary of the results obtained and conclusions along with future work is presented in Section 4.
Methods
Prioritization Workflow
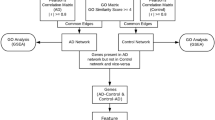
The schematic workflow of our computational integrative approach for disease gene prioritization is shown in Figure 1. Firstly, PPIN and gene-expression are integrated to provide an initial list of prioritized genes. Secondly, we incorporate additional data in the form of network topology and ontology-based semantic similarity to provide further prioritization. This list of genes is then evaluated using enrichment analysis and measurement of tissue specificity.

Overview of Computational Framework to Prioritize Disease Gene Candidates.
AD gene expression data
Human AD gene expression data was obtained from the Gene Expression Omnibus (GEO) database. (http://www.ncbi.nlm.nih.gov/geo/). The selected profile GSE4757 was generated using the platform GPL570: Affymetrix Human Genome U133 Plus 2.0 Array. The study by Dunckley et al. [20] examined the transcriptome of entorhinal neurons from six cortical areas with or without neurofibrillary tangles (a histopathology feature of AD) using Laser capture microdissection. The dataset consists of gene expression profiles of NFT-bearing entorhinal cortex neurons from 10 mid-stage AD patients (Disease) compared with 10 histopathologically normal neurons (Control) from the same patients and brain region. These represent the different stages of AD according to the pattern of disease spread. Using the MAS5.0 function in R the CEL files were firstly normalized. Probes in expression profile were then mapped to corresponding NCBI Gene IDs. The average expression value was calculated in cases where the Gene ID related to more than one probe resulting in 20,539 unique Gene IDs.
A total of 10,106 significant genes were obtained using the significance analysis of microarrays (SAM) [21] technique, a regularized t-test approach, using the false discovery rate (FDR = 0.98). Differentially expressed (DE) genes are genes whose expression levels are significantly different between two groups of experiments.
Human protein-protein interaction network
The PPIN was constucted using data from literature curated sources along with the recent Y2H screening by Rolland et al. [22]. The literature based dataset consists of 11,045 binary human protein pairs extracted from seven publically available databases including BioGRID [23], DIP [24], Biomolecular Interaction Network Database (BIND) [25], HPRD [26], InACT [27], Protein Data Bank (PDB) [28] and Molecular INTeraction database (MINT) [29]. The protein pairs were filtered on evidence where pairs that are only supported by two or more pieces of evidence are included. Protein pairs were also obtained from Y2H experimentation whereby 15,517 opening read frames (ORFs) were systematically screened using the platform hORFeome v5.1 (Space II) resulting in 13,944 pairwise interactions [22]. Both these data are integrated to produce a PPIN consisting of 7,783 nodes and 22,621 protein pairs.
Toplogical analysis
AD gene expression data was mapped to the PPIN via NCBI geneIDs using Cytoscape version 3.2.1 [30]. This resulted in an AD disease specific network consisting of 5,457 nodes and 10,852 protein pairs. Hub genes were defined based on the measurement of network topological features (1) degree and (2) betweenness centrality using Cytoscape version 3.2.1 [30].
Degree connectivity
Degree is a measure of the number of edges that connects a node. Genes with a high degree of connectivity within a network have large numbers of interacting partners. In PPINs it has been observed that genes with high degrees of connectivity are more likely to be essential as genes. Furthermore, many interacting partners in a network tend to be involved in important cellular processes [1]. Using this assumption, the top genes with the highest degree distribution were selected as hub proteins. This approach has previously been applied by Taylor et al. [7]. The degree cut-off threshold for selecting hubs is defined as:
where AVG is the average degree across all DE genes in the PPINs and Std, the standard deviation [31].
Betweenness centrality
Betweenness is a topological feature of a network measuring information flow through the network. In biological networks, betweenness measures the paths through which signals can pass through the interaction network. Yu et. al. [32]. identified betweenness as an important topological property of a network where nodes with high betweenness control most of the information flow. The betweenness centrality C b (n) of a node n is computed as follows:
where s and t are nodes in the network different from n, θ st denoting the number of shortest paths from s to t, and θ st (n) is the number of shortest paths from s to t that n lies on. Betweenness centraility is calculated in Cytoscape. Using the node betweenness distribution, genes located in the top 50% are selected as hub genes.
