
Abstract
Background
Mycobacterium avium subsp. avium (Maa) and M. avium subsp. hominissuis (Mah) are environmental mycobacteria and significant opportunistic pathogens. Mycobacterium avium infections in humans and pigs are mainly due to Mah. It is not known whether this is caused by a difference in virulence or difference in exposure to the two subspecies. The aim of the present study was to investigate the ability of the M. avium subspecies to replicate intracellularly and to characterise the gene expression program triggered by infection of human primary macrophages.
Results
All isolates were able to invade and persist within human macrophages. However, intracellular replication was only evident in cells infected with the two Maa isolates. Transcriptional responses to the isolates were characterized by upregulation of genes involved in apoptosis, immune- and inflammatory response, signal transduction and NF-kB signaling, cell proliferation and T-cell activation. Although similar pathways and networks were perturbed by the different isolates, the response to the Maa subspecies was exaggerated, and there was evidence of increased activation of type I and II interferon signaling pathways.
Conclusion
Mycobacterium avium isolates of different genetic characteristics invaded monocytes and induced different degree of macrophage activation. Isolates of Maa were able to replicate intracellularly suggesting that differences in exposure, uptake or induction of adaptive immunity are more likely explanations for the difference in prevalence between M. avium subspecies.
Similar content being viewed by others

Background
The Mycobacterium avium complex comprises a group of environmental and opportunistic intracellular pathogens[1] with the ability to persist within macrophages and escaping the host’s killing mechanisms[2]. The complex is constituted by the subspecies of M. avium together with M. intracellulare[3]. Two subspecies of clinical importance for humans and animals are M. avium subsp. hominissuis (Mah) and M. avium subsp. avium (Maa)[4]. Mah is the subspecies mainly isolated from porcine and human cases of MAC infection[4], while Maa is the causative agent of avian tuberculosis, but is occasionally responsible for M. avium infections in mammals[5]. Little is known about the routes of transmission and the virulence mechanisms of M. avium, and a zoonotic potential has not been ruled out[6].
Definite reasons for Mah being the dominating subspecies in humans and pigs infected with M. avium have not been identified, although differences in exposure could be considered a contributing factor. Explanations to the higher prevalence of Mah in the human and porcine population might also be found in differences between Mah and Maa at the molecular level of interaction between the host and mycobacterium, a process crucial to the outcome of infection, which therefore might affect the prevalence of the subspecies. However, little has been done to investigate whether Mah and Maa interact differently with their target cells.
Mycobacterial virulence depends on the ability of these bacteria to invade, persist and replicate within the hostile environment in macrophages of the host organism[7]. This is achieved by the protective properties of the mycobacterial cell wall and by interference with the host’s immune response[8, 9]. To assess the ability of Mah and Maa to grow intracellularly, the use of primary cells will give a more representative result compared to immortalised macrophage cell-lines. Tissue resident macrophages represent a heterogeneous population and it is not clear whether M. avium infect a particular phenotype. Monocyte derived macrophages matured in absence of any stimuli give rise to a mixed culture of both the M1 and M2 phenotype[10] and such cultures may thus be considered appropriate for infection studies with M. avium. Finally, using macrophages derived from the relevant target species is important since the global inflammatory response evoked in mice recently was demonstrated to be vastly different from humans[11]. To the authors’ best knowledge, comparative studies of the ability of Maa and Mah to invade, replicate and trigger immune responses in human primary macrophages have not been published. The aim of the present study was therefore to assess the growth of M. avium isolates in human primary macrophages and to characterise the response triggered in infected macrophages by selected isolates. The isolates used in the present study represented the two different subspecies, were derived from different host species and differed in biofilm abilities, glycopeptidolipid (GPL) genes, and in the presence of the insertion sequence (IS) element ISMpa1[12]. The present data showed that Maa replicated to a larger extent in human macrophages suggesting that an inability to grow intracellularly is not likely the cause of the discrepancy of the prevalence of the two subspecies in humans. Furthermore, the response induced in the macrophages could not entirely explain the difference in growth rate.
Methods
Mycobacterial isolates
Four clinical isolates of M. avium subsp. hominissuis (Mah VI101, Mah 1655, Mah H1 and Mah H38) and two clinical isolates of M. avium subsp. avium (Maa 1794 and Maa 1553) were used in the present study. Details on origin, biofilm producing abilities, presence of ISMpa1, nsGPL- and ssGPL genes were described by Johansen et al.[12], and are summarised in Table 1. The human isolates were from a strain collection at the The Norwegian Institute of Public Health. All isolates were grown in Middlebrook 7H9 (BD Diagnostics, Sparks MD) w/OADC (BD Diagnostics), 1% Tween 80 and 10% glycerol under agitation for two weeks before aliquots were stored at −70°C in an equivalent medium containing 10% glycerol. The concentration of bacteria in the stock suspensions was determined by colony forming unit (CFU) counts. Bacterial inocula for the gene expression assay were prepared from seven days subcultures grown on plates of Middlebrook 7H10 (BD Diagnostics) with 10% OADC (BD Diagnostics) at 37°C. To adjust the number of bacteria in the inoculum, hsp 65 real-time PCR[13] was performed on serial dilutions in HBSS (Invitrogen, Oslo, Norway) of a suspension adjusted to McFarland standard 2.0, homogenised through a 23 G needle to minimise clumping.
