
Abstract
Background
Understanding mechanisms that contribute to viral dissemination in mosquito vectors will contribute to our ability to interfere with the transmission of viral pathogens that impact public health. The expression of genes in two Culex pipiens quinquefasciatus populations from Florida with known differences in vector competence to West Nile virus (WNV) were compared using high throughput sequencing.
Results
A total of 15,176 transcripts were combined for comparison of expression differences between the two populations and 118 transcripts were differentially expressed (p < 0.05). The fold change in expression of the differentially expressed genes ranged from -7.5 – 6.13. The more competent population for WNV (Gainesville) over expressed 77 genes and down regulated 44 genes, compared with the less competent population for WNV (Vero Beach). Also, splicing analysis identified 3 transcripts with significantly different splice forms between the two populations. The functional analysis showed that the largest proportion of transcripts was included in the catalytic activity and transporter activity groups except for those in the unknown group. Interestingly, the up- regulated gene set contained most of the catalytic activity function and the down- regulated gene set had a notable proportion of transcripts with transporter activity function. Immune response category was shown in only the down regulated gene set, although those represent a relatively small portion of the function. Several different vitellogenin genes were expressed differentially. Based on the RNAseq data analysis, ovary development was compared across the populations and following WNV infection. There were significant differences among the compared groups.
Conclusions
This study suggests that ovary development is correlated to vector competence in two Culex populations in Florida. Both populations control energy allocations to reproduction as a response to WNV. This result provides novel insight into the defense mechanism used by Culex spp. mosquitoes against WNV.
Similar content being viewed by others

Background
West Nile virus was successfully introduced to the Western Hemisphere in 1999 and has since spread westward across the United States, southward into Central America and the Caribbean and northward into Canada [1]. West Nile virus (WNV) is transmitted to humans by the bite of mosquitoes that previously acquired the virus by feeding on infected birds. Since WNV was detected in New York in 1999, the CDC has reported over 39,000 human cases [2, 3] and has been detected in 59 mosquito species throughout North America, although less than 10% are considered principal WNV vectors [4]. Mosquitoes of the genus Culex are the primary vectors of WNV in the US due to their vector competence and host preference [5]. There is geographic variation in Culex WNV vectors, due to the presence of different, locally important Culex spp.; such as Culex pipiens pipiens in the Northeast, Cx. tarsalis in the West, and Cx. p. quinquefasciatus and Cx. nigripalpus in the South. Additionally, refractoriness can be attributed to the type of virus and variation in the genetics of the virus, which can affect an individual mosquito or mosquito species. Different populations of the same mosquito species may also show differential vector competence under similar environmental conditions [6–9] due to genetic differences contributing to vector competence [10]. Several studies have shown that local populations of Culex spp. vary in their capacity to transmit WNV [11, 12]. Recent studies have supported and extended the concept by investigating certain aspects of vector competence including susceptibility of different mosquito species to WNV, permissiveness of various mosquito tissues to infection, and release of virus from infected salivary glands into the saliva [13, 14]. Differences in vector competence could be attributed to barriers to infection, including midgut infection, midgut escape and salivary gland invasion barriers [15, 16], as well as the effects of extrinsic and intrinsic factors [17–19]. Vector competence is also likely affected by the innate immune response, which can result in gene expression differences [20–22].
Although the work cited above proposes that vector competence involves complex interactions between arbovirus and mosquito, the differences in the ability of one population to disseminate WNV better than another population was not addressed nor was the modification in competence attributed to changes in the transcriptome. For example the work by Sanders et al. [23] reported alphavirus-induced mosquito midgut transcription changes in genes involved in vesicle transport and terminal kinase immune cascades, but stopped short at assigning them a role in alphavirus infection and/or dissemination. Mercado-Curiel et al. [24] discovered two midgut cell proteins that bind all four serotypes of dengue virus. These proteins have been linked to Aedes aegypti vector competence for dengue virus [25]. Although these findings represent a step in understanding vector competence, the expression of these molecules post-virus exposure was not shown, and their potential as antiviral molecules was only shown in tissue culture.
