
Abstract
Background
The chicken (Gallus gallus) is an important model organism that bridges the evolutionary gap between mammals and other vertebrates. Copy number variations (CNVs) are a form of genomic structural variation widely distributed in the genome. CNV analysis has recently gained greater attention and momentum, as the identification of CNVs can contribute to a better understanding of traits important to both humans and other animals. To detect chicken CNVs, we genotyped 475 animals derived from two broiler chicken lines divergently selected for abdominal fat content using chicken 60 K SNP array, which is a high-throughput method widely used in chicken genomics studies.
Results
Using PennCNV algorithm, we detected 438 and 291 CNVs in the lean and fat lines, respectively, corresponding to 271 and 188 CNV regions (CNVRs), which were obtained by merging overlapping CNVs. Out of these CNVRs, 99% were confirmed also by the CNVPartition program. These CNVRs covered 40.26 and 30.60 Mb of the chicken genome in the lean and fat lines, respectively. Moreover, CNVRs included 176 loss, 68 gain and 27 both (i.e. loss and gain within the same region) events in the lean line, and 143 loss, 25 gain and 20 both events in the fat line. Ten CNVRs were chosen for the validation experiment using qPCR method, and all of them were confirmed in at least one qPCR assay. We found a total of 886 genes located within these CNVRs, and Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses showed they could play various roles in a number of biological processes. Integrating the results of CNVRs, known quantitative trait loci (QTL) and selective sweeps for abdominal fat content suggested that some genes (including SLC9A3, GNAL, SPOCK3, ANXA10, HELIOS, MYLK, CCDC14, SPAG9, SOX5, VSNL1, SMC6, GEN1, MSGN1 and ZPAX) may be important for abdominal fat deposition in the chicken.
Conclusions
Our study provided a genome-wide CNVR map of the chicken genome, thereby contributing to our understanding of genomic structural variations and their potential roles in abdominal fat content in the chicken.
Similar content being viewed by others

Background
Recently, genome-wide association studies (GWAS) have been successfully used in uncovering key genes or markers associated with complex diseases in humans and economically important traits in domestic animals [1, 2]. Based on genotyping data collected from high-throughput SNP chips, new genomic structural variations have been found in the human genome [3]. Copy number variations (CNVs) are a form of genomic structural variation, defined by DNA segments ranging from kilobases (kb) to megabses (Mb) in size, exhibiting differences in copy numbers when comparing two or more genomes [3, 4]. This type of variation includes submicroscopic insertions, deletions and segmental duplications, as well as inversions and translocations [4–6].
CNV detection methods include the use of comparative genomic hybridization (CGH) arrays, SNP arrays, and next-generation sequencing (NGS), and efficient algorithms and softwares are developed to analyze the generated large-scale data. SNP array genotyping offers a number of advantages, low cost, dense coverage and high throughput, therefore, many studies have focused on designing efficient algorithms and softwares to detect reliable CNVs using SNP array data [7, 8], including CNVPartition (http://www.illumina.com), QuantiSNP [9], PennCNV [10], Birdsuite [11], Cokgen [12], Gada [13], and CONAN [14]. These programs have their own strengths and weaknesses [15, 16]. PennCNV distinguishes itself from other algorithms by incorporating multiple information sources (allele frequency, signal intensity, allelic intensity ratio, and distances between SNPs), and by fitting regression models with GC content, it can also overcome the issue of “genomic waves” [17, 18].
CNV can affect gene expression levels, since it contains or disrupts multiple gene coding regions or regulatory elements, which could then lead to phenotypic variation [19]. Not only have CNV studies been performed in humans [19–21], but also in domestic animals, including dogs [22–26], cattle [27–31], swine [32–36], sheep [37–39] and goats [40, 41]. In humans, CNV studies focus mainly on disease development [20], and a multi-allelic CNV encompassing the salivary amylase gene (AMY1) was found to be significantly associated with body mass index (BMI) and obesity [21]. In dogs, detection of CNVs responsible for infertility found two genomic regions harboring two important genes for spermatogenesis, DNM2 and TEKT1 [26]. However, in domestic animals, most studies were limited on the identification of CNVs and the construction of CNV maps [27–31, 35].
Chicken (Gallus gallus) is a classical avian model, and an economically important farm animal, too. The chicken is the first livestock species to have its genome sequenced, and a large number of SNPs have been identified since then. Besides these SNPs, other genomic structural variations are also detected in the chicken genome, such as CNVs. The pea-comb phenotype is caused by a CNV in intron 1 of SOX5 on chicken chromosome 1 (GGA1) [42]; the late feathering locus includes a partial duplication of the PRLR and SPEF2 genes on GGAZ [43]; and the dark brown plumage color on GGA1 [44] and the dermal hyperpigmentation on GGA20 [45] are also associated with CNVs. Additional large number of CNVs had been detected in the chicken using CGH array, SNP chip and genome sequencing. Using CGH arrays, Wang et al. examined ten birds and identified 96 CNVs corresponding to approximately 1.3% of the chicken genome [46]; another study detected 130 CNVRs in four chicken breeds (Cobb broiler, White Leghorn, Chinese Dou and Chinese Dehong) [47]; and Crooijmans et al. detected 3,154 CNVs, grouped into 1,556 CNVRs in a variety of chicken breeds [48]. Using the SNP chip, Jia et al. identified 209 CNVRs in two distinct chicken lines (White Leghorn and dwarf) [49]. Using the sequencing method, Fan et al. identified 8,839 CNVs in two domestic chickens (Silkie and the Taiwanese native chicken L2) [50]. Two other papers reported several CNVs putatively associated with chicken diseases [51, 52]. As in other domestic animals, CNV studies in chickens are also limited in their power. Majority of CNVs detected are relatively large in size, of low resolution, and could contain a high amount of false positives. Genome-wide analysis of CNVs in chicken populations from different genetic backgrounds could help validate CNV regions detected in various studies.
In the present study, the chicken 60 k SNP chip was used to perform genome-wide CNV detection in a population of 475 birds from two broiler lines divergently selected for abdominal fat content (lean and fat lines) [53]. The two lines were selected for more than ten generations and abdominal fat percentage of the lean and fat lines was significantly different from the 4th generation and onwards. We used two methods, PennCNV and CNVPartition, to carry out CNV analysis. Our study provided a comprehensive map of CNVs, which is helpful in understanding genomic variation in the chicken genome, validating CNVs detected in previous studies, and providing preliminary data for investigating the association between CNVs and various phenotypes of economical importance, e.g. abdominal fat content.
Methods
Ethics statement
All animal work was conducted according to the guidelines for the Care and Use of Experimental Animals established by the Ministry of Science and Technology of the People’s Republic of China (approval number: 2006–398), and approved by the Laboratory Animal Management Committee of Northeast Agricultural University.
Animals
In total, 475 birds (203 and 272 individuals from the lean and fat lines, respectively) from the 11th generation population of Northeast Agricultural University broiler lines divergently selected for abdominal fat content (NEAUHLF) were used. Detailed information regarding NEAUHLF has been published previously [53]. Briefly, after 11 generations of divergent selection for abdominal fatness, the abdominal fat percentage of the fat broiler line at 7 weeks of age was 3.59 times more than that of the lean line. All birds were kept in similar environmental conditions and had free access to feed and water.
Genotyping and quality control
Genomic DNA samples were extracted from blood using a standard phenol/chloroform method, and DNA sample quality was determined using spectrophotometry and agarose gel electrophoresis. The Illumina chicken 60 k SNP chip [54] containing 57,636 SNPs was used, and genotyping data were generated using BeadStudio (Version 3.2.2). Quality control was performed using the following default cutoffs: LRR standard deviation, 0.30; BAF drift, 0.01; and waviness factor, 0.05 [47]. After having removed SNPs of low quality, a total of 475 birds, and 48,035 SNPs on 28 autosomes and the Z sex chromosome were kept for CNV detection.