Cell culture
Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coat drawn from healthy human blood donors. The material was commercially obtained from a blood donation centre, and the study was approved by the Regional Committee for Medical Research Ethics, South-East Norway. Fifty mL buffy coat was diluted 1:1 with RPMI 1640 (Sigma-Aldrich, Oslo, Norway) with 1% L-glutamine (Sigma-Aldrich). PBMCs were harvested by density gradient centrifugation with Lymphoprep (Medinor, Bryn, Norway) and resuspended in PBS with 0.5% BSA and 2 mM EDTA at pH 7.2. Isolation of CD14+ cells was performed using CD14 MACS magnetic beads (Milteny Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. The cells were resuspended in RPMI 1640 with 1% L-glutamine supplemented with 10% human serum to the desired concentration of the respective infection assay, added to the appropriate Costar® cell culture plates (Sigma-Aldrich) and incubated at 37°C with 5% CO2. The CD14+ cells were incubated overnight before used in the uptake and replication assays and allowed to mature into macrophages by incubation for five days before used in the gene expression study.
Uptake and intracellular replication
Uptake and intracellular replication of the six M. avium isolates in human blood derived CD14+ cells were assessed. The assays were performed in 96-well cell culture plates (4 × 105 cells/mL, 125 μl per well), which in our hands gave a stable monolayer for most donors. Each isolate was tested in triplicates at an MOI of 5:1. The cells were washed twice with pre-warmed medium to remove extracellular bacteria before the quantification of intracellular bacteria was performed. Intracellular uptake of the bacterial isolates was assayed after three, six, 12 and 24 hours in cells from three donors.
Intracellular replication of the same isolates was assayed in six other donors, following the protocol described above. Based on the results from the uptake studies, CD14+ cells were incubated with the bacterial isolates for six hours. After removal of extracellular bacteria, fresh medium was added and the cells were incubated further. Cell lysates were harvested at six hours, and after one, four and seven days of infection.
Quantitation of bacteria and cells
Numbers of intracellular bacteria and cells was determined by enumeration of mycobacterial and human genomes in cell lysates. Replication of bacterial isolates in the supernatant had been tested and found insignificant. Lysis of cells was achieved through adding milliQ water to the wells. After 30 minutes, lysates were transferred into 2 mL O-ring vials (Biospec products, Techtumlab, Umeå, Sweden) containing 200 μL silica beads (Biospec products) and stored at −20°C. Lysates from triplicate wells were pooled. After completing all sampling, lysates were thawed and inactivated at 100°C for 30 min, prior to mechanical disruption using the MiniBeadBeater (Techtumlab) for 45 sec.
Enumeration of mycobacteria and cells was achieved by performing a duplex real time PCR of a 103 bp long segment of the mycobacterial hsp65 gene and a 150 bp long segment of the human β-globin gene on the lysates, as described by Salte et al.[13]. Briefly, the segment of the mycobacterial gene hsp65 (GroEl2) was amplified using the primers MycoFP1 (5′CGAGGCGATGGACAAGGT-3′) and TB 12 (5′CTTGTCGAACCGCATACCCT-3′), and the VIC tagged TaqMan-MGB probe MycoPr1 (5′-AACGAGGGCGTCACCGTCG-3′)[14]. The segment of the human β-globin gene was amplified by using the primers BG-F (5′-TGCCTATCAGAAAGTGGTGGCT-3′) and BG-R (5′-GCTCAAGGCCCTTCATAATATCC-3′), and the FAM tagged TaqMan-MGB probe BG-TAQ (5′-TGGCTAATGCCCTGGCCCACAA-3′)[13, 15]. Dilutions of known concentrations of commercially available mycobacterial DNA (ATCC-19015D-5, LGC Standards, Middlesex, UK) and human DNA (Applied Biosystems, Foster City, CA, USA) were run together with the samples and used to create standard curves. The conversion of the known amount of standard DNA to number of bacterial and human genomes were described by Salte et al.[13], as well as the validation of the number of mycobacterial targets against CFU counts. Total volume of each reaction mixture was 20 μl, consisting of 8 μl lysate, 10 μl Quantitect® mastermix (Quiagen, West Sussex, UK) and 1 μl of a primer/probe mix for each target, with a stock concentration of 8 μM for each primer and 4 μM for the probe, resulting in a final primer- and probe concentration of 0.4 μM and 0.2 μM, respectively. Samples and standard DNA dilutions were run on a Stratagene Mx3005P MxPro v 4.10 (Stratagene, La Jolla, CA, USA) and data analysed by the Stratagene MxPro v 4.10 software (Stratagene) with the following thermal cycling conditions: 95°C for 15 min, followed by 40 cycles of 95°C for 1 min and 60°C for 1 min.