The study demonstrates the relationship between the differing vector competence of two populations of Culex p. quinquefasciatus and gene expression changes that result from exposure to WNV. We know that these populations differ in their dissemination rates for WNV even when infection rates are the same [19] therefore changes in gene expression can be attributed to WNV infection processes after initial infection. Findings such as these would support our hypothesis that the difference in dissemination rates may be due to one of the infection barriers, such as midgut escape, in the less competent population. However this study extends this theory to include differences in the mosquito body transcriptome that might influence antiviral immune response, transcription and translation machinery sequestration and which may ultimately lead to modification of dissemination and proposes the involvement of other organ-tissues in their vector competence differences. These findings are expected to help elucidate a potential arbovirus vector competence mechanism.
Results and discussion
Sample preparation and RNA sequencing
The expression of genes from two Culex pipiens quinquefasciatus populations [Vero Beach (CQV) and Gainesville (CQG)] from Florida with known differences in vector competence to West Nile virus (WNV) were compared using high throughput sequencing. The previous vector competence study with the same WNV infected CQV and CQG populations indicated significant difference with 44% and 82% dissemination rate, respectively, after 12 days of exposure [19]. In this study, four-day-old female mosquitoes were fed a blood meal containing 5.3 log10 pfu/ml of WNV. The titration of WNV in 6 mosquitoes in each population was tested (Table 1). The titer of the freshly fed mosquitoes was significantly lower than in the blood meal, but was not significantly different between mosquito populations indicating that genes that are differentially expressed between the two mosquito populations will not be attributable to differences in the amount of virus imbibed (Table 2). By 5 dpi, titer in the two populations increased to the same level as the blood meal, suggesting WNV replication. Five days following infection, a time that falls within the extrinsic incubation period for WNV of 3-6 days [26], RNA from female mosquito bodies was extracted and sent for transcriptome analysis using Illumina high throughput sequencing by the Sequencing Core in the Hussman Institute for Human Genomics, at the University of Miami. A total six RNA-seq libraries including triplication of each population were generated and sequenced (Table 2). A total 220,388,641 and 202,416,138 reads were generated from Vero Beach and Gainesville populations and a total of 88,402 transcripts hit on annotated transcript sequences of Cx. quinquefasciatus (Table 2). The data from each replicate was combined for differential analyses.
Differential expression between WNV-infected Cx. quinquefasciatus females from two Florida populations
A total of 15,176 transcripts were combined for comparison of expression differences between the two populations and 118 transcripts were differentially expressed (p < 0.001) (Table 3). Of these, 41 transcripts showed decreased expression in CQG compared to CQV, and 77 transcripts increased abundance in CQG compared to CQV, 5 days after a WNV-infected blood meal. In order to validate the RNAseq data analysis, several genes were randomly selected and their gene expression level tested by quantitative real time PCR. All the selected genes showed significantly different expression levels between the two CQ populations, supporting the RNAseq data analysis (Figure 1).

Validation of the expression of transcripts between two Cx. quinquefasciatus populations by qRT-PCR. *< 0.05. **< 0.01.
Comparison of the expression of the118 transcripts with significant expression differences revealed that only 5 genes were not expressed in the CQV but had greater than 3 fold change in expression in the CQG population; while only 3 of the 118 genes were not expressed in CQG and were down regulated between -3 and -7 fold. Some of these genes were included in the validation by qRT-PCR (CPIJ003600 and CPIJ004426).
Investigation of transcripts with expression levels of less than 1 fold change in expression in either of the two Cx. quinquefasciatus populations was performed to determine if the differences in dissemination was due to absence of expressed products. Of the 5 transcripts expressed only in the more permissive population CQG, one transcript (CPIJ003600-RA) codes for a ctl transporter and its functional parental attribute suggest it is a cellular component involved in membrane transport and is 4-fold more highly expressed. Two transcription products, the first encoding a putative alpha-xylosidase (CPIJ019915-RA) and the second of unknown function (CPIJ010313-RA) have a role in catalysis and are 18-fold and 6-fold higher in expression in the CQG population, respectively. A transcript (CPIJ004426-RA) involved in binding or interacting with zinc is 4-fold higher in expression. Another transcript (CPIJ005240-RA) is novel and does not match any functional groups but is 17-fold higher in expression in the CQG population. The three transcripts expressed only in the less permissive mosquito population, CQV (CPIJ002311-RA, CPIJ013631-RA, CPIJ008969-RA), were novel and therefore functional parental group assignments could not be made.