Network variation of hub genes
For each hub protein in the PPIN the average of Pearson correlation coefficients (PCC) between the hub and each of its respective partners was calculated for both disease and control groups. This method has previously been applied by Taylor et al. [7] to measure network variations among candidate genes and their interacting genes. To determine if interactions are varied, the difference of AD gene expression correlations of PPIs in disease and control samples is calculated. The average hub difference (AvgPCC) off correlation (Pearson's correlation co-efficient (PCC)) values between the disease and control groups was calculated as follows:
where D i and C i represent the correlations of a hub and its interactors for the disease and control groups respectively and n the number of i interactors for a given hub.
Genes that are significantly different between the disease and control groups were selected as follows: (1) labels from the AD expression data were randomly assigned to either the disease or control group. (2) The AvgPCC was recalculated as RandomPCC by repeating the analysis defined in equation × 1000 times in order to calculate random distribution values. (3) P values for each hub was calculated by:
A network of significant hub genes was generated using significant cut-off threshold of P <= 0.05. P values are adjusted using Bonferroni correction. Random reassignment of the expression data was taken by randomly shuffling the expression data gene labels. This method of random reassignment retains the topological network structure of the interactome during the randomization.
Ontology based semantic similarity
Genes involved in phenotypically similar diseases are often functionally related on the molecular level [33]. Based on this observation, the semantic similarity between hub genes and their interactors has been selected to analyze hub genes based on the Gene Ontology (GO) [34]. The GO is a controlled vocabulary describing the charateristics of gene products. Semantic similarity measures evaluate information two genes share. The functional similarity between two proteins is estimated using encoded information in the GO hierarchies. In this study Wang's measure of similarity [35] is applied to the Biological Process hierarchy. This measure determines the semantic similarity of two GO terms based on the locations of terms in the GO graph and their semantic relations with their ancestor terms. Given a GO term A, T A denotes the set of all its ancestor terms including term A itself. S A (t) can be defined as the contribution of a term to the semantics of A based on the relative locations of t and A in the graph. Given GO terms A and B respectively, the semantic similarity between these two terms, S GO (A,B), is defined as:
As one gene may be annotated by many GO terms, similarity between two genes Sim(G1,G2), is then calculated by taking the average semantic similarity scores for all pairs of their associated terms. The similarity score can range between (0,1), whereby a value closer to 1 indicates close relatedness of the two genes in biological process. Wang's measure was implemented using the GOSemSim package in R [36], taking the median semantic similarity between a hub protein and its interactors.
Construction of AD reference dataset
A reference dataset containing known and susceptible AD genes was constructed using the OMIM 'morbid map' table [5]. Known and recently discovered AD susceptibility genes in detailed in the study by Lamberet et al. [19] were also included. This resulted in a list of 52 AD related genes.
Tissue specific gene expression data
Candidate genes were filtered using tissue-specific gene expression data retrieved from BioGPS [37] to determine if these genes are expressed in tissues where AD is observed including the tissue locations: whole brain and prefrontal cortex. This dataset contains the transcription levels of 84 human tissues and cell lines and was processed using the method described by Lopes et al. [38]. A list of 570 housekeeping genes were also included, obtained from [39].
Classification of disease outcome
Construction of feature sets for individual samples were generated using the prioritized hub genes in order to classify sample outcomes applying the approach proposed by Taylor et al. [17]. Using the gene co-expression values for each sample in the AD dataset, the absolute difference in gene co-expression CoeDiff values of the prioritized genes and their interactors was calculated by:
where the difference between the gene expression values of each prioritized gene G and its interactors Im is calculated. This was evaluated for all prioritized proteins across each sample in the AD dataset.
Results and discussion
Using an integrative approach defined in the Method section, diverse heteregenous data obtained from AD gene expression, network topological analysis, along with semantic similarity and tissue-specific information are combined to to generate and analyze candidate AD genes.