Gene expression profiling
Isolates Mah VI101, Mah 1655 and Maa 1794, were used to infect macrophages for gene expression analysis. CD14+ cells were obtained as described above and allowed to mature into macrophages by incubation for five days in duplicate wells (6 well plates, 1 × 106 cells/mL, 2 ml/well). The cells were then infected with the three isolates at a MOI of 10:1 for four hours. Total RNA was isolated of the adherent macrophages using the RNeasy Mini Kit® (Qiagen, Hilden, Germany), following the manufacturer’s instructions. RNA from the different donors within each assay was pooled per bacterial isolate. The first assay was performed in macrophages from three donors and subsequently repeated twice in two new donors per assay. The quality and integrity of RNA was confirmed by Agilent 2100 Bioanalyzer (Agilent Technologies) and Nanodrop™ (Thermo Fisher Scientific, Wilmington, DE, USA). Gene expression levels were profiled using Illumina HumanHT-12 Expression Bead Chip (Illumina Inc., San Diego, CA, USA). The gene expression data was exported from Illumina’s GenomeStudio software package without background subtraction or normalization. The raw data was preprocessed in the R language and environment for statistical computing[16] employing the normexp by control (neqc) algorithm[17]. Probes that were not significantly expressed above background (detection p-value < 0.01 in at least one sample) were filtered out of the analysis. Differentially expressed genes were identified employing Bayesian/moderated t-statistics with False Discovery Rate (FDR) control for multiple testing[18]. Differentially expressed genes were identified at FDR < 0.05. The raw microarray data have been made publicly available in the Array Express repository (http://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-1101/).
Bioinformatics
Biological functions enriched in the differentially expressed genes were identified using modular enrichment analysis with DAVID (Database for Annotation, Visualization and Integrated Discovery)[19, 20]. Enrichment scores > 1.3 were considered statistically significant. Biological pathways enriched in the data were identified using IPA software (Ingenuity Systems, Redwood City, CA). Upstream regulator analysis was performed in IPA software to infer the putative pathways that give rise to the observed gene expression changes. Molecular interaction networks were reconstructed from the differentially expressed genes in IPA software using mechanistic data from prior studies[21]. In this analysis differentially expressed genes were defined at FDR < 0.01.
Protein analysis
CD14+ cells were isolated and matured as described above for gene expression studies. Cell lawns were infected at an MOI of 5:1 for 24 hours with Maa 1794, Mah VI101 and Mah 1655. Supernatants were removed for cytokine profiling, and subsequently the adherent cells were washed twice in PBS and collected with a cell scraper in 100 μl of 1x SDS Sample Buffer for immunoblotting. All samples were stored at −70°C until further processing.
SDS-PAGE and immunoblotting
Proteins were separated by horizontal SDS-PAGE with precast gradient ExcelGel™ SDS Gradient 8 – 18% (GE Healthcare Life Sciences, Uppsala, Sweden) and ExcelGel™ SDS Buffer strips (GE Healthcare) in a Multiphor™ II 2117 (GE Healthcare), prior to diffusion blotting onto nitrocellulose membranes with pore size 0.2 μm (Schleicher & Schuell, Dassel Germany)[22]. Proteins remaining within the gel were visualized by Coomassie brilliant blue staining. Membranes were blocked with PBS with 2% BSA and incubated over night with primary antibodies (Cell Signalling Technology, Danvers, MA) recognizing the following selected proteins involved in apoptosis: Cleaved caspase-3 (Asp175), cleaved caspase-8 (Asp391), cleaved caspase-9 (Asp315), cleaved caspase-9 (Asp330), Bcl-2, Phospho-Bcl-2 (Ser70). Detection of bound antibodies was performed by incubaction with ECL™ Anti-rabbit IgG, Horseradish peroxidise labelled linked F(ab’)2 fragment from donkey (GE Healthcare) and visualization by Immun-Star™ WesternC™ Kit (Bio-Rad, Hercules, CA).
Cytokine profiling
The concentration of TNF-α, IL-23(p19), IL-10, IL-8 and IL-6 was measured in supernatants by using Bio-Plex™ Cytokine Assay (Bio-Rad) within the Bio-Plex suspension array system (Bio-Rad), following the manufacturer’s instructions. Differences between isolates were tested for significance using the Wilcoxon matched pairs signed-rank test, considering p-values ≤ 0.05 significant.
Results
Uptake and intracellular replication
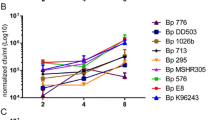
All the isolates were taken up by the CD14+ cells, and intracellular bacteria were detected after 3 hours (Figure 1). Maa 1553, Mah H38 and Maa 1794 appeared to be taken up to a greater extent, while the Mah isolate VI101 showed the lowest degree of uptake in macrophages. Between six and 12 hours of infection some of the isolates showed an exponential increase in the number of intracellular bacteria, suggesting the event of onset of intracellular replication. Six hours was therefore chosen as the time between inoculation and removal of extracellular bacteria for the subsequent study of intracellular replication over time. At this point in time VI101 had significantly (p < 0.05) lower numbers of intracellular bacteria compared to the other isolates except for Mah H1, while no significant difference were observed between the other isolates using the paired t-test.

Uptake of six clinical isolates of Mycobacterium avium in human CD14+ cells. Cells were inoculated for three, six, 12 and 24 hours. Numbers represent the mean values from three donors, showing the ratio of numbers of mycobacterial to human genomes (A) and absolute numbers of mycobacteria per well (B), measured by real-time PCR of single copy genes in cell lysates.
Intracellular growth was only observed for the two Maa isolates. There was a significant increase in the ratio of mycobacteria to human genomes at day four for isolate Maa 1794 (p = 0.04) and at day 7 for Maa 1794 (p = 0.01) and Maa 1553 (p = 0.02) and in absolute numbers of bacteria for isolate Maa 1794 (p = 0.01) at day 7 using the paired t-test (Figure 2). The remaining isolates, which were all Mah, did not show significant increase in intracellular numbers throughout the duration of infection, however the count of intracellular bacteria was quite stable, indicating that the bacteria were able to persist within the macrophages.