Identification of chicken CNVs using PennCNV
PennCNV software [33] was used to identify chicken CNVs. The PennCNV algorithm incorporates multiple information sources, including LRR and BAF of each SNP marker, and the population frequency of B allele (PFB). Both LRR and BAF were exported from BeadStudio using default clustering files for each SNP. PFB was calculated based on the BAF of each marker. Furthermore, PennCNV integrates a computational approach, by fitting regression models with GC content to overcome the issue of “genomic waves”. Chicken gcmodel files were generated by calculating the GC content of 1 Mb genomic regions surrounding each marker (500 kb on each side), and genomic waves were then adjusted using the option -gcmodel. CNV was chosen based on two criteria: first, it must contain three or more consecutive SNPs, and second, it must be present in at least one animal. CNV regions (CNVRs) were determined by combining overlapping CNVs.
CNV calling using CNVPartition
The CNVPartition software [55] was employed to analyze the same data set, with the confidence score threshold set at 35, in order to verify CNVs detected by PennCNV. In addition, the CNVRs detected by both CNVPartition and PennCNV were cross-checked.
Validation of CNVR by qPCR
The quantitative real-time PCR (qPCR) method was used to validate ten CNVRs identified by both PennCNV and CNVPartition. For each of the ten CNVRs, we selected animals predicted by PennCNV to have different status of CNVs (loss, gain or both) for the validation experiment. Together with three other birds predicted by PennCNV to be normal, a total of 65 birds were used. All qPCR experiments were conducted on an ABI Prism 7500 sequence detection system (Applied Biosystems), using SYBR green chemistry in three replicates. Each reaction was performed with a volume of 10 μl. Primers were designed using Primer premier 5.0 and Oligo 6.0, by limiting product sizes in a range from 100 to 200 bp (Additional file 1: Table S1). The vimentin (VIM) gene, with a single copy in the chicken genome, was chosen as the reference [56]. For every CNVR, three samples without CNVRs were used as negative controls. The condition for thermal cycle was as follows: 2 min at 50°C, 10 min at 95°C and 40 cycles of 15 s at 95°C and 1 min at 60°C. The 2-ΔΔCt method was used to calculate the copy numbers [57].
Gene detection and functional annotation
Genes located in the identified CNVRs were retrieved from Ensembl (http://www.ensembl.org) (Galgal4). Functional annotation of genes was performed using DAVID bioinformatics resources 6.7 (http://david.abcc.ncifcrf.gov/summary.jsp) [58] for Gene Ontology (GO) terms [59] and Kyoto Encyclopedia of Genes and Genomes (KEGG) [60] pathway analysis. Statistical significance was determined using a P-value < 0.05.
Results
Genome-wide identification of CNVs
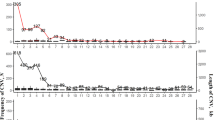
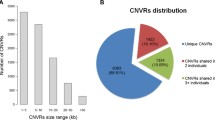
Using 28 autosomal chromosome and the Z sex chromosome, PennCNV identified 438 and 291 CNVs in the lean and fat lines, respectively. Among these CNVs, 17 were common to both lean and fat lines. Combining overlapping CNVs, we identified a total of 271 and 188 CNVRs across the whole genome, covering 40.26 and 30.60 Mb (3.92% and 2.98% of whole genome length) in the lean and fat lines, respectively (Figure 1). Among CNVRs in the lean line, there were 177 loss, 68 gain and 27 both (i.e. loss and gain within the same region) events, while in the fat line, there were 143 loss, 25 gain and 20 both events. In the lean line, the length of CNVRs ranged from 6.23 to 932.14 kb with a mean of 148.77 kb and median of 107.81 kb. In the fat line, CNVRs ranged from 0.33 to 1442.99 kb, with a mean of 163.43 kb and median of 99.81 kb. CNVR locations and characteristics across all 28 autosomal and the Z sex chromosomes were summarized (Figure 1, Additional file 2: Table S2, Additional file 3: Table S3). It is apparent that CNVRs are not uniformly distributed across different chromosomes. No CNVRs were detected on chromosomes 16 and 19 in the lean line, and on chromosomes 15, 16, 19, 20, 23 and 24 in the fat line. Chromosome 1 harbored the largest number of CNVRs, with 53 and 35 in the lean and fat lines, respectively.

Distribution of CNVRs identified in the lean and fat lines.
The CNVPartition program implemented in Illumina GenomeStudio software was also used to verify the CNVRs detected by PennCNV. There were only 4 and 3 CNVRs, out of 271 and 188 CNVRs in the lean and fat lines respectively, missed by CNVPartition (Additional file 2: Table S2, Additional file 3: Table S3). When compared the results obtained from the PennCNV and CNVPartition programs, we saw that most of the CNVRs identified by the CNVPartition were larger in size, and can contain multiple CNVRs identified by PennCNV.
CNV validation by quantitative PCR
The quantitative real-time PCR experiments were performed to validate the CNVRs detected in the current study. Ten putative CNVRs were selected, and represented different status of copy numbers (predicted to be from gain, loss, or both events) (Table 1). We performed 333 qPCR assays in 65 animals. Out of the 333 qPCR assays, 204 (61%) were in agreement with the CNV prediction made by PennCNV. Direct counting of the CNVRs confirmed that all 10 CNVRs had copy number variations in at least one qPCR assay (Figure 2, Table 1).

Verification of 10 detected CNVRs by qPCR. CNVR1-10 represents 10 CNVRs selected for qPCR validation. X-axis shows the individuals used in the validation experiment. Y-axis shows the relative quantification (RQ) values obtained by qPCR. Different shapes denote samples with different RQ values. Asterisk: samples with RQ values about 1 denote normal individuals (two copies); Box: samples with RQ values below 0.59 (ln1.5) denote copy number loss individuals; Triangle: samples with RQ values about 1.59 (ln3) or more denote copy number gain individuals (>three copies) [61].
Genes located in CNVRs
Within the 271 CNVRs identified in the lean line, 204 (75.3%) CNVRs contained 626 genes retrieved from Ensembl, and the remaining 67 CNVRs did not contain any annotated genes. Within the 188 CNVRs identified in the fat line, 140 (74.5%) CNVRs had 374 genes, and no annotated genes located in the other 48 CNVRs, either. There were 114 common genes detected in both chicken lines. Therefore, a total of 886 genes were identified within the detected CNVRs.
Functional enrichment analyses found that a total of 1744 GO terms for the 886 genes, with eight GO terms statistically significant (P < 0.05) (Table 2). Significant GO terms were mainly involved in interleukin-1 binding, interleukin-1 receptor activity, hydrogen antiporter activity, calcium ion binding, solute antiporter activity, plasma membrane, antiporter activity, and cation antiporter activity. GO terms with marginal significance were mainly involved in cell-cell adhesion, cytokine receptor activity, dystrophin-associated glycoprotein complex, homophilic cell adhesion and hydrogen antiporter activity (monovalent cation and sodium). KEGG pathway analyses showed that genes in CNVRs were enriched in 95 pathways, two of them statistically significant (calcium signaling and N-Glycan biosynthesis pathways) (Table 2).