Selecting differentially expressed genes
The microarray GSE4757 was analyzed using Significance Analysis of Microarrays (SAM) to identify significantly expressed AD related genes in R using the SAM 5.0 package from [40]. A total of 10,107 significantly positive differentially expressed (DE) genes were observed from 20,539 genes in the AD microarray dataset based on the two class (disease and control) unpaired t-test, using the false discovery rate (FDR = 0.98). These DE genes were selected for the construction of the AD specific PPIN. An overview of the top 10 DE genes is presented in Table1 along with the SAM score based on the t-statistic value.
Construction of AD PPIN
The protein pairs in the human PPIN applied in this study have been derived from both small-scale studies described in literature and large-scale high-throughput Y2H experimentation. We integrate both these data in the construction of the PPIN to improve interactome coverage. Protein pairs obtained from small-scale studies are considered high quality, however, it has been observed by Rolland et al. [22] that their coverage of the interactome is limited to a narrow dense zone. Protein pairs obtained via high-throughput Y2H experimentation have demonstrated distributed homogeneously across the interactome [22].
AD gene expression data was firstly mapped to the PPIN via NCBI geneIDs using Cytoscape version 3.2.1 [30]. This network was further filtered to include only the 10,107 DE genes identified from the SAM analysis. The mapping of DE genes with the PPIN resulted in an AD disease specific network consisting of 3,795 nodes and 5,410 protein pairs.
Identification of hubs through network topology analysis
Hub genes were firstly defined based on network topological features using Cytoscape version 3.2.1 [30]. The AD PPIN is represented as an undirected graphs, G = (V,E), whereby V represents a set of nodes (proteins) and , the set of edges connecting the nodes. An overview of the global properties of the network is presented in Table2. Using Cytoscape, a total of 174 hub genes were selecting based upon the topological analysis measures degree distribution and betweenness centrality. Genes with a high degree of connectivity and genes with low connectivity but high betweenness were selected using the cut-off thresholds defined in the Methods section. Table3 presents the number of hubs obtained using these measures. These two measures have been selected as studies of model organisms have observed that proteins with high degree of connectivity tend to be encoded by essential genes [41]. Furthermore, detection of these genes leads to larger numbers of phenotypic outcomes compared to genes with lower connectivity [42]. However, not all disease genes in humans are essential genes. Goh et al. [8] found that non-essential disease genes tend to be tissue specific located at the functional periphery of the interactome and do not necessarily encode hubs. Both Yu et al. [32] and Joy et al. [43] demonstrated how nodes with a low degree of centrality but high betweenness are important in a network. Taking this into consideration, we also include betweenness as an indicator of centrality.
Prioritization of hubs
Step 1: Integration of co-expression and PPIN
To further filter the list of 174 identified hub genes and select significant hubs we integrated the AD gene expression data with the PPIN. The network variation of the hub genes and their interactors was calculated using the AvgPCC equation defined in Methods. PCC values between the hubs and their interactors were calculated for both the disease and control groups. Significant hub genes were selecting using the Bonferroni corrected cut-off threshold of P < 0:05. A total of 22 genes were identified.
Step 2: Gene ontology semantic similarity analysis
The semantic similarity between hub genes and their interactors has also been selected to prioritize hub genes based on the Gene Ontology (GO). It has been observed that genes involved in phenotypically similar diseases are often functionally related on the molecular level [33]. In this study, the semantic similarity between a gene hub and it's interacting partners was performed using Wang's [35] measure of similarity detailed in equation 4. This was applied to the GO Biological Process hierarchy as a quantitative measure of functionality similarity between gene pairs using the R package GOSemSim [36]. To obtain the similarity value for the hub and all its interactors, the median similarity was taken across all protein pairs. The semantic similarity values obtained ranged between 0 and 1. The hub gene semantic similarity distribution was sorted in ascending order and hubs with GO semantic similarity greater than 0.5 selected. This resulted in a total of 10 prioritized hubs. Combining candidate genes output from analysis in Step 1 and Step 2 resulted in a list of 32 prioritized hub genes summarized in Table4.