Intracellular replication of six clinical isolates of Mycobacterium avium in human CD14+ cells. Sampling was performed after six hours of inoculation with added bacteria, then one, four and seven days after removal of extracellular bacteria. Numbers represent the mean values from six donors, showing the ratio of numbers of mycobacterial to human genomes (A) and absolute numbers of mycobacteria per well (B), measured by real-time PCR of single copy genes in cell lysates. There was a significant increase in the ratio of mycobacteria at day four for isolate Maa 1794 (p = 0.04) and at day 7 for Maa 1794 (p = 0.01) and Maa 1553 (p = 0.02) and in absolute numbers of bacteria for isolate Maa 1794 (p = 0.01) at day 7 using the paired t-test.
Enumeration of both cells and intracellular mycobacteria was performed by real-time PCR of single-copy genes. This method does not distinguish between viable and non-viable bacteria and cells, but it has the advantage that the number of bacteria is not influenced by the degree of clumping which varies between Maa and Mah. Furthermore it enabled the measurement of the number of intracellular bacteria per cell in addition to absolute counts of bacteria. Some degree of cell death and loss of adherence were observed in both infected and non-infected cells, and varied considerably between donors. Similar results were observed whether absolute numbers of bacteria or the bacteria per cell ratio were used, however the difference in cell death between donors may explain why the absolute counts of bacteria only reached a significant level of p < 0.05 for Maa 1794 at day 7, while the ratio was significant for both Maa isolates at day 7 and for Maa 1794 at day 4.
Gene expression analysis and bioinformatics
Based on the results from the initial replication assays, three isolates were selected for gene expression studies in macrophages. Maa 1794, which replicated intracellularly, and Mah VI101, which only persisted within cells, were selected, in addition to Mah 1655, which, as opposed to the other two isolates, contains the IS element ISMpa1 and belong to an RFLP type that has been demonstrated to be particularly virulent in pigs[23, 24].
The Illumina® HumanHT-12 v3 Expression Bead allows whole-genome expression analysis of 12 samples on the same chip, each array targeting genomewide with more than 48,000 probes. Of the 14,581 probe sets with signal intensity above background, 2,766 probe sets were differentially expressed (FDR < 0.05) in the cells infected with M. avium (1,229 up-regulated, 1,537 down-regulated), regardless of the individual isolate, compared to uninfected control cells (Additional file1: Table S1). The largest response was induced by Maa 1794 where 2,439 probe sets (representing 2,147 unique genes) were differentially expressed. Of these probe sets, 1,077 were up-regulated, and 1,362 were down-regulated. Mah VI101 modulated expression of 1,520 probe sets (810 up-regulated, 710 down-regulated) representing 1,348 unique genes. In comparison, Mah 1665 altered the expression of only 341 probe sets (239 up-regulated 102 down-regulated) representing 298 unique genes. A large overlap was observed in the responses elicited by the three isolates (Figure 3). There were virtually no genes (5 transcripts only) that were uniquely modulated by Isolate Mah 1655. In contrast, hundreds of genes were uniquely differentially expressed in response to infection with Maa 1794 and Mah VI101.

Numbers of differentially expressed genes in human macrophages infected with different isolates of Mycobacterium avium . The Venn diagrams of numbers of up-regulated (A) and down-regulated (B) genes show unique and common differential expression of transcripts in human macrophages in response to four hours of incubation by three different isolates of Mycobacterium avium. Maa 1794 is an intracellularly replicating isolate of M. avium subsp. avium, whereas Mah VI101 and Mah 1655 are isolates of M. avium subsp. hominissuis that only invade and persist within cells.
Bioinformatics analyses demonstrated that the major biological functions up-regulated in response to the three isolates were apoptosis, inflammatory response, cytokine/chemokine activity, signal transduction, cell-proliferation and regulation of T-cell activation (Table 2). Biological pathways up-regulated in the responses were related to apoptosis and innate immunity and cytokine signaling (Table 3). In addition, communication pathways between innate and adaptive immune responses and IL-17 signaling were up-regulated. Upstream regulator analysis was performed to infer the putative molecular mechanisms that give rise to the observed gene expression changes, and is illustrated in Table 4. This analysis revealed major roles for TNF, NFkB, IL-1B, IFNg and others in the regulation of the responses. Altogether these analyses revealed a strong overlap between the impacts of all the three isolates.
To identify the putative molecular mechanisms that are regulating the responses, we constructed molecular interaction networks for each isolate using mechanistic findings from previous studies. This analysis showed that Maa 1794 induced the largest gene network in human macrophages, followed by Mah VI101, then by Mah 1655 (Figure 4). The networks were organized around a series of hyper-connected hubs, and the most important hubs (based on number of interactions) are listed in the Table 5. Notably, the network connectivity patterns were dominated by TNF, IL-1B, IL-6, NFkB1, NFkBIA, PTGS2 and others, and many of these pathways were also highlighted by the upstream regulator analysis.