Integrative analysis of CNVRs, QTLs, and selective sweeps for abdominal fat content
In the chicken QTL database (http://www.animalgenome.org/cgi-bin/QTLdb/GG/index; May 21, 2013), there were 3,808 quantitative trait loci (QTLs) affecting 296 traits, including 291 QTLs for abdominal fat weight and percentage. In the lean line, 160 CNVRs overlapped with 74 QTLs affecting abdominal fat weight or percentage. Furthermore, for the 271 CNVRs in the current study and 5357 core regions detected in the previous seletive sweep study [62], we found that 225 CNVRs overlapped with 311 core regions, after comparing the CNVR and selective sweep results. In the 311 core regions, 10 regions reached statistical significance of P < 0.01. These 10 core regions related to 10 CNVRs, with six CNVRs located in QTL regions for abdominal fat content. Eight genes were found within these six CNVRs, including solute carrier family 9 member 3 (SLC9A3), guanine nucleotide binding protein (G-protein), alpha activating activity polypeptide, olfactory type (GNAL), sparc/osteonectin, cwcv and kazal-like domains proteoglycan (testican) 3 (SPOCK3), annexin A10 (ANXA10), IKAROS family zinc finger 2 (HELIOS), myosin light chain kinase (MYLK), coiled-coil domain containing 14 (CCDC14), and sperm-associated antigen 9 (SPAG9).
In the fat line, 112 CNVRs overlapped with 66 QTLs affecting abdominal fat weight or percentage in the chicken QTL database. In addition, for the 188 CNVRs in the current study and 5593 core regions detected in the previous seletive sweep study [62], we found that 140 CNVRs overlapped with 203 core regions, and in the 203 core regions, 6 regions reached statistical significance of P < 0.01. These 6 core regions related to six CNVRs, with four CNVRs located in abdominal fat QTLs. Six genes were found within these four CNVRs, including SRY (sex determining region Y)-box 5 (SOX5), visinin-like 1 (VSNL1), structural maintenance of chromosomes 6 (SMC6), GEN endonuclease homolog 1 (GEN1), mesogenin 1 (MSGN1) and zona pellucida protein (ZPAX).
Discussion
The chicken 60 k SNP array was originally developed for high-throughput SNP genotyping in GWAS studies. Although CNV detection is feasible with this SNP panel, it is of less power due to low marker density, non-uniform SNP distribution along chicken chromosomes, and a lack of non-polymorphic probes specifically designed for CNV identification [63]. Thus, using this array, typically only large CNVRs could be identified.
Several algorithms have been developed to identify CNVs, including CNVPartition, QuantiSNP, PennCNV, Birdsuite, Cokgen, Gada, and CONAN [9–14]. Each algorithm has its own strength and weakness [15, 16]. In the present study, the PennCNV algorithm was used for CNV detection, and CNVPartition was employed to verify the CNVs detected by PennCNV. We found that 99% of the CNVRs detected by PennCNV could be verified by the CNVPartition program. This high ratio indicated that the CNVRs detected in this study were credible and the following discussion was based on the PennCNV results.
We used two lines divergently selected for abdominal fat content to detect CNVs in the chicken, and found the lean line had more CNVs than the fat line (438 vs 291). One of the reason could be due to different number of animals (203 and 272 individuals in the lean and fat lines, respectively). Additionally, these two lines have different selection signatures as reported previously [62], which suggests that artificial selection for abdominal fat could also lead to CNV alterations between these two lines.
We compared our results with several previous reports on chicken CNVs. The first study was reported by Griffin et al. [64]. They used the CGH array and detected 12 CNVs in broiler and layer genomes, compared with the Red Jungle Fowl. Two of these 12 CNVs overlapped with our results (Additional file 4: Table S4). Wang et al. detected 96 CNVs in three chicken lines (Cornish Rock broiler, Leghorn, and Rhode Island Red) using whole-genome tiling arrays [46]. Of these 96 CNVs, 14 CNVs overlapped with our results (Additional file 4: Table S4). In 2012, Wang et al. detected 130 CNVRs in four chicken breeds (Cobb broiler, White Leghorn, Chinese Dou and Chinese Dehong) using CGH arrays, with 16 overlapping CNVs (Additional file 4: Table S4) [47]. In the same year, Jia et al. identified 209 CNVRs in two distinct chicken lines (White Leghorn and dwarf) using chicken 60 k SNP arrays, with 47 overlapping CNVRs (Additional file 4: Table S4) [49]. Luo et al. identified 45 CNVs in four chicken lines (L63, L72, RCS-L, and RCS-M), with two CNVs overlapping with our CNVRs [51]. Crooijmans et al. detected 1556 CNVRs using the CGH arrays in a wide variety of chicken breeds, with 140 overlapping CNVRs with our current study [48]. In total, 181 of 459 CNVRs (271 and 188 CNVRs in lean and fat lines, respectively) (39%) detected in our study were also detected in previous studies (Additional file 4: Table S4). Potential reasons for the observed differences include the following three considerations. Firstly, the populations are of different sizes and genetic background; Secondly, different array platforms are used, either SNP genotyping or CGH arrays; Thirdly, genomic waves can interfere with accurate CNV detection [41, 65]. Genomic waves refer to signal intensity patterns across all chromosomes, with different samples showing highly variable magnitudes of waviness [41]. In our study, we adjusted for genomic waves using the -gcmodel option in PennCNV. Genomic waves were generally not considered in other studies. Apart from low overlapping rates between different chicken CNV studies, the same issue was also encountered in other animals [65–67].
In previous observations, CNVs are preferentially located in gene-poor regions [68, 69]. It is speculated that CNVs present in gene-rich regions may be deleterious and under purifying selection [70]. In the chicken genome, there are approximately 28,000 genes (data from the GeneChip®Chicken Genome Array Profile), and 886 (3.16%) annotated genes located in the 271 and 188 CNVRs in the lean and fat lines, respectively, were identified in our current study. These CNVRs covered 3.92 and 2.98% of the chicken genome in the lean and fat lines, respectively. Therefore, we can not state that these CNVRs locate in gene-poor or gene-rich regions.
QPCR is often used to validate novel CNVRs, but confirmation rates are usually not very high [11, 20, 21]. For instance, Fadista et al. [20] and Hou et al. [65] confirmed 50 and 60% of CNVRs selected for validation, respectively. Our validation rate was 61% (204 out of the 333 qPCR assays), comparable to the results of other studies.
Comparing CNVs detected in our current study with known QTLs (in the QTL database) and selective sweeps for abdominal fat content [62], we identified 14 genes (8 and 6 in the lean and fat lines, respectively). For the eight genes in the lean line, we found SLC9A3, GNAL, ANXA10, MYLK, CCDC14, and SPAG9 expressed in chicken pre-adipocytes, and SLC9A3, GNAL, ANXA10, HELIOS, MYLK, CCDC14, and SPAG9 expressed in both chicken abdominal fat and liver tissues (data not published). For the six genes in the fat line, we found SOX5, VSNL1, SMC6, and GEN1 expressed in chicken pre-adipocytes, GEN1, SMC6, SOX5, and VSNL1 expressed in chicken abdominal fat tissue, and SOX5, VSNL1, and SMC6 expressed in chicken liver tissue (data not published). Basic functions of these 14 genes are described as follows.
SLC9A3 is also known as sodium–hydrogen antiporter 3, or sodium–hydrogen exchanger 3 (NHE3) [71]. SLC9A3 is expressed in human intestine, stomach, respiratory tract, kidneys, glandular and epithelial cells [72]. SLC9A3 is present in the brush-border of intestinal Na + -absorptive cells and renal proximal tubules, playing an important role in gastrointestinal and renal Na + absorption [73], and suggesting it may be involved in food digestion and nutrient absorption, and in turn, abdominal fat deposition.