Evaluation
Functional annotation enrichment
GO and pathway enrichment analysis using the DAVID resource [44] was applied to investigate the biological implications of the prioritized hub gene list. Functional annotation was obtained by extracting the most over-representative GO terms (Biological Process, Cellular Component and Molecular Function) for the groups of genes under observation with respect to the whole genome taken as the reference background set (p-value <0.05). We applied this approach to measure if the prioritized hub genes are more enriched in GO terms or involved in pathways than what would be expected by chance. The number of GO terms and KEGG pathways are summarized in Table5.
Enrichment analysis of the prioritized hub genes identified significant biological processes modulated in AD pathogenesis including: neuron differentiation, neuron projection morphogenesis, neuron projection development and regulation of neuron apoptosis. Furthermore, significant KEGG pathways including: the Wnt signaling pathway and the TGF-beta signaling pathway both of which have been implicated in neurodegenerative diseases [45, 46] These results highlight the potential of this approach in using prioritized hubs for the prediction of AD biomarkers.
Reference dataset comparison
The list of prioritized hub genes were compared to the reference dataset consisting of 52 AD related genes. A total of 9 AD susceptible genes from the list of hub genes identified were identified summarized in Table6. Mutations in PSEN1 are the most common cause of early onset of AD. TRAF1, is a critical regulator of cerebral ischemia-reperfusion injury and neuronal death [47]. LZTS2 has shown associated with late onset AD [48].
Tissue analysis
The prioritized gene hubs were analyzed to determine if gene hubs are expressed in tissues in whereby by AD is observed including the prefrontal cortex and whole brain. Tissue specificity is an important component of network analysis as genetic diseases often target specific tissue(s). Therefore, perturbations of pathways or proteins may have differential effects among diverse tissues [49]. Using tissue specific expression data from BioGPS [37] along with housekeeping genes obtained from Lopes et al. [38], we identified that 4 of the prioritized gene hubs are located in the whole brain and/or the prefrontal cortex tissues.
Disease outcome classification
Using the prioritized gene-candidate list along with gene expression values from the GEO dataset GSE4757, we constructed a feature vector applying the approach described in Methods using Equation 6. These features measure the differences in absolute co-expression values between the prioritized genes and their interactors across each sample in the dataset. The aim of this approach is to determine if measured differences in co-expression values of prioritized genes and their interactors can predict sample outcome. A total of 32 feature values (obtained using each of the genes in the prioritized list) were generated for each instance in the AD dataset. The AD dataset has a total of 20 individual samples, 10 AD and 10 normal. We use these as labels when measuring the classification performance. Classification of sample outcome using this vector was performed using the Random Forest classifier [50] in the WEKA toolbox [51]. Leave one out cross validation (LOOCV) was implemented, whereby, for a dataset with n samples, n experiments are performed. Each experiment uses n-1 samples for training and the remaining sample for testing. The performance was assessed by a receiver operating characteristics (ROC) curve which plots the true positive rate against the false positive rate at various threshold settings. The area under the ROC curve (AUC) was estimated for each of the prioritization approaches described in Table7. Reasonable AUC values were observed for the various prioritization approaches ranging from 0.73-0.70 as presented in Table7 and illustrated using ROC curves in Figure 2. This analysis suggests that the integration of diverse data in the prioritization of protein hubs are indeed useful for sample outcome prediction. Interestingly, we can see a slight decrease in classification performance when gene hubs prioritized using topological features are integrated with gene hubs prioritized using GO semantic similarity. This is compared to using only the topological or GO semantic similarity prioritization approach only.

ROC Curves illustrating the predictive performance of the Gene Hub Prioritization Approches.