Gene networks triggered in human macrophages infected with different isolates of Mycobacterium avium . Cells have been incubated with M. avium subsp. avium 1794 (A), M. avium subsp. hominissuis VI101 (B) and M. avium subsp. hominissuis 1655 (C) for four hours. Blue hubs indicate up-regulation, yellow indicate down-regulation of the respective gene. The size of the gene name reflects the number of links to other genes in the network, and genes with < 5 genes are minimized. For this analysis the number of genes was reduced, in order to comply with the limitation of the IPA software, by increasing the stringency of the statistical analysis to FDR < 0.01 from FDR < 0.05.
Differential gene expression elicited by Maa 1794, Mah VI101 and Mah 1655
At FDR < 0.05, no significant differences were observed between cells infected with Maa 1794 and Mah VI101, while only two genes (IL6, GPR109A) were upregulated in Maa 1794 compared to Mah 1655 and one gene (IL-6) was upregulated in Mah VI101 compared to Mah 1655.
Protein analysis
Gene expression analysis pointed to apoptosis and immune response as two major biological functions upregulated in response to all three M. avium isolates, albeit at lower levels in Mah 1655 compared to Maa 1794 and Mah VI101. We thus attempted to look at proteins involved in apoptosis by western blot and cytokine by a multiplex assay. However, differences between the isolates with regards to apoptotic proteins were not detected, probably due to the lower sensitivity in this assay compared to the microarray analyses (data not shown).
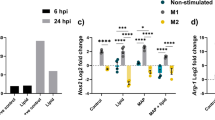
Macrophages infected with all three isolates consistently produced more IL-10, IL-23 and TNF-α than uninfected controls (Figure 5). The production of IL-6 and IL-8 in uninfected cells was often high, and in some donors infection led to a decrease in cytokine production. The decrease in cytokine production was particularly evident for IL-8 after infection with Maa 1794, to a lesser extent by infection with Mah 1655 and rarely by Mah VI101 (Figure 5). There was a large donor to donor variation in the amount of cytokines produced, and this variation surpassed the difference induced by the isolates. Nevertheless, Mah VI101 consistently induced more IL-8, IL-23 and TNF-α than Maa 1794 (p < 0.05) and more IL-23 than Mah 1655 (p < 0.05).

Cytokine expression from primary monocyte-derived macrophages. CD14+ cells were obtained from six donors and allowed to mature into macrophages by incubation for five days. The cells were then infected with the three isolates, Maa 1794, Mah 1655 and Mah VI101, at a MOI of 10:1 for 24 hours or left uninfected (neg). The supernatant was removed and assessed for IL-6, IL-8, IL-10, IL-23 and TNF-α by the Bio-plex assay. The six donors are represented with individual symbols. Each point is the median of triplicate wells. The horizontal line represents the mean response of the six donors. Mah VI101, consistently produced more IL-8, IL-23 and TNF-α than Maa 1794 and more IL-23 than Mah 1655. The difference was significant (p < 0.05) using the non-parametric Wilcoxon signed rank test.
Discussion
The present study describes the uptake and intracellular growth of Maa and Mah isolates in human monocytes, and characterises genome-wide transcriptional responses elicited by these isolates in macrophages. Mycobacterium avium isolates of different genotypic characteristics were all able to enter and persist within human primary monocytes. Intracellular replication was only seen by Maa suggesting that the differences in the ability to survive and replicate in macrophages cannot explain why humans are more often infected with Mah. The gene expression program induced by infection with Maa and Mah isolates was broadly comparable in terms of the biological functions, pathways, and gene networks that were perturbed. However, the degree of perturbation varied considerably. The different level of expression led to an apparent discrepancy in the number of unique genes induced by the various isolates and the lack of differences when comparing the isolates with each other. Many of the genes significantly induced by Maa 1794 were also induced at a lower level by the other isolates (Additional file2: Figure S1). This intermediate expression was neither significantly different from uninfected cells nor from Maa 1794. A larger samples size might have given more significant differences between the isolates since large donor to donor variation is present when using primary cell lines.
In general, responses to Maa 1794 were excessive, and were characterized by increased expression of genes downstream of type I and II interferon signaling. Further studies will be required to determine if variations in these pathways account for the exaggerated responses and/or differences in prevalence. Similar pathways were induced in ruminant monocyte derived macrophages infected with the closely related Mycobacterium avium subspecies paratuberculosis (Map)[25–27], while a study using the murine cell line RAW264.7 showed that Maa and Mah induced higher levels of pro-inflammatory genes compared to Map[28]. Together with our data, these studies suggest that similar pathways were induced by all M. avium subspecies. A direct comparison of the level of macrophage activation is however challenging considering the variation in cell cultures and isolates used in the various studies.
Macrophage activation can be interpreted as an appropriate reaction to an invading pathogen or as a failed attempt of the mycobacteria to silence the host’s defences. Maa 1794 and Mah VI101 differed completely in their ability to replicate within host cells, although both isolates induced a strong activation of macrophages and innate immune responses on the transcriptional level. Analyses on the protein level revealed that the final synthesis of inflammatory cytokines were higher in response to Mah VI101 compared to Maa 1794. One could thus argue that the inflammatory response was sufficient to limit growth of Mah VI101, but not Maa 1794. The complexity of the outcome of an infection, and in vitro culture, was illustrated by the fact that despite a low activation of the innate immune response Mah 1655 did not proliferate intracellularly. The microarray data provided no clear explanation to the lower level of inflammatory cytokines induced by Maa 1794 on the protein level compared to Mah VI101. However, regulation of inflammation on the post-transcriptional level has been demonstrated to be important[29], and a down-regulation of immune responses to avoid immunopathology can occur in chronic infections. The presence of unidentified virulence factors in Maa 1794 more actively shutting down the innate responses cannot be excluded. The genomes of the three isolates did for instance differ in the presence of GPL genes, and the involvement of GPLs in M. avium colony morphology and of ssGPLs in pathogenicity is increasingly recognised[30–32].