Functional analysis of differentially expressed transcripts by GO
Functional analysis using GO terms was conducted on all 118 transcripts with significant expression difference between the CQG and CQV Cx. quinquefasciatus populations. The largest proportion of the total number of differentially expressed transcripts (34%) was of unknown function (Figure 2). Of the transcripts that were up regulated in CQG, 31% were of unknown function; while 41% of the down-regulated transcripts were of unknown function. Of the 118 transcripts, 22% had a role in catalysis, where a large majority (33%) were up regulated in the more permissive mosquito population, CQG. Gene products with catalytic activity included those involved in blood digestion such as trypsin (CPIJ017964) and chymotrypsin (CPIJ000835) both showing >2 fold change in expression in the CQG population. There were a number of transcripts encoding proteins such as carboxypeptidase and kinase present and the expression of an alpha- glucosidase, an enzyme involved in sugar feeding (CPIJ019915), was 6-fold higher in the CQG population. There were a number of transcripts encoding salivary gland catalytic enzymes, such as salivary lipase and alpha amylase. A number of transcripts encoding glutathione S-transferase, with roles in detoxification of xenobiotics, were present with fold expression changes ranging from 1.7-2.0 in the more WNV-permissive population.

Functional characterization of significantly differentially expressed transcripts between two Cx. quinquefasciatus populations. *UP are up-regulated genes in CQG; Down are down-regulated.
Most of the transcripts involved in signal transduction, transporter activity and immune response were down regulated in the CQG population, 7%, 24%, 7%, respectively, compared to the CQV population. Signal transduction pathways involve the binding of extracellular signaling molecules to receptors on the cell surface, which in turn activates intracellular molecules eliciting a physiological response. In mosquitoes, these pathways are integral parts of their physiology and include cascades that control the innate immune response, odorant detection, and gene activation. Of the 5% transcripts with signal transduction activity, 3% had increased and 7% had decreased expression in the CQG population. Of the transcripts with signaling function, three with up-regulated expression in the CQG population had fold change in expression ranging from 1.6 – 2.1, of these two were similar to hedgehog receptor activity proteins and one was similar to G-protein coupled receptor proteins. Three transcripts had approximately -1.7-fold change in expression and were involved in cell function or activity, such as regulation of transcription.
Proteins with transporter activity are involved in the movement of ions and small molecules into, out of or within a cell, or between cells. Of the transcripts differentially expressed between the two WNV-infected Cx quinquefasciatus populations assigned the transporter activity GO term, 24% were down-regulated in CQG and 6% were upregulated. Transcripts with a 2-4 fold decrease in expression included those involved in lipid transport such as a vitellogenin precursor (CPIJ010191) and oxygen transport such as arylphorin subunit precursor (CPIJ007783, CPIJ009033) and larval serum protein 2 precursor (CPIJ001822). A number of transcripts with transport activity increased in expression, such as for transport of calcium, vacuolar and transmembrane transport, also a transcript involved in mitochondrial electron transport (CPIJ019847) showed a 5-fold increase in expression in the CQG population.
The immune response genes with fold change ranging from -3.0 to -1.8, are surprisingly all with similarity to the Cecropin family of antimicrobial genes of the innate immune response pathway.
Transcripts with binding, electron carrier, structural molecule activity, biosynthesis and cell adhesion GO term assignments were also represented, with 11% of the total number of transcripts, and 12% of the up-regulated transcripts, involved in binding. The other transcripts made up only ≤ 4%. Proteins involved in binding molecules could be used in transport of virus into cells (receptors) or in other molecular processes from blood detection to virus replication. Of the 11% with binding capabilities, 12% were upregulated in the CQG population, while 7% were suppressed in this population.