G-proteins are divided into four subfamilies according to their α-subunits (Gαs, Gαi/o, Gαq, and Gα12) [74]. Gα subunits interact with both receptor and effect or molecules, and are considered the functional component of G-proteins. GNAL shares 88% amino acid homology with Gαs, and is considered a member of the Gαs family [74]. Although GNAL was originally discovered in olfactory neuroepithelium and striatum, it is also present in pancreatic β-cells, testis, spleen, lung, and heart [75]. In addition, this gene is highly expressed in adipose tissue (http://www.genatlas.org/), indicating it may be associated with abdominal fat deposition.
SPOCK3 encodes a member of the novel family of calcium-binding proteoglycan proteins that contain thyroglobulin type-1 and Kazal-like domains. Encoded SPOCK3 protein may play a key role in adult T-cell leukemia by inhibiting membrane-type matrix metalloproteinase activity [76]. SPOCK3 is expressed in the mouse nervous system [77].
ANXA10 belongs to the annexin family, and is over-expressed in oral squamous cell carcinoma-derived cell lines [78]. ANXA10 plays an important role in cellular functioning of endocytosis and exocytosis, anticoagulant activity, cytoskeletal interactions, differentiation, and cellular proliferation [79, 80]. Moreover, ANXA10 shows relevant malignancy in Barrett’s esophagus, gastric cancer, and bladder cancer [81–83]. ANXA10 is expressed in the digestive system including liver and stomach tissues (http://www.genatlas.org/), indicating it may affect food digestion and absorption, and consequently be associated with fat deposition.
Helios is a member of the Ikaros transcription factor family, and preferentially expressed by regulatory T cells [84]. Previous work has shown that obese patients with insulin resistance have decreased HELIOS but increased FOXP3 mRNA expression in visceral adipose tissue [85]. Helios is expressed in ectodermal and neuroectodermal-derived tissues [86].
MYLK is a muscle member of the immunoglobulin gene superfamily, and encodes myosin light chain kinase, a calcium/calmodulin dependent enzyme. Genetic and functional studies show that heterozygous loss-of-function mutations in MYLK are associated with aortic dissection [87]. MYLK is highly expressed in heart, prostate, trachea tissues, and the digestive system (including esophagus and small intestine), suggesting this gene is involved in food digestion and absorption, and consequently associated with fat deposition.
CCDC14 is a protein-coding gene with unknown function. CCDC14 is expressed in male testis tissue (http://www.genatlas.org/).
SPAG9 is a novel member of c-Jun NH2 -terminal kinase (JNK) interacting proteins, exclusively expressed in testis [88]. SPAG9 may play a key role in reproductive processes, and tumor growth and development [88, 89].
SOX5 is a member of the SOX (SRY-related HMG-box) family, and involved in regulation of embryonic development and cell fate determination [90]. In chicken, CNV in intron 1 of SOX5 can cause the Pea-comb phenotype [42]. SOX5 is expressed in brain, spinal cord, testis, lung, and kidney, and can control cell cycle progression in neural progenitors by interfering with the WNT-beta-catenin pathway [91]. A recent study indicated SOX5 may play an important role in left ventricular mass regulation, a disease that may be affected by abdominal obesity [92].
VSNL1 is a member of the visinin/recoverin subfamily of neuronal calcium sensor proteins, and highly expressed in human heart and brain [93, 94]. Previous results suggest VSNL1 regulates heart natriuretic peptide receptor B [93]. The VSNL1 gene also plays a critical role in regulating cell adhesion and migration via downregulation of fibronectin receptor expression [95]. The VSNL1 gene is highly expressed in the nervous system.
Structural maintenance of chromosomes (SMC) proteins are a family of related proteins that form the core of three protein complexes. Smc1 and 3 ensure sister chromatids remain associated after DNA replication, as well as playing roles in gene expression and DNA repair [96]. Smc2 and 4 are responsible for chromosome condensation during mitosis [97]. The Smc5-6 complex is required for DNA repair by homologous recombination, although its exact role is not fully understood [98].
GEN1 is a member of the Rad2/XPG family of monomeric, structure-specific nucleases [99]. This protein family includes N-terminal and internal XPG nuclease motifs, and a helix–hairpin–helix domain [100]. The GEN1 gene is expressed in pancreas, thymus, brain, testis, lung, and kidney, and has Holliday junction resolvase activity in vitro, presumably functioning in homology-driven repair of DNA double-strand breaks [101].
Msgn1 is a basic helix–loop–helix transcription factor, specifically expressed in the presomitic mesoderm (psm). Msgn1 controls differentiation and movement of psm progenitor cells, and mouse embryos lacking Msgn1 exhibit a severely reduced psm and an absence of trunk somites [102, 103].
The vertebrate egg envelope is constructed by a set of related proteins encoded by the zona pellucida (ZP) genes [104]. Vertebrate ZP genes have six subfamilies: ZPA/ZP2, ZPB/ZP4, ZPC/ZP3, ZP1, ZPAX, and ZPD [105]. The Zpb pseudogene was identified in the mouse genome, Zp1 pseudogene in the dog and bovine genomes, and Zpax pseudogene in the human, chimpanzee, macaque, and bovine genomes [105]. ZP genes may play an important role in sperm-egg recognition [105].
All these genes are located in QTLs for abdominal fat weight or percentage in the chicken. From the known functions of these genes, GNAL, HELIOS, and SOX5 are directly related to adipose tissue metabolism or obesity, while SLC9A3, SPOCK3, ANXA10, MYLK, and VSNL1 may be indirectly related. The function of CCDC14, SPAG9, SMC6, GEN1, MSGN1, and ZPAX on adipose tissue development is unknown. However, further investigation are still needed to examine their functional implications in chicken adipogenesis.
Conclusions
We have constructed a CNVR map for the broiler chicken using two lines divergently selected for abdominal fat content. In total, 271 and 188 CNVRs in the lean and fat lines were identified, respectively. Integrating detected CNVRs, and results of QTLs and selection signatures for abdominal fat content, 14 genes (including SLC9A3, GNAL, SPOCK3, ANXA10, HELIOS, MYLK, CCDC14, SPAG9, SOX5, VSNL1, SMC6, GEN1, MSGN1, and ZPAX) were identified as putatively important for chicken abdominal fat content.
Availability of supporting data
The data sets supporting the results of this article are included within the article and its additional files. The chicken 60 k SNP data presented in this paper have been deposited into Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) with the identifier GSE58551.
Abbreviations
- AFP:
-
Abdominal fat percentage
- CGH:
-
Comparative genomic hybridization
- CNV:
-
Copy number variation
- CNVR:
-
CNV region
- GO:
-
Ontology
- GWAS:
-
Genome-wide association studies
- kb:
-
Kilobases
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- Mb:
-
Megabases
- NEAUHLF:
-
Northeast Agricultural University broiler lines divergently selected for abdominal fat content
- PFB:
-
Population frequency of B allele
- SNP:
-
Single nucleotide polymorphism
- VLDL:
-
Plasma very low-density lipoprotein.
References
Nelson CL, Pelak K, Podgoreanu MV, Ahn SH, Scott WK, Allen AS, Cowell LG, Rude TH, Zhang Y, Tong A, Ruffin F, Sharma-Kuinkel BK, Fowler VG: A genome-wide association study of variants associated with acquisition of Staphylococcus aureus bacteremia in a healthcare setting. BMC Infect Dis. 2014, 14 (1): 83-10.1186/1471-2334-14-83.
Hayes B, Goddard M: Genome-wide association and genomic selection in animal breeding. Genome. 2010, 53 (11): 876-883. 10.1139/G10-076.