The induction of pro-apoptotic genes, such as RIPK2, BID and tBID was seen after infection with all three isolates, however apoptotic pathways were affected to a lesser degree by Mah 1655 than by Maa 1794 and Mah VI101. The correlation between apoptosis and virulence of mycobacterial strains is debated. Inhibition of apoptosis has been described as a virulence factor in mycobacteria[33–35], but it has also been suggested that M. avium can use apoptosis of macrophages as a tool for spreading[36]. One of the other more pronounced differences between the isolates was the increased expression of genes involved in activation of T cells like CD25, CD40, CD274 and IL23 in Maa 1794 and Mah VI101. CD25, CD40 and CD274 are transmembrane surface proteins involved in activation of macrophages and T cells upon binding, while IL-23 is essential for the persistence and function of Th17 cells[37, 38]. These findings suggest that differential activation of apoptotic pathways and/or activation of T cells may occur in humans after infection with the different isolates. The lower activation of macrophages by Mah 1655 may be a useful strategy to avoid the induction of an adaptive immune response in the host. This isolate was included because it harbours ISMpa1 and has a similar IS1245 RFLP pattern to strains shown to be associated with virulence[24, 39]. Further studies, including experimental infection models, could provide an answer to whether the “silencing approach” of Mah 1655 might be the most successful strategy.
Other possible explanations for the low prevalence of Maa observed in the human population, and also in pigs, include the lack of exposure or a reduced ability to cross the human intestinal barrier. In the Norwegian pig population only Mah has been isolated[24], but the virulence of Maa in pigs has been demonstrated by others[40], and previous research demonstrated that Maa 1794 and Mah VI101 were equally able to infect piglets[41]. It thus appears that a lack of exposure is the most likely explanation for the difference in prevalence in pigs. The virulence of Maa has also been demonstrated in mice[42]. Although the study of Pedrosa et al. was performed prior to the division of M. avium into subspecies, one could assume that the bird isolates used in the older studies are equivalent with Maa, and the human isolates with Mah. The virulence of M. avium subspecies in humans can obviously not be determined in vivo. However, altogether the studies in human primary cell lines and infection studies in other species, suggest that different exposure to Mah and Maa in humans is a plausible explanation for the difference in prevalence.
Conclusion
In conclusion, our findings demonstrate that the ability to replicate in macrophages cannot explain the difference in prevalence of the two subspecies in humans. Genome-wide expression profiling studies further showed that similar biological pathways and networks were elicited by the two subspecies, although responses to the Maa subspecies were exaggerated.
Availability of supporting data
The raw microarray data have been made publicly available in the Array Express repository http://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-1101/. The supporting data are included in the additional file.
References
Ashford DA, Whitney E, Raghunathan P, Cosivi O: Epidemiology of selected mycobacteria that infect humans and other animals. Rev Sci Tech. 2001, 20: 325-337.
Thorel MF, Huchzermeyer HF, Michel AL: Mycobacterium avium and Mycobacterium intracellulare infection in mammals. Rev Sci Tech. 2001, 20: 204-218.
Inderlied CB, Kemper CA, Bermudez LE: The Mycobacterium avium complex. Clin Microbiol Rev. 1993, 6: 266-310.
Mijs W, de Haas P, Rossau R, Van der Laan T, Rigouts L, Portaels F, et al: Molecular evidence to support a proposal to reserve the designation Mycobacterium avium subsp. avium for bird-type isolates and ‘M. avium subsp. hominissuis’ for the human/porcine type of M. avium. Int J Syst Evol Microbiol. 2002, 52: 1505-1518. 10.1099/ijs.0.02037-0.
Thorel MF, Huchzermeyer H, Weiss R, Fontaine JJ: Mycobacterium avium infections in animals. Literature review. Vet Res. 1997, 28: 439-447.
Biet F, Boschiroli ML, Thorel MF, Guilloteau LA: Zoonotic aspects of Mycobacterium bovis and Mycobacterium avium-intracellulare complex (MAC). Vet Res. 2005, 36: 411-436. 10.1051/vetres:2005001.
Sturgill-Koszycki S, Schaible UE, Russell DG: Mycobacterium-containing phagosomes are accessible to early endosomes and reflect a transitional state in normal phagosome biogenesis. EMBO J. 1996, 15: 6960-6968.
Rastogi N, Frehel C, Ryter A, Ohayon H, Lesourd M, David HL: Multiple drug resistance in Mycobacterium avium: is the wall architecture responsible for exclusion of antimicrobial agents?. Antimicrob Agents Chemother. 1981, 20: 666-677. 10.1128/AAC.20.5.666.
Rocco JM, Irani VR: Mycobacterium avium and modulation of the host macrophage immune mechanisms. Int J Tuberc Lung Dis. 2011, 15: 447-452. 10.5588/ijtld.09.0695.