Conclusions
In this study, we have proposed a novel computational framework that integrates diverse heterogeneous including gene expression, network topological features: degree and betweenness along with GO semantic similarity and tissue specificity information to prioritize and analyze disease-gene candidates. Our proposed pipeline provides the flexibility to integrate other heterogeneous data sources. To illustrate our approach, AD was applied as a Case Study. AD is a chronic neurodegenerative disorder characterized by a progressive decline in memory and cognitive abilities. It is estimated that 4-8% of the population over 65 years of age suffers from this disease with the rate of incidence increasing with age. Currently, only the symptoms of AD are treated with no effective therapeutic strategy available. Using our pipeline, 22 prioritized candidate AD disease genes were generated. Enrichment analysis revealed GO Biological Process terms were enriched by the prioritized gene-list such as regulation of neurogenesis and generation of neurons which are linked to AD pathogenesis. Analysis of KEGG pathways identified prioritized genes involved in pathways including Wnt signaling pathway and the TGF-beta signaling pathway. AD susceptible genes: PSEN1 and TRAF1 extracted from OMIN [5] and a recent study by Lambert et al. [19] were identified using the prioritization approach. A reasonable predictive performance (AUC: 0.70) was achieved when classifying AD and normal gene expression profiles from individuals using a feature set generated from the prioritized gene-list along with supervised classification using Random forest and LOOCV. These results demonstrate that the integration of PPINs along with disease datasets and contextual information is an important tool in unraveling the molecular basis of diseases. It is important to note that network based approaches for candidate disease gene priortization need to be viewed with care as the map of the binary human PPIN is still incomplete. However, coverage of the human interactome is increasing through systematic Y2H and transcriptional interaction screens. This increased coverage, quality, and diversity of human PPIN data will provide further opportunities for the molecular characterization and understanding of human disease [1].
References
Barabási AL, Gulbahce N, Loscalzo J: Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011, 12 (1): 56-68. 10.1038/nrg2918.
Chan SY, Loscalzo J: The emerging paradigm of network medicine in the study of human disease. Circ Res. 2012, 111 (3): 359-374. 10.1161/CIRCRESAHA.111.258541.
Goñi J, Esteban FJ, de Mendizábal NV, Sepulcre J, Ardanza-Trevijano S, Agirrezabal I, Villoslada P: A computational analysis of protein-protein interaction networks in neurodegenerative diseases. BMC Syst Biol. 2008, 2: 52-10.1186/1752-0509-2-52.
Xu J, Li Y: Discovering disease-genes by topological features in human protein-protein interaction network. Bioinformatics. 2006, 22 (22): 2800-2805. 10.1093/bioinformatics/btl467.
Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA: Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005, 33 (Database issue): D514-D517.
Chuang HY, Lee E, Liu YT, Lee D, Ideker T: Network-based classification of breast cancer metastasis. Mol Syst Biol. 2007, 3: 140-
Taylor IW, Linding R, Warde-Farley D, Liu Y, Pesquita C, Faria D, et al: Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nat Biotechnol. 2009, 27 (2): 199-204. 10.1038/nbt.1522.
Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabási AL: The human disease network. Proc Natl Acad Sci USA. 2007, 104 (21): 8685-8690. 10.1073/pnas.0701361104.
Gandhi TKB, Zhong J, Mathivanan S, Karthick L, Chandrika KN, Mohan SS, et al: Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet. 2006, 38 (3): 285-293. 10.1038/ng1747.
Liu ZP, Wang Y, Zhang XS, Chen L: Network-based analysis of complex diseases. IET Systems Biology. 2012, 6 (1): 22-33. 10.1049/iet-syb.2010.0052.
Ala U, Piro RM, Grassi E, Damasco C, Silengo L, Oti M, et al: Prediction of human disease genes by human-mouse conserved coexpression analysis. PLoS Comput Biol. 2008, 4 (3): e1000043-10.1371/journal.pcbi.1000043.
Navlakha S, Kingsford C: The power of protein interaction networks for associating genes with diseases. Bioinformatics. 2010, 26 (8): 1057-1063. 10.1093/bioinformatics/btq076.
Chen J, Bardes EE, Aronow BJ, Jegga AG: ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37 (Web Server issue): W305-W311.
Köhler S, Bauer S, Horn D, Robinson PN: Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet. 2008, 82 (4): 949-958. 10.1016/j.ajhg.2008.02.013.
Vanunu O, Magger O, Ruppin E, Shlomi T, Sharan R: Associating genes and protein complexes with disease via network propagation. PLoS Comput Biol. 2010, 6 (1): e1000641-10.1371/journal.pcbi.1000641.