Feuk L, Carson AR, Scherer SW: Structural variation in the human genome. Nat Rev Genet. 2006, 7 (2): 85-97. 10.1038/nrm1837.
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, González JR, Gratacòs M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, et al: Global variation in copy number in the human genome. Nature. 2006, 444 (7118): 444-454. 10.1038/nature05329.
Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C: Detection of large-scale variation in the human genome. Nat Genet. 2004, 36 (9): 949-951. 10.1038/ng1416.
Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Månér S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M: Large-scale copy number polymorphism in the human genome. Science. 2004, 305 (5683): 525-538. 10.1126/science.1098918.
Zöllner S, Su G, Stewart WC, Chen Y, McInnis MG, Burmeister M: Bayesian EM algorithm for scoring polymorphic deletions from SNP data and application to a common CNV on 8q24. Genet Epidemiol. 2009, 33 (4): 357-368. 10.1002/gepi.20391.
Diskin SJ, Hou C, Glessner JT, Attiyeh EF, Laudenslager M, Bosse K, Cole K, Mossé YP, Wood A, Lynch JE, Pecor K, Diamond M, Winter C, Wang K, Kim C, Geiger EA, McGrady PW, Blakemore AI, London WB, Shaikh TH, Bradfield J, Grant SF, Li H, Devoto M, Rappaport ER, Hakonarson H, Maris JM: Copy number variation at 1q21.1 associated with neuroblastoma. Nature. 2009, 459 (7249): 987-991. 10.1038/nature08035.
Colella S, Yau C, Taylor JM, Mirza G, Butler H, Clouston P, Bassett AS, Seller A, Holmes CC, Ragoussis J: QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007, 35 (6): 2013-2025. 10.1093/nar/gkm076.
Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M: PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 17 (11): 1665-1674. 10.1101/gr.6861907.
Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins PJ, Darvishi K, Lee C, Nizzari MM, Gabriel SB, Purcell S, Daly MJ, Altshuler D: Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet. 2008, 40 (10): 1253-1260. 10.1038/ng.237.
Yavas G, Koyutürk M, Ozsoyoğlu M, Gould MP, LaFramboise T: An optimization framework for unsupervised identification of rare copy number variation from SNP array data. Genome Biol. 2009, 10 (10): R119-10.1186/gb-2009-10-10-r119.
Forer L, Schönherr S, Weissensteiner H, Haider F, Kluckner T, Gieger C, Wichmann HE, Specht G, Kronenberg F, Kloss-Brandstätter A: CONAN: copy number variation analysis software for genome-wide association studies. BMC Bioinformatics. 2010, 11: 318-10.1186/1471-2105-11-318.
Pique-Regi R, Cáceres A, González JR: R-Gada: a fast and flexible pipeline for copy number analysis in association studies. BMC Bioinformatics. 2010, 11: 380-10.1186/1471-2105-11-380.
Winchester L, Yau C, Ragoussis J: Comparing CNV detection methods for SNP arrays. Brief Funct Genomic Proteomic. 2009, 8 (5): 353-366. 10.1093/bfgp/elp017.
Pinto D, Darvishi K, Shi X, Rajan D, Rigler D, Fitzgerald T, Lionel AC, Thiruvahindrapuram B, Macdonald JR, Mills R, Prasad A, Noonan K, Gribble S, Prigmore E, Donahoe PK, Smith RS, Park JH, Hurles ME, Carter NP, Lee C, Scherer SW, Feuk L: Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat Biotechnol. 2011, 29 (6): 512-520. 10.1038/nbt.1852.
Marioni JC, Thorne NP, Valsesia A, Fitzgerald T, Redon R, Fiegler H, Andrews TD, Stranger BE, Lynch AG, Dermitzakis ET, Carter NP, Tavaré S, Hurles ME: Breaking the waves: improved detection of copy number variation from microarray-based comparative genomic hybridization. Genome Biol. 2007, 8 (10): R228-10.1186/gb-2007-8-10-r228.
Diskin SJ, Li M, Hou C, Yang S, Glessner J, Hakonarson H, Bucan M, Maris JM, Wang K: Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. Nucleic Acids Res. 2008, 36 (19): e126-10.1093/nar/gkn556.
Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, Fitzgerald T, Hu M, Ihm CH, Kristiansson K, Macarthur DG, Macdonald JR, Onyiah I, Pang AW, Robson S, Stirrups K, Valsesia A, Walter K, Wei J, Tyler-Smith C, Carter NP, Lee C, Scherer SW, Hurles ME, Wellcome Trust Case Control Consortium: Origins and functional impact of copy number variation in the human genome. Nature. 2010, 464 (7289): 704-712. 10.1038/nature08516.
Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, Krauss RM, Myers RM, Ridker PM, Chasman DI, Mefford H, Ying P, Nickerson DA, Eichler EE: Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009, 84 (2): 148-161. 10.1016/j.ajhg.2008.12.014.
Falchi M, El-Sayed Moustafa JS, Takousis P, Pesce F, Bonnefond A, Andersson-Assarsson JC, Sudmant PH, Dorajoo R, Al-Shafai MN, Bottolo L, Ozdemir E, So HC, Davies RW, Patrice A, Dent R, Mangino M, Hysi PG, Dechaume A, Huyvaert M, Skinner J, Pigeyre M, Caiazzo R, Raverdy V, Vaillant E, Field S, Balkau B, Marre M, Visvikis-Siest S, Weill J, Poulain-Godefroy O, et al: Low copy number of the salivary amylase gene predisposes to obesity. Nat Genet. 2014, doi:10.1038/ng.2939
Chen WK, Swartz JD, Rush LJ, Alvarez CE: Mapping DNA structural variation in dogs. Genome Res. 2009, 19 (3): 500-509.
Nicholas TJ, Cheng Z, Ventura M, Mealey K, Eichler EE, Akey JM: The genomic architecture of segmental duplications and associated copy number variants in dogs. Genome Res. 2009, 19 (3): 491-499.
Nicholas TJ, Baker C, Eichler EE, Akey JM: A high-resolution integrated map of copy number polymorphisms within and between breeds of the modern domesticated dog. BMC Genomics. 2011, 12: 414-10.1186/1471-2164-12-414.
Alvarez CE, Akey JM: Copy number variation in the domestic dog. Mamm Genome. 2012, 23 (1–2): 144-163.
Cassatella D, Martino NA, Valentini L, Guaricci AC, Cardone MF, Pizzi F, Dell'Aquila ME, Ventura M: Male infertility and copy number variants (CNVs) in the dog: a two-pronged approach using Computer Assisted Sperm Analysis (CASA) and Fluorescent In Situ Hybridization (FISH). BMC Genomics. 2013, 14: 921-10.1186/1471-2164-14-921.
Fadista J, Thomsen B, Holm LE, Bendixen C: Copy number variation in the bovine genome. BMC Genomics. 2010, 11: 284-10.1186/1471-2164-11-284.
Liu GE, Bickhart DM: Copy number variation in the cattle genome. FunctIntegr Genomics. 2012, 12 (4): 609-624.
Jiang L, Jiang J, Wang J, Ding X, Liu J, Zhang Q: Genome-wide identification of copy number variations in Chinese Holstein. PLoS One. 2012, 7 (11): e48732-10.1371/journal.pone.0048732.
Jiang L, Jiang J, Yang J, Liu X, Wang J, Wang H, Ding X, Liu J, Zhang Q: Genome-wide detection of copy number variations using high-density SNP genotyping platforms in Holsteins. BMC Genomics. 2013, 14: 131-10.1186/1471-2164-14-131.