Eligini S, Crisci M, Bono E, Songia P, Tremoli E, Colombo GI, et al: Human monocyte-derived macrophages spontaneously differentiated in vitro show distinct phenotypes. J Cell Physiol. 2013, 228: 1464-1472. 10.1002/jcp.24301.
Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al: Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013, 110: 3507-3512. 10.1073/pnas.1222878110.
Johansen TB, Agdestein A, Olsen I, Nilsen SF, Holstad G, Djonne B: Biofilm formation by Mycobacterium avium isolates originating from humans, swine and birds. BMC Microbiol. 2009, 9: 159-10.1186/1471-2180-9-159.
Salte T, Pathak S, Wentzel-Larsen T, Asjo B: Increased intracellular growth of Mycobacterium avium in HIV-1 exposed monocyte-derived dendritic cells. Microbes Infect. 2011, 13: 276-283. 10.1016/j.micinf.2010.11.001.
Baba K, Pathak S, Sviland L, Langeland N, Hoosen AA, Asjo B, et al: Real-time quantitative PCR in the diagnosis of tuberculosis in formalin-fixed paraffin-embedded pleural tissue in patients from a high HIV endemic area. Diagn Mol Pathol. 2008, 17: 112-117. 10.1097/PDM.0b013e31814ceac3.
Eishi Y, Suga M, Ishige I, Kobayashi D, Yamada T, Takemura T, et al: Quantitative analysis of mycobacterial and propionibacterial DNA in lymph nodes of Japanese and European patients with sarcoidosis. J Clin Microbiol. 2002, 40: 198-204. 10.1128/JCM.40.1.198-204.2002.
R Development Core Team: R: A Language and Environment for Statistical Computing. 2011, Vienna, Austria: R Foundation for Statistical Computing
Shi W, Oshlack A, Smyth GK: Optimizing the noise versus bias trade-off for Illumina whole genome expression BeadChips. Nucleic Acids Res. 2010, 38: e204-10.1093/nar/gkq871.
Smyth GK: Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Bio. 2004, 3: Article3-
Huang DW, Sherman BT, Lempicki RA: Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37: 1-13. 10.1093/nar/gkn923.
Huang DW, Sherman BT, Lempicki RA: Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009, 4: 44-57.
Bosco A, Ehteshami S, Panyala S, Martinez FD: Interferon regulatory factor 7 is a major hub connecting interferon-mediated responses in virus-induced asthma exacerbations in vivo. J Allergy Clin Immunol. 2012, 129: 88-94. 10.1016/j.jaci.2011.10.038.
Olsen I, Wiker HG: Diffusion blotting for rapid production of multiple identical imprints from sodium dodecyl sulfate polyacrylamide gel electrophoresis on a solid support. J Immunol Methods. 1998, 220: 77-84. 10.1016/S0022-1759(98)00147-1.
Olsen I, Johansen TB, Billman-Jacobe H, Nilsen SF, Djonne B: A novel IS element, ISMpa1, in Mycobacterium avium subsp. paratuberculosis. Vet Microbiol. 2004, 98: 297-306. 10.1016/j.vetmic.2003.10.025.
Johansen TB, Olsen I, Jensen MR, Dahle UR, Holstad G, Djonne B: New probes used for IS1245 and IS1311 restriction fragment length polymorphism of Mycobacterium avium subsp. avium and Mycobacterium avium subsp. hominissuis isolates of human and animal origin in Norway. BMC Microbiol. 2007, 7: 14-10.1186/1471-2180-7-14.
Marfell BJ, O'Brien R, Griffin JF: Global gene expression profiling of monocyte-derived macrophages from red deer (Cervus elaphus) genotypically resistant or susceptible to Mycobacterium avium subspecies paratuberculosis infection. Dev Comp Immunol. 2013, 40: 210-217. 10.1016/j.dci.2013.02.004.
Kabara E, Kloss CC, Wilson M, Tempelman RJ, Sreevatsan S, Janagama H, et al: A large-scale study of differential gene expression in monocyte-derived macrophages infected with several strains of Mycobacterium avium subspecies paratuberculosis. Brief Funct Genomics. 2010, 9: 220-237. 10.1093/bfgp/elq009.
MacHugh DE, Taraktsoglou M, Killick KE, Nalpas NC, Browne JA, DE Park S, et al: Pan-genomic analysis of bovine monocyte-derived macrophage gene expression in response to in vitro infection with Mycobacterium avium subspecies paratuberculosis. Vet Res. 2012, 43: 25-10.1186/1297-9716-43-25.
Basler T, Geffers R, Weiss S, Valentin-Weigand P, Goethe R: Mycobacterium avium subspecies induce differential expression of pro-inflammatory mediators in a murine macrophage model: evidence for enhanced pathogenicity of Mycobacterium avium subspecies paratuberculosis. Immunobiology. 2008, 213: 879-888. 10.1016/j.imbio.2008.07.009.
Stoecklin G, Anderson P: Posttranscriptional mechanisms regulating the inflammatory response. Adv Immunol. 2006, 89: 1-37.
Bhatnagar S, Schorey JS: Elevated mitogen-activated protein kinase signalling and increased macrophage activation in cells infected with a glycopeptidolipid-deficient Mycobacterium avium. Cell Microbiol. 2006, 8: 85-96. 10.1111/j.1462-5822.2005.00602.x.