There were 7 proteins (CPIJ005910, CPIJ010700, CPIJ014550, CPIJ018300, CPIJ018735, CPIJ800096, and CPIJ800099) identified as having similarity to the salivary gland D7 precursor proteins with 1-3 fold increase in expression in the CQG population and similarity to odorant binding proteins (CPIJ014553, CPIJ800101, and CPIJ018736). Salivary gland products are known to be involved in defense mechanisms in mosquito biology and the 7 different salivary gland genes and immune response genes were differentially expressed in this study. Two transcripts assigned to binding had the highest fold expression. One transcript (CPIJ004426) with a 4-fold expression change was similar to a family of zinc ion binding proteins and another transcript (CPIJ013626) showing a 3 fold increase in expression was similar to chitin binding proteins. The over represented functional terms were analyzed with hyper geometric analysis with bonferroni correction (p < 0.05, Table 4), supporting the proportional functional analysis in Figure 2. Most of the GO terms from the differentially expressed genes were over represented in the functional enrichment analysis. The immune response function was not over represented in the differentially expressed gene list compared to the genome-wide functions but the over represented functions of “Detection of chemical stimulus involved in sensory perception of smell” and “odorant binding” identified in the dataset were associated with the immune response.
Cufflinks and Cuffdiff analyses measured the abundance of the expressed genes in each group and identified differences in Transcript Start Site (TSS) and alternative splicing in the two CQ populations (Table 5). In both the CQV and CQG populations, a total of thirteen transcripts showed different TSS. The expression ratio in different TSS of each transcript was calculated in the two CQ populations from Florida and compared. The expression of these genes from different start sites, we suggest, may be influenced by infection with WNV, although the transcripts do not show differential expression, and have some role in vector competence. Out of the total TSS, 5 transcripts were not annotated and the other 6 annotated transcripts were mostly related to binding to nucleus structure.
Three transcripts showed different isoforms in CQV and CQG but their expression levels were not significant. The transcripts encode vitellogenin (CPIJ00927), tripartite (CPIJ00966), and pdz (post synaptic density protein, also known as Drosophila discs large tumor suppressor and zona occludens 1 protein, CPIJ01791). This vitellogenin is different from the other two vitellogenin precursors already identified in this study as significantly differentially down regulated but they all have the same lipid transport function. The tripartitie and pdz proteins play a role in pathogen recognition, including viruses, and cell signaling assembly, respectively.
As mentioned, there were two vitellogenin-A1 precursor genes differentially down regulated in CQG, -2.45 and -2.64. Another vitellogenin transcript was spliced differently between the two Culex populations. Vitellogenin genes, including vitellogenin-A1 in mosquitoes are known as egg yolk precursor proteins, which are used in ovary development and are regulated by juvenile hormone. The mosquito is known to synthesize vitellogenin in the fat body after a blood meal [27]. The fat body is one of the tissues in the mosquito that is well known to play a role in defense mechanisms. Results from this study suggest that the two populations showing different vectorial capacities appear to have functional differences in the lipid transport pathway between the fat body and ovary, and this difference might involve defense against WNV.
Ovary development between the two CQ populations
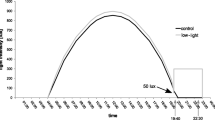
We identified a number of vitellogenin precursors as differentially expressed genes between the two populations and also found different splicing forms between CGV and CQG. Generally vitellogenin is a precursor protein of lipoprotein and phospho protein in egg yolk and functions as a lipid transporter from fat body to ovary in invertebrates [27]. Ovary development allows increased reproductive capacity but results in a trade-off with allocation of resources in the fat body, impacting production of molecules that are essential for defense functions in insects [28]. Immune related genes and transport functions were enriched among differentially expressed genes in this study (Figure 2). Because of the correlation between the expression of vitellogenin and other co-regulated genes involved in defense and signal transduction as revealed in the RNAseq data, the size of the ovary in female mosquitoes from each population was measured after providing a blood meal, with or without WNV (Figure 3).