Wu X, Liu Q, Jiang R: Align human interactome with phenome to identify causative genes and networks underlying disease families. Bioinformatics. 2009, 25 (1): 98-104. 10.1093/bioinformatics/btn593.
Erten S, Bebek G, Koyutürk M: Vavien: an algorithm for prioritizing candidate disease genes based on topological similarity of proteins in interaction networks. J Comput Biol. 2011, 18 (11): 1561-1574. 10.1089/cmb.2011.0154.
Jacobsen JS, Reinhart P, Pangalos MN: Current concepts in therapeutic strategies targeting cognitive decline and disease modification in Alzheimer's disease. NeuroRx. 2005, 2 (4): 612-626. 10.1602/neurorx.2.4.612.
Lambert JC, Ibrahim-Verbaas C a, Harold D, Naj aC, Sims R, Bellenguez C, et al: Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013, 45 (12): 1452-1458. 10.1038/ng.2802.
Dunckley T, Beach TG, Ramsey KE, Grover A, Mastroeni D, Walker DG, et al: Gene expression correlates of neurofibrillary tangles in Alzheimer's disease. Neurobiol Aging. 2006, 27 (10): 1359-1371. 10.1016/j.neurobiolaging.2005.08.013.
Tusher VG, Tibshirani R, Chu G: Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001, 98 (9): 5116-5121. 10.1073/pnas.091062498.
Rolland T, Taşan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, et al: A Proteome-Scale Map of the Human Interactome Network. Cell. 2014, 159 (5): 1212-1226. 10.1016/j.cell.2014.10.050.
Chatr-Aryamontri A, Breitkreutz BJ, Heinicke S, Boucher L, Winter A, Stark C, et al: The BioGRID interaction database: 2013 Update. Nucleic Acids Res. 2013, 41 (Database issue):
Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D: The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004, 32 (Database issue): D449-D451.
Bader GD, Betel D, Hogue CW V: BIND: the Biomolecular Interaction Network Database. Nucleic Acids Res. 2003, 31 (1): 248-250. 10.1093/nar/gkg056.
Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, et al: Human Protein Reference Database--2009 update. Nucleic Acids Res. 2009, 37 (Database issue): D767-D772.
Kerrien S, Aranda B, Breuza L, Bridge A, Broackes-Carter F, Chen C, et al: The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012, 40 (Database issue): D841-D846.
Rose PW, Prlić A, Bi C, Bluhm WF, Christie CH, Dutta S, et al: The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. Nucleic Acids Res. 2014, 43 (Database issue): D345-D356.
Chatr-aryamontri A, Ceol A, Palazzi LM, Nardelli G, Schneider MV, Castagnoli L, Cesareni G: MINT: the Molecular INTeraction database. Nucleic Acids Res. 2007, 35 (Database issue): D572-D574.
Yeung N, Cline MS, Kuchinsky A, Smoot ME, Bader GD: Exploring biological networks with cytoscape software. Current Protocols in Bioinformatics. 2008, Chapter 8: Unit 8.13-
Rakshit H, Rathi N, Roy D: Construction and analysis of the protein-protein interaction networks based on gene expression profiles of Parkinson's disease. PLoS One. 2014, 9 (8): e103047-10.1371/journal.pone.0103047.
Yu H, Kim PM, Sprecher E, Trifonov V, Gerstein M: The importance of bottlenecks in protein networks: Correlation with gene essentiality and expression dynamics. PLoS Comput Biol. 2007, 3 (4): 713-720.
Schlicker A, Lengauer T, Albrecht M: Improving disease gene prioritization using the semantic similarity of gene ontology terms. Bioinformatics. 2010, 26 (18): i561-i567. 10.1093/bioinformatics/btq384.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al: Gene Ontology : tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000, 25 (1): 25-29. 10.1038/75556.
Wang JZ, Du Z, Payattakool R, Yu PS, Chen CF: A new method to measure the semantic similarity of GO terms. Bioinformatics. 2007, 23 (10): 1274-1281. 10.1093/bioinformatics/btm087.