Krzywinska E, Bhatnagar S, Sweet L, Chatterjee D, Schorey JS: Mycobacterium avium 104 deleted of the methyltransferase D gene by allelic replacement lacks serotype-specific glycopeptidolipids and shows attenuated virulence in mice. Mol Microbiol. 2005, 56: 1262-1273. 10.1111/j.1365-2958.2005.04608.x.
Schorey JS, Sweet L: The mycobacterial glycopeptidolipids: structure, function, and their role in pathogenesis. Glycobiology. 2008, 18: 832-841. 10.1093/glycob/cwn076.
Briken V, Miller JL: Living on the edge: Inhibition of host cell apoptosis by Mycobacterium tuberculosis. Future Microbiol. 2008, 3: 415-422. 10.2217/17460913.3.4.415.
Toossi Z, Wu M, Rojas R, Kalsdorf B, Aung H, Hirsch CS, et al: Induction of serine protease inhibitor 9 by Mycobacterium tuberculosis inhibits apoptosis and promotes survival of infected macrophages. J Infect Dis. 2012, 205: 144-151. 10.1093/infdis/jir697.
Kabara E, Coussens PM: Infection of primary bovine macrophages with mycobacterium avium subspecies paratuberculosis suppresses host cell apoptosis. Front Microbiol. 2012, 3: 215-
Early J, Fischer K, Bermudez LE: Mycobacterium avium uses apoptotic macrophages as tools for spreading. Microb Pathog. 2011, 50: 132-139. 10.1016/j.micpath.2010.12.004.
Stockinger B, Veldhoen M: Differentiation and function of Th17 T cells. Curr Opin Immunol. 2007, 19: 281-286. 10.1016/j.coi.2007.04.005.
Khader SA, Pearl JE, Sakamoto K, Gilmartin L, Bell GK, Jelley-Gibbs DM, et al: IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J Immunol. 2005, 175: 788-795.
Oliveira RS, Sircili MP, Oliveira EM, Balian SC, Ferreira-Neto JS, Leao SC: Identification of Mycobacterium avium genotypes with distinctive traits by combination of IS1245-based restriction fragment length polymorphism and restriction analysis of hsp65. J Clin Microbiol. 2003, 41: 44-49. 10.1128/JCM.41.1.44-49.2003.
Slana I, Kaevska M, Kralik P, Horvathova A, Pavlik I: Distribution of Mycobacterium avium subsp. avium and M. a. hominissuis in artificially infected pigs studied by culture and IS901 and IS1245 quantitative real time PCR. Vet Microbiol. 2010, 144: 437-443. 10.1016/j.vetmic.2010.02.024.
Agdestein A, Johansen TB, Kolbjornsen O, Jorgensen A, Djonne B, Olsen I: A comparative study of Mycobacterium avium subsp. avium and Mycobacterium avium subsp. hominissuis in experimentally infected pigs. BMC Vet Res. 2012, 8: 11-10.1186/1746-6148-8-11.
Pedrosa J, Florido M, Kunze ZM, Castro AG, Portaels F, McFadden J, et al: Characterization of the virulence of Mycobacterium avium complex (MAC) isolates in mice. Clin Exp Immunol. 1994, 98: 210-216.
Acknowledgements
The project was funded by the Research Council of Norway and the Norwegian Veterinary Institute. AB is supported by a BrightSpark Foundation McCusker Fellowship. Birgitta Åsjø and Sharad Pathak at Gades Insitute, Haukeland Hospital, Bergen, are greatly acknowledged for sharing their experience with real-time PCR for determination of the number of mycobacteria per cell in infected macrophage cultures. The microarray service was provided by the Norwegian Microarray Consortium (NMC) at the national technology platform, and supported by the functional genomics program (FUGE) in the Research Council of Norway.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors state that there are no competing interests related to the present study.
Authors’ contributions
AA was responsible for conception and design of the experiments, contributed to work on immunoblotting, performed work with cell-culture and bacterial quantification, data analysis and drafted the manuscript. AJ contributed to analysis of microarray data and critical revision of the manuscript. AF contributed to microarray analysis and critical revision of the manuscript. TBJ contributed to conception and design of the experiment and critical revision of the manuscript. IAH contributed to cell-culture, immunoblotting and critical revision of the manuscript. BD contributed to conception and design and critical revision of the manuscript. AB contributed to analysis of microarray data and drafting of the manuscript. IO was involved in conception and design of the experiment, performed cytokine profiling and contributed to drafting of the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2013_5699_MOESM1_ESM.xlsx
Additional file 1: Table S1: List of genes differentially expressed in strain Maa 1794, Mah VI101 and Mah 1655 compared to uninfected cells. (XLSX 252 KB)
12864_2013_5699_MOESM2_ESM.pdf
Additional file 2: Figure S1: Volcano plots comparing gene expression patterns in human marophages cultured in the presence or absence of different isolates of Mycobacterium avium. A: Maa 1794. B: Mah VI101. C: Mah 1655. In all three plots, the red data points show the location of genes that were upregulated in responses to isolate Maa 1794. (PDF 3 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Agdestein, A., Jones, A., Flatberg, A. et al. Intracellular growth of Mycobacterium avium subspecies and global transcriptional responses in human macrophages after infection. BMC Genomics 15, 58 (2014). https://doi.org/10.1186/1471-2164-15-58
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-58