Ovary growth after blood feeding. Standard deviation is calculated from replicated samples tested at each time point and shows significantly different growth rates (ANOVA, p < 0.001). Vero = CQV; GN = CQG; ctrl = control.
Female mosquitoes from both the CQG and CQV population significantly lessen the rate of ovary development around 18 hour post WNV infection. In the normal condition without WNV, CQG population forms larger ovaries than CQV but after the WNV infectious bloodmeal there is no significant difference between the two populations (Figure 3). This result agrees with the RNAseq data, where transport functions including vitellogenin are significantly decreased in CQG. This observation showed that Culex populations control the rate of ovary development when infected with WNV. Also, the WNV permissive population, CQG, appears to contribute more energy to reproduction in general. Once CQG is exposed to WNV, the CQG population alters the lipid transport to levels similar to the levels in CQV. Although there is no evidence about whether immune response genes influence ovary development or ovary development affects immune response, this decreased ovary development suggests that in CQ mosquitoes, vectorial capacity might result from the inability to properly route signals involved in lipid transport. The difference of ovary development rate before the WNV infection trial can be the influence of the vectorial capacity. Also, the drastic change in ovary development can be due to defense against WNV. These conclusions are speculative, at best, as no experiments have been done to directly measure resource allocation in these populations. Therefore there may be other underlying factors, such as a difference in dissemination rate, that contribute to the differences in ovary development in WNV infected mosquitoes in these populations.
Conclusions
This study suggests that a correlation exists between ovary development and vector competence to WNV. Between two populations, the size of an ovary was significantly different 24 hours after bloodfeeding without the infection. This difference is almost impossible to notice when we observed these two populations during normal rearing under the same conditions. Interestingly, the size of the ovaries after WNV exposure was not statistically different between the two populations, supporting the RNAseq data that showed two vitellogenin precursor genes down regulating after WNV exposure. In mosquitoes, fat body and salivary glands play important roles in several immune pathways and in antimicrobial peptide production controlling infection by pathogens including virus. It has been demonstrated that a change in gene expression related to fat body and salivary gland function affects vectorial capacity [29, 30]. In the illumina data, a number of transcripts related to salivary proteins were also differentially expressed. This study suggests that energy allocation may affect defense mechanisms supported by transport function enrichment and warrants further study. The candidate genes including vitellogenin precursor, salivary protein, and immune response need to be tested by RNAi to verify the involvement in vector competence and to discover the pathway responsible for vector competence.
These results will help elucidate potential arbovirus vector competence mechanisms that will enhance our understanding of the genetic interactions between WNV and Culex spp. mosquitoes and help guide development of strategies for controlling the spread of vector-borne disease.
Methods
Mosquito sample preparation for RNAseq
Populations of Culex pipiens quinquefasciatus were collected from Gainesville, Florida and Vero Beach, Florida, then reared and maintained under standard insectary conditions (26°C, 14 h/10 h light/dark period, 70% relative humidity) and fed chicken blood for oviposition. The use of chickens to maintain colony mosquitoes described herein complies with the strict guidelines in the approved protocol (Project #201207682) awarded by the University of Florida Institutional Animal Care and Use Committee. Four-day-old female mosquitoes from two populations were fed on chicken blood (Hemostat, Dixon, CA) containing 5.3 log10 pfu/ml of West Nile virus. Five days following infection two hundred female mosquito bodies were collected and immediately frozen for RNA extraction. Extracted total RNA from each population was sent to the Center for Genome Technology Sequencing Core, John P. Hussman Institute for Human Genomics in University of Miami Miller School of Medicine for transcriptome analysis using Illumina high throughput sequencing. Six RNA-seq libraries were generated (Table 2). Cluster generation took place on the Illumina cBot according to the manufacturer’s recommendations. Sequencing took place on the Illumina HiSeq2000 using the reagents provided in the Illumina TruSeq PE Cluster Kit v3 and the TruSeq SBS Kit – HS (200 cycle) kit. Six RNAseq libraries were generated from the two populations of Cx. quinquefasciatus, showing different vector competence for WNV. Each of the replicates showed a consistent number of sequence reads and transcripts (Table 2). The reads were mapped on the Vectorbase Cx. quinquefasciatus database by using TopHat [31, 32].