Choi JW, Lee KT, Liao X, Stothard P, An HS, Ahn S, Lee S, Lee SY, Moore SS, Kim TH: Genome-wide copy number variation in Hanwoo, Black Angus, and Holstein cattle. Mamm Genome. 2013, 24 (3–4): 151-163.
Fadista J, Nygaard M, Holm LE, Thomsen B, Bendixen C: A snapshot of CNVs in the pig genome. PLoS One. 2008, 3 (12): e3916-10.1371/journal.pone.0003916.
Ramayo-Caldas Y, Castelló A, Pena RN, Alves E, Mercadé A, Souza CA, Fernández AI, Perez-Enciso M, Folch JM: Copy number variation in the porcine genome inferred from a 60 k SNP BeadChip. BMC Genomics. 2010, 11: 593-10.1186/1471-2164-11-593.
Wang J, Jiang J, Fu W, Jiang L, Ding X, Liu JF, Zhang Q: A genome-wide detection of copy number variations using SNP genotyping arrays in swine. BMC Genomics. 2012, 13: 273-10.1186/1471-2164-13-273.
Chen C, Qiao R, Wei R, Guo Y, Ai H, Ma J, Ren J, Huang L: A comprehensive survey of copy number variation in 18 diverse pig populations and identification of candidate copy number variable genes associated with complex traits. BMC Genomics. 2012, 13: 733-10.1186/1471-2164-13-733.
Fowler KE, Pong-Wong R, Bauer J, Clemente EJ, Reitter CP, Affara NA, Waite S, Walling GA, Griffin DK: Genome wide analysis reveals single nucleotide polymorphisms associated with fatness and putative novel copy number variants in three pig breeds. BMC Genomics. 2013, 14: 784-10.1186/1471-2164-14-784.
Norris BJ, Whan VA: A gene duplication affecting expression of the ovine ASIP gene is responsible for white and black sheep. Genome Res. 2008, 18 (8): 1282-1293. 10.1101/gr.072090.107.
Fontanesi L, Beretti F, Martelli PL, Colombo M, Dall'olio S, Occidente M, Portolano B, Casadio R, Matassino D, Russo V: A first comparative map of copy number variations in the sheep genome. Genomics. 2011, 97 (3): 158-165. 10.1016/j.ygeno.2010.11.005.
Liu J, Zhang L, Xu L, Ren H, Lu J, Zhang X, Zhang S, Zhou X, Wei C, Zhao F, Du L: Analysis of copy number variations in the sheep genome using 50 K SNP BeadChip array. BMC Genomics. 2013, 14: 229-10.1186/1471-2164-14-229.
Fontanesi L, Beretti F, Riggio V, Gómez González E, Dall'Olio S, Davoli R, Russo V, Portolano B: Copy number variation and missense mutations of the agouti signaling protein (ASIP) gene in goat breeds with different coat colors. Cytogenet Genome Res. 2009, 126 (4): 333-347. 10.1159/000268089.
Fontanesi L, Martelli PL, Beretti F, Riggio V, Dall'Olio S, Colombo M, Casadio R, Russo V, Portolano B: An initial comparative map of copy number variations in the goat (Capra hircus) genome. BMC Genomics. 2010, 11: 639-10.1186/1471-2164-11-639.
Wright D, Boije H, Meadows JRS, Bed’hom B, Gourichon D, Vieaud A, Tixier-Boichard M, Rubin C-J, Imsland F, Hallböök F, Andersson L: Copy number variation in intron 1 of SOX5 causes the Pea-comb phenotype in chickens. PLoS Genet. 2009, 5: e1000512-10.1371/journal.pgen.1000512.
Elferink MG, Vallee AAA, Jungerius A, Crooijmans RPMA, Groenen MAM: Partial duplication of the PRLR and SPEF2 genes at the late feathering locus in chicken. BMC Genomics. 2008, 9: 391-399. 10.1186/1471-2164-9-391.
Gunnarsson U, Kerje S, Bed’hom B, Sahlqvist A-S, Ekwall O, Tixier-Boichard M, Kämpe O, Andersson L: The dark brown plumage color in chickens is caused by an 8.3-kb deletion upstream of SOX10. Pigment Cell Melanoma Res. 2011, 24: 268-274. 10.1111/j.1755-148X.2011.00825.x.
Dorshorst B, Molin A-M, Rubin CJ, Johansson AM, Strömstedt L, Pham M-H, Chen C-F, Hallböök F, Ashwell C, Andersson L: A complex genomic rearrangement involving the endothelium 3 locus causes dermal hyperpigmentation in the chicken. PLoS Genet. 2011, 7: e1002412-10.1371/journal.pgen.1002412.
Wang X, Nahashon S, Feaster TK, Bohannon-Stewart A, Adefope N: An initial map of chromosomal segmental copy number variations in the chicken. BMC Genomics. 2010, 11: 351-10.1186/1471-2164-11-351.
Wang Y, Gu X, Feng C, Song C, Hu X, Li N: A genome-wide survey of copy number variation regions in various chicken breeds by array comparative genomic hybridization method. Anim Genet. 2012, 43 (3): 282-289. 10.1111/j.1365-2052.2011.02308.x.
Crooijmans RP, Fife MS, Fitzgerald TW, Strickland S, Cheng HH, Kaiser P, Redon R, Groenen MA: Large scale variation in DNA copy number in chicken breeds. BMC Genomics. 2013, 14: 398-10.1186/1471-2164-14-398.
Jia X, Chen S, Zhou H, Li D, Liu W, Yang N: Copy number variations identified in the chicken using a 60 K SNP BeadChip. Anim Genet. 2013, 44 (3): 276-284. 10.1111/age.12009.
Fan WL, Ng CS, Chen CF, Lu MY, Chen YH, Liu CJ, Wu SM, Chen CK, Chen JJ, Mao CT, Lai YT, Lo WS, Chang WH, Li WH: Genome-wide patterns of genetic variation in two domestic chickens. Genome Biol Evol. 2013, 5 (7): 1376-1392. 10.1093/gbe/evt097.
Luo J, Yu Y, Mitra A, Chang S, Zhang H, Liu G, Yang N, Song J: Genome-wide copy number variant analysis in inbred chickens lines with different susceptibility to Marek's disease. G3 (Bethesda). 2013, 3 (2): 217-223. 2013.
Abe H, Nagao K, Inoue-Murayama M: Short copy number variations potentially associated with tonic immobility responses in newly hatched chicks. PLoS One. 2013, 8 (11): e80205-10.1371/journal.pone.0080205.
Zhang H, Hu X, Wang Z, Zhang Y, Wang S, Wang N, Ma L, Leng L, Wang S, Wang Q, Wang Y, Tang Z, Li N, Da Y, Li H: Selection signature analysis implicates the PC1/PCSK1 region for chicken abdominal fat content. PLoS One. 2012, 7 (7): e40736-10.1371/journal.pone.0040736.
Groenen MA, Megens HJ, Zare Y, Warren WC, Hillier LW, Crooijmans RP, Vereijken A, Okimoto R, Muir WM, Cheng HH: The development and characterization of a 60K SNP chip for chicken. BMC Genomics. 2011, 12 (1): 274-10.1186/1471-2164-12-274.
Dellinger AE, Saw SM, Goh LK, Seielstad M, Young TL, Li YJ: Comparative analyses of seven algorithms for copy number variant identification from single nucleotide polymorphism arrays. Nucleic Acids Res. 2010, 38 (9): e105-10.1093/nar/gkq040.
Zehner ZE, Paterson BM: Characterization of the chicken vimentin gene: single copy gene producing multiple mRNAs. Proc Natl Acad Sci U S A. 1983, 80: 911-915. 10.1073/pnas.80.4.911.