Yu G, Li F, Qin Y, Bo X, Wu Y, Wang S: GOSemSim: An R package for measuring semantic similarity among GO terms and gene products. Bioinformatics. 2010, 26 (7): 976-978. 10.1093/bioinformatics/btq064.
Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, et al: BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009, 10 (11): R130-10.1186/gb-2009-10-11-r130.
Lopes TJS, Schaefer M, Shoemaker J, Matsuoka Y, Fontaine JF, Neumann G, et al: Tissue-specific subnetworks and characteristics of publicly available human protein interaction databases. Bioinformatics. 2011, 27 (17): 2414-2421. 10.1093/bioinformatics/btr414.
Eisenberg E, Levanon EY: Human housekeeping genes are compact. Trends in Genetics. 2003, 19 (7): 362-365. 10.1016/S0168-9525(03)00140-9.
Chu G, Li J, Narasimhan B, Tibshirani R, Tusher V: SAM - Significance Analysis of Microarrays - Users guide and technical document. Policy. 2011, 1-42.
Jeong H, Mason SP, Barabási AL, Oltvai ZN: Lethality and centrality in protein networks. Nature. 2001, 411 (6833): 41-42. 10.1038/35075138.
Yu H, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, et al: High-quality binary protein interaction map of the yeast interactome network. Science. 2008, 322 (5898): 104-110. 10.1126/science.1158684.
Joy MP, Brock A, Ingber DE, Huang S: High-betweenness proteins in the yeast protein interaction network. J Biomed Biotechnol. 2005, 2005 (2): 96-103. 10.1155/JBB.2005.96.
Huang DW, Sherman BT, Lempicki RA: Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009, 4 (1): 44-57.
Inestrosa NC, Varela-Nallar L: Wnt signaling in the nervous system and in Alzheimer's disease. J Mol Cell Biol. 2014, 6 (1): 64-74. 10.1093/jmcb/mjt051.
Das P, Golde T: Dysfunction of TGF-beta signaling in Alzheimer's disease. J Clin Invest. 2006, 116 (11): 2855-2857. 10.1172/JCI30284.
Lu YY, Li ZZ, Jiang DS, Wang L, Zhang Y, Chen K, et al: TRAF1 is a critical regulator of cerebral ischaemia-reperfusion injury and neuronal death. Nat Commun. 2013, 4: 2852-
Beecham GW, Martin ER, Li YJ, Slifer MA, Gilbert JR, Haines JL, Pericak-Vance MA: Genome-wide Association Study Implicates a Chromosome 12 Risk Locus for Late-Onset Alzheimer Disease. Am J Hum Genet. 2009, 84 (1): 35-43. 10.1016/j.ajhg.2008.12.008.
Guan Y, Gorenshteyn D, Burmeister M, Wong AK, Schimenti JC, Handel MA, et al: Tissue-specific functional networks for prioritizing phenotype and disease genes. PLoS Comput Biol. 2012, 8 (9): e1002694-10.1371/journal.pcbi.1002694.
Breiman L: Random Forests. Mach Learn. 45 (): 5-32.
Hall M, Frank E, Holmes G, Pfahringer B, Reutemann P, Witten IH: The WEKA data mining software. ACM SIGKDD Explor Newsl. 2009, 11 (1): 10-18. 10.1145/1656274.1656278.
Acknowledgements
The funding for the publication comes from the Ulster University PVC Challenge Award.
This article has been published as part of BMC Genomics Volume 16 Supplement 9, 2015: Selected articles from the IEE International Conference on Bioinformatics and Biomedicine (BIBM 2014): Genomics. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcgenomics/supplements/16/S9.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
FB, HW and HZ conceived and designed the framework. FB performed the experiments and analyzed the data. FB, HW and HZ wrote the paper.
Fiona Browne contributed equally to this work.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Browne, F., Wang, H. & Zheng, H. A computational framework for the prioritization of disease-gene candidates. BMC Genomics 16 (Suppl 9), S2 (2015). https://doi.org/10.1186/1471-2164-16-S9-S2
Published:
DOI: https://doi.org/10.1186/1471-2164-16-S9-S2