The quantity of WNV RNA in the blood meal, bodies from freshly fed mosquitoes, and mosquito bodies collected 5 days post-infection was determined using quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) and WNV specific primers. RT reactions were performed using Enhanced Avian Reverse Transcriptase (Sigma, St. Louis, MO) and quantitative real time PCR performed using SsoAdvanced SYBR Green Supermix (BIO-RAD, Hercules, CA) and following the included protocols. The standard curve was generated based on 10-fold serial dilutions of WNV and quantified by qRT-PCR as described above. WNV titer was determined from triplicate cycle threshold (Ct) values using Bio-Rad CFX manager software. The data was normalized by Log10 transformation and regression analysis was used to determine a qPCR-derived titer (Qpfu/ml).
Differentially expressed gene analysis
The Sequencing Core of the University of Miami Miller School of Medicine provided the raw data from the Illumina analysis. The Illumina raw data was subjected to RNAseq analysis by the Center for Genetic Epidemiology and Statistical Genetics Core that is part of the Hussman Institute for Human Genomics at the University of Miami to determine differentially expressed genes between two populations after WNV oral infection (Table 1). Samples were barcoded to allow for multiplexing of 3 samples per HiSeq2000 PE100 lane. The sequencing data is deposited in Short Read Archive at NCBI (Accession number SRS515667, SRS516159). The mapped reads were assembled into transcripts and the abundance and differential expression of the reads between the two populations, CQV and CQG, measured by Cufflinks and Cuffdiff [33, 34].
The differentially expressed genes from RNAseq data analysis were validated by Real-Time quantitative RT-PCR. A total of 8 genes were randomly selected from the differentially expressed transcript list from the Cuffdiff analysis (q < 0.05, Table 3). The primer sets were designed (Integrated DNA Technologies), total RNA was extracted by Trizol from female mosquito bodies, which were prepared in the same way as described above and three biological replications were used for the validation. cDNA synthesis was carried out using the Enhanced Avian HS RT-PCR kit (Sigma, Saint Louis, MO) with reverse transcription completed using oligo dT. Actin protein gene was used as a control endogenous gene and the randomly selected genes were quantified with Real-Time quantitative RT-PCR.
The functions of the putative proteins encoded by the Cx. quinquefasciatus transcripts are theoretical and based on nucleotide sequence similarity to other orthologous organisms as described in the literature. Taking this into account, Gene Ontology analysis was performed (BioMart.org, Vector Base.org).
Ovary development study
We used the same aged females from each population as previously described. After bloodfeeding with/without WNV, ovaries were dissected (10, 12, 14, 16, 18, 24, 36, and 48 hours post blood-feeding) and fixed on glass slides with one drop of Canadian balsam and cover glass added. At least 10 females were used for ovary dissections in each time point per population with or without WNV infection. The fixed samples were visualized under an Olympus SZX12 microscope and pictures taken under the microscope with 16 times magnification. The pictures were analyzed with imageJ (from http://rsbweb.nih.gov/ij/) to determine the area occupied by each ovary. We conducted statistical analyses for ovary growth based on measurements of the size by visual inspection of the digital photograph (Figure 3). Analysis of variance (PROC GLM, SAS 9.22) was used to evaluate differences in ovary growth in each group. If significant differences were observed, then a Duncan test was used to determine differences in the means.
References
Dauphin G, Zientara S, Zeller H, Murgue B: West Nile: worldwide current situation in animals and humans. Comp Immunol Microb. 2004, 27: 343-355.
Hayes EB, Komar N, Nasci RS, Montgomery SP, O’Leary DR, Campbell GL: Epidemiology and transmission dynamics of West Nile virus disease. Emerg Infec Dis. 2005, 11: 1167-1173.