Yu S, Kielt M, Stegner AL, Kibiryeva N, Bittel DC, Cooley LD: Quantitative real-time polymerase chain reaction for the verification of genomic imbalances detected by microarray-based comparative genomic hybridization. Genet Test Mol Biomarkers. 2009, 2009 (13): 751-760.
da Huang W, Sherman BT, Lempicki RA: Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009, 4 (1): 44-57.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000, 25 (1): 25-29. 10.1038/75556.
Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M: KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38 (Database issue): D355-360.
Wang L, Liu X, Zhang L, Yan H, Luo W, Liang J, Cheng D, Chen S, Ma X, Song X, Zhao K, Wang L: Genome-wide copy number variations inferred from SNP genotyping arrays using a Large White and Minzhu intercross population. PLoS One. 2013, 8 (10): e74879-10.1371/journal.pone.0074879.
Zhang H, Wang SZ, Wang ZP, Da Y, Wang N, Hu XX, Zhang YD, Wang YX, Leng L, Tang ZQ, Li H: A genome-wide scan of selective sweeps in two broiler chicken lines divergently selected for abdominal fat content. BMC Genomics. 2012, 13: 704-10.1186/1471-2164-13-704.
Ramos AM, Crooijmans RP, Affara NA, Amaral AJ, Archibald AL, Beever JE, Bendixen C, Churcher C, Clark R, Dehais P, Hansen MS, Hedegaard J, Hu ZL, Kerstens HH, Law AS, Megens HJ, Milan D, Nonneman DJ, Rohrer GA, Rothschild MF, Smith TP, Schnabel RD, Van Tassell CP, Taylor JF, Wiedmann RT, Schook LB, Groenen MA: Design of a high density SNP genotyping assay in the pig using SNPs identified and characterized by next generation sequencing technology. PLoS One. 2009, 4 (8): e6524-10.1371/journal.pone.0006524.
Griffin DK, Robertson LB, Tempest HG, Vignal A, Fillon V, Crooijmans RP, Groenen MA, Deryusheva S, Gaginskaya E, Carré W, Waddington D, Talbot R, Völker M, Masabanda JS, Burt DW: Whole genome comparative studies between chicken and turkey and their implications for avian genome evolution. BMC Genomics. 2008, 9: 168-10.1186/1471-2164-9-168.
Hou Y, Liu GE, Bickhart DM, Cardone MF, Wang K, Kim ES, Matukumalli LK, Ventura M, Song J, VanRaden PM, Sonstegard TS, Van Tassell CP: Genomic characteristics of cattle copy number variations. BMC Genomics. 2011, 12: 127-10.1186/1471-2164-12-127.
Liu GE, Hou Y, Zhu B, Cardone MF, Jiang L, Cellamare A, Mitra A, Alexander LJ, Coutinho LL, Dell'Aquila ME, Gasbarre LC, Lacalandra G, Li RW, Matukumalli LK, Nonneman D, Regitano LC, Smith TP, Song J, Sonstegard TS, Van Tassell CP, Ventura M, Eichler EE, McDaneld TG, Keele JW: Analysis of copy number variations among diverse cattle breeds. Genome Res. 2010, 20 (5): 693-703. 10.1101/gr.105403.110.
Matsuzaki H, Wang PH, Hu J, Rava R, Fu GK: High resolution discovery and confirmation of copy number variants in 90 Yoruba Nigerians. Genome Biol. 2009, 10 (11): R125-10.1186/gb-2009-10-11-r125.
Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK: A high-resolution survey of deletion polymorphism in the human genome. Nat Genet. 2006, 38 (1): 75-81. 10.1038/ng1697.
Freeman JL, Perry GH, Feuk L, Redon R, McCarroll SA, Altshuler DM, Aburatani H, Jones KW, Tyler-Smith C, Hurles ME, Carter NP, Scherer SW, Lee C: Copy number variation: new insights in genome diversity. Genome Res. 2006, 16 (8): 949-961. 10.1101/gr.3677206.
Conrad DF, Hurles ME: The population genetics of structural variation. Nat Genet. 2007, 39 (7 Suppl): S30-36.
Brant SR, Bernstein M, Wasmuth JJ, Taylor EW, McPherson JD, Li X, Walker S, Pouyssegur J, Donowitz M, Tse CM, Jabs EW: Physical and genetic mapping of a human apical epithelial Na+/H + exchanger (NHE3) isoform to chromosome 5p15.3. Genomics. 1993, 15 (3): 668-672. 10.1006/geno.1993.1122.
Xu H, Zhang B, Li J, Chen H, Wang C, Ghishan FK: Transcriptional inhibition of intestinal NHE8 expression by glucocorticoids involves Pax5. Am J Physiol Gastrointest Liver Physiol. 2010, 299 (4): G921-927. 10.1152/ajpgi.00227.2010.
Tse CM, Brant SR, Walker MS, Pouyssegur J, Donowitz M: Cloning and sequencing of a rabbit cDNA encoding an intestinal and kidney-specific Na+/H + exchanger isoform (NHE-3). J Biol Chem. 1992, 267 (13): 9340-9346.
Wackym PA, Cioffi JA, Erbe CB, Popper P: G-protein Golfalpha (GNAL) is expressed in the vestibular end organs and primary afferent neurons of Rattusnorvegicus. J Vestib Res. 2005, 15 (1): 11-15.
Régnauld K, Nguyen QD, Vakaet L, Bruyneel E, Launay JM, Endo T, Mareel M, Gespach C, Emami S: G-protein alpha(olf) subunit promotes cellular invasion, survival, and neuroendocrine differentiation in digestive and urogenital epithelial cells. Oncogene. 2002, 21 (25): 4020-4031. 10.1038/sj.onc.1205498.
Kamioka M, Imamura J, Komatsu N, Daibata M, Sugiura T: Testican 3 expression in adult T-cell leukemia. Leuk Res. 2009, 33 (7): 913-918. 10.1016/j.leukres.2008.11.006.
Hartmann U, Hülsmann H, Seul J, Röll S, Midani H, Breloy I, Hechler D, Müller R, Paulsson M: Testican-3: a brain-specific proteoglycan member of the BM-40/SPARC/osteonectin family. J Neurochem. 2013, 125 (3): 399-409. 10.1111/jnc.12212.
Yamano Y, Uzawa K, Shinozuka K, Fushimi K, Ishigami T, Nomura H, Ogawara K, Shiiba M, Yokoe H, Tanzawa H: Hyaluronan-mediated motility: a target in oral squamous cell carcinoma. Int J Oncol. 2008, 32 (5): 1001-1009.
Gerke V, Moss SE: Annexins: from structure to function. Physiol Rev. 2002, 82 (2): 331-371.
Gerke V, Creutz CE, Moss SE: Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005, 6 (6): 449-461. 10.1038/nrm1661.
van Baal JW, Milano F, Rygiel AM, Bergman JJ, Rosmolen WD, van Deventer SJ, Wang KK, Peppelenbosch MP, Krishnadath KK: A comparative analysis by SAGE of gene expression profiles of Barrett's esophagus, normal squamous esophagus, and gastric cardia. Gastroenterology. 2005, 129 (4): 1274-1281. 10.1053/j.gastro.2005.07.026.
Kim JK, Kim PJ, Jung KH, Noh JH, Eun JW, Bae HJ, Xie HJ, Shan JM, Ping WY, Park WS, Lee JY, Nam SW: Decreased expression of annexin A10 in gastric cancer and its overexpression in tumor cell growth suppression. Oncol Rep. 2010, 24 (3): 607-612.