Epidemic/Epizootic West Nile virus in the United States: guidelines for surveillance, Prevention, and Control. [http://stacks.cdc.gov/view/cdc/7271]
West Nile virus: Statistics, surveillance, and control. [http://www.cdc.gov/ncidod/dvbid/westnile/surv&controlCaseCount07_detailed.htm]
Kramer LD, Ebel GD: Dynamics of flavivirus infection in mosquitoes. Adv Virus Res. 2003, 60: 187-232.
Gubler DJ, Nalim S, Tan R, Saipan H, Sulianti Saroso J: Variation in susceptibility to oral infection with dengue virus among geographic strains of Aedes aegypti. Am J Trop Med. 1979, 28: 1045-1062.
Gubler DJ, Rosen L: Variation among geographic strains of Aedes albopictus is susceptibility to infection with dengue virus. Am J Trop Med. 1976, 25: 318-346.
Tabachnick WJ, Wallis GP, Aitken THG, Miller BR, Amato GD, Lorenz L, Powell JR, Beaty BJ: Oral infection of Aedes aegypti with yellow fever virus: Geographic variation and genetic considerations. Am J Trop Med. 1985, 34: 1219-1224.
Lozano-Fuentes S, Fernandez-Salas I, de Lourdes Munoz M, Garcia-Rejon J, Olson KE, Beaty BJ, Black WC: 4th: The neovolcanic axis is a barrier to gene flow among Aedes aegypti populations in Mexico that differ in vector competence for dengue 2 virus. PLoS Neglect Trop D. 2009, 3 (6): e468-
Beerntsen BT, James AA, Christensen BM: Genetics of mosquito vector competence. Microbiol Mol Biol R. 2000, 64: 115-137.
Blackmore CG, Stark LM, Jeter WC, Oliveri RL, Brooks RG, Conti LA, Wiersma ST: Surveillance results from the first West Nile virus transmission season in Florida, 2001. Am J Trop Med. 2003, 69: 141-150.
Godsey MS, Nasci R, Savage HM, Aspen S, King R, Powers AM, Burkhalter K, Colton L, Charnetzky D, Lasater S, Taylor V, Palmisano CT: West Nile virus-infected mosquitoes, Louisiana, 2002. Emerg Infect Dis. 2005, 11: 1399-1404.
Girard YA, Popov V, Wen J, Han V, Higgs S: Ultrastructural study of West Nile virus pathogenesis in Culex pipiens quinquefasciatus (Diptera: Culicidae. J Med Entomol. 2005, 42: 429-444.
Colton L, Nasci RS: Quantification of West Nile virus in the saliva of Culex species collected from the southern United States. J Am Mosquito Contr. 2006, 22: 57-63.
Romoser WS, Turell MJ, Lerdthusnee K, Neira M, Dohm D, Ludwig G, Wasieloski L: Pathogenesis of Rift Valley fever virus in mosquitoes–tracheal conduits & the basallamina as an extra-cellular barrier. Archiv Virol. 2005, 19: 89-100.
Mahmood F, Chiles RE, Fang Y, Green EN, Reisen WK: Effects of time after infection, mosquito genotype, and infectious viral dose on the dynamics of Culex tarsalis vector competence for western equine encephalomyelitis virus. J Am Mosquito Contr. 2006, 22: 272-281.
Richards SL, Mores CN, Lord CC, Tabachnick WJ: Impact of extrinsic incubation temperature and virus exposure on vector competence of Culex pipiens quinquefasciatus (Diptera: Culicidae) for West Nile virus. Vector-borne and Zoonot. 2007, 7: 629-636.
Richards SL, Lord CC, Pesko K, Tabachnick WJ: Environmental and biological factors influencing Culex pipiens quinquefasciatus Say (Diptera: Culicidae) vector competence for Saint Louis encephalitis virus. Am J Trop Med. 2009, 81: 264-272.