Munksgaard PP, Mansilla F, BremsEskildsen AS, Fristrup N, Birkenkamp-Demtröder K, Ulhøi BP, Borre M, Agerbæk M, Hermann GG, Orntoft TF, Dyrskjøt L: Low ANXA10 expression is associated with disease aggressiveness in bladder cancer. Br J Cancer. 2011, 105 (9): 1379-1387. 10.1038/bjc.2011.404.
Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, Shevach EM: Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. 2010, 184 (7): 3433-3441. 10.4049/jimmunol.0904028.
Eller K, Kirsch A, Wolf AM, Sopper S, Tagwerker A, Stanzl U, Wolf D, Patsch W, Rosenkranz AR, Eller P: Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes. 2011, 60 (11): 2954-2962. 10.2337/db11-0358.
Martín-Ibáñez R, Crespo E, Esgleas M, Urban N, Wang B, Waclaw R, Georgopoulos K, Martínez S, Campbell K, Vicario-Abejón C, Alberch J, Chan S, Kastner P, Rubenstein JL, Canals JM: Helios transcription factor expression depends on Gsx2 and Dlx1&2 function in developing striatal matrix neurons. Stem Cells Dev. 2012, 21 (12): 2239-2251. 10.1089/scd.2011.0607.
Wang L, Guo DC, Cao J, Gong L, Kamm KE, Regalado E, Li L, Shete S, He WQ, Zhu MS, Offermanns S, Gilchrist D, Elefteriades J, Stull JT, Milewicz DM: Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet. 2010, 87 (5): 701-707. 10.1016/j.ajhg.2010.10.006.
Jagadish N, Rana R, Mishra D, Kumar M, Suri A: Sperm associated antigen 9 (SPAG9): a new member of c-Jun NH2 -terminal kinase (JNK) interacting protein exclusively expressed in testis. Keio J Med. 2005, 54 (2): 66-71. 10.2302/kjm.54.66.
Garg M, Kanojia D, Khosla A, Dudha N, Sati S, Chaurasiya D, Jagadish N, Seth A, Kumar R, Gupta S, Gupta A, Lohiya NK, Suri A: Sperm-associated antigen 9 is associated with tumor growth, migration, and invasion in renal cell carcinoma. Cancer Res. 2008, 68 (20): 8240-8248. 10.1158/0008-5472.CAN-08-1708.
Morales AV, Perez-Alcala S, Barbas JA: Dynamic Sox5 protein expression during cranial ganglia development. DevDyn. 2007, 236 (9): 2702-2707.
Martinez-Morales PL, Quiroga AC, Barbas JA, Morales AV: SOX5 controls cell cycle progression in neural progenitors by interfering with the WNT-beta-catenin pathway. EMBO Rep. 2010, 11 (6): 466-472. 10.1038/embor.2010.61.
Della-Morte D, Beecham A, Rundek T, Wang L, McClendon MS, Slifer S, Blanton SH, Di Tullio MR, Sacco RL: A follow-up study for left ventricular mass on chromosome 12p11 identifies potential candidate genes. BMC Med Genet. 2011, 12: 100-10.1186/1471-2350-12-100.
Buttgereit J, Qadri F, Monti J, Langenickel TH, Dietz R, Braunewell KH, Bader M: Visinin-like protein 1 regulates natriuretic peptide receptor B in the heart. RegulPept. 2010, 161 (1–3): 51-57.
Li C, Pan W, Braunewell KH, Ames JB: Structural analysis of Mg2+ and Ca2+ binding, myristoylation, and dimerization of the neuronal calcium sensor and visinin-like protein 1 (VILIP-1). J Biol Chem. 2011, 286 (8): 6354-6366. 10.1074/jbc.M110.173724.
Gonzalez Guerrico AM, Jaffer ZM, Page RE, Braunewell KH, Chernoff J, Klein-Szanto AJ: Visinin-like protein-1 is a potent inhibitor of cell adhesion and migration in squamous carcinoma cells. Oncogene. 2005, 24 (14): 2307-2316. 10.1038/sj.onc.1208476.
Dorsett D, Ström L: The ancient and evolving roles of cohesin in gene expression and DNA repair. Curr Biol. 2012, 22 (7): R240-250. 10.1016/j.cub.2012.02.046.
Losada A, Hirano T: Dynamic molecular linkers of the genome: the first decade of SMC proteins. Genes Dev. 2005, 19 (11): 1269-1287. 10.1101/gad.1320505.
Pebernard S, Wohlschlegel J, McDonald WH, Yates JR, Boddy MN: The Nse5-Nse6 dimer mediates DNA repair roles of the Smc5-Smc6 complex. Mol Cell Biol. 2006, 26 (5): 1617-1630. 10.1128/MCB.26.5.1617-1630.2006.
Johnson RE, Kovvali GK, Prakash L, Prakash S: Role of yeast Rth1 nuclease and its homologs in mutation avoidance, DNA repair, and DNA replication. Curr Genet. 1998, 34 (1): 21-29. 10.1007/s002940050362.
Hosfield DJ, Mol CD, Shen B, Tainer JA: Structure of the DNA repair and replication endonuclease and exonuclease FEN-1: coupling DNA and PCNA binding to FEN-1 activity. Cell. 1998, 95 (1): 135-146. 10.1016/S0092-8674(00)81789-4.
Rass U, Compton SA, Matos J, Singleton MR, Ip SC, Blanco MG, Griffith JD, West SC: Mechanism of Holliday junction resolution by the human GEN1 protein. Genes Dev. 2010, 24 (14): 1559-1569. 10.1101/gad.585310.
Fior R, Maxwell AA, Ma TP, Vezzaro A, Moens CB, Amacher SL, Lewis J, Saúde L: The differentiation and movement of presomitic mesoderm progenitor cells are controlled by Mesogenin 1. Development. 2012, 139 (24): 4656-4665. 10.1242/dev.078923.
Yoon JK, Moon RT, Wold B: The bHLH class protein pMesogenin1 can specify paraxial mesoderm phenotypes. Dev Biol. 2000, 222 (2): 376-391. 10.1006/dbio.2000.9717.
Hughes DC: ZP genes in avian species illustrate the dynamic evolution of the vertebrate egg envelope. Cytogenet Genome Res. 2007, 117 (1–4): 86-91.
Goudet G, Mugnier S, Callebaut I, Monget P: Phylogenetic analysis and identification of pseudogenes reveal a progressive loss of zonapellucida genes during evolution of vertebrates. Biol Reprod. 2008, 78 (5): 796-806. 10.1095/biolreprod.107.064568.
Acknowledgments
The authors would like to acknowledge members of the Poultry Breeding Group of the College of Animal Science and Technology in Northeast Agricultural University for managing the birds and collecting data. This research was supported by the National 863 project of China (No. 2013AA102501), the China Agriculture Research System (No. CARS-42), and the National Natural Science Foundation (No. 31301960).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HZ contributed to the conception and design of the experiments, participated in data interpretation, and prepared the manuscript. ZQD participated in data interpretation and contributed to writing the manuscript. JQD participated in data analysis and interpretation. HXW and HYS carried out the validation experiment. NW participated in the study design and contributed to writing the manuscript. SZW participated in the study design, data interpretation, and contributed to writing the manuscript. HL led the conception and design of the study, participated in data interpretation, and contributed to writing the manuscript. All authors commented on the draft, read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Zhang, H., Du, ZQ., Dong, JQ. et al. Detection of genome-wide copy number variations in two chicken lines divergently selected for abdominal fat content. BMC Genomics 15, 517 (2014). https://doi.org/10.1186/1471-2164-15-517
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-517