Anderson SL, Richards SL, Tabachnick WJ, Smartt CT: Effects of West Nile virus dose and extrinsic incubation temperature on temporal progression of vector competence in Culex pipiens quinquefasciatus. J Am Mosquito Contr. 2010, 26: 103-107.
Keene KM, Foy BD, Sanchez-Vargas I, Beaty BJ, Blair CD, Olsen KE: RNA interference acts as a natural antiviral response to O’nyong-nyong virus (Alphavirus:Togaviridae) infection of Anopheles gambiae. PNAS. 2004, 101: 17240-17245.
Sanchez-Vargas I, Travanty EA, Keene KM, Franz AW, Beaty BJ, Blair CD, Olson KE: RNA interference, arthropod-borne viruses, and mosquitoes. Virus Res. 2004, 102: 65-74.
Souza-Neto JA, Sim S, Dimopoulos G: An evolutionary conserved function of the JAKSTAT pathway in anti-dengue defense. PNAS. 2009, 106: 17841-17846.
Sanders HR, Foy BD, Evans AM, Ross LS, Beaty BJ, Olson KE, Gill SS: Sindbis virus induces transport processes and alters expression of innate immunity pathway genes in the midgut of the disease vector, Aedes aegypti. Insect Biochem Molec. 2005, 35: 1293-1307.
Mercado-Curiel RF, Esquinca-Aviles HA, Tovar R, Diaz-Badillo A, Camacho-Nuez M, Munoz Mde L: The four serotypes of dengue recognize the same putative receptors in Aedes aegypti midgut and Ae. albopictus cells. BMC Microbiol. 2006, 6: 85-
Mercado-Curiel RF, Black WC, Muñoz ML: A dengue receptor as possible genetic marker of vector competence in Aedes aegypti. BMC Microbiol. 2008, 8: 118-
Girard YA, Klingler KA, Higgs S: West Nile virus dissemination and tissue tropisms in orally infected Culex pipiens quinquefasciatus. Vector Borne Zoonot D. 2004, 4: 109-122.
TR F, Hagedorn HH: Vitellogenin synthesis in the mosquito: the role of juvenile hormone in the development of responsiveness to ecdysone. Physiol Entomol. 1977, 2: 173-178.
Arrese EL, Soulages JL: Insect fat body: energy, metabolism, and regulation. Annu Rev Entomol. 2010, 55: 207-225.
Anderson JR, Rico-Hesse R: Aedes aegypti vectorial capacity is determined by the infecting genotype of dengue virus. Am J Trop Med Hyg. 2006, 75 (5): 886-892.
Price DP, Nagarajan V, Churbanov A, Houde P, Milligan B, Drake LL, Gustafson JE, Hansen IA: The fat body transcriptomes of the yellow fever mosquito Aedes aegypti, pre- and post- blood meal. PLoS One. 2011, 6 (7): e22573-
Trapnell C, Pachter L, Salzberg SL: TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009, 25: 1105-1111.
Langmead B, Trapnell C, Pop M, Salzberg SL: Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10: R25-
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G: Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010, 28: 511-515.
Jiang H, Wong WH: Statistical inferences for isoform expression in RNA-Seq. Bioinformatics. 2009, 25: 1026-1032.
Acknowledgements
We thank the Sequencing Core and Bioinformatics Core, John P. Hussman Institute for Human Genomics, University of Miami, Miller School of Medicine for running Illumina HiSeq 2000 and generating raw reads, and for providing the use of the TopHat Tools. The project is supported by NIH (5-U54-AI-057157-09)/UNC SERCEB) subcontract to C.T.S.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DS carried out the molecular biology studies, the data analysis and the statistical analysis, and participated in the design of the study and drafted the manuscript. AC and CA participated the molecular biology studies including ovary dissection. CTS conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Shin, D., Civana, A., Acevedo, C. et al. Transcriptomics of differential vector competence: West Nile virus infection in two populations of Culex pipiens quinquefasciatus linked to ovary development. BMC Genomics 15, 513 (2014). https://doi.org/10.1186/1471-2164-15-513
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-513