
Abstract
Background
Leprosy has afflicted humankind throughout history leaving evidence in both early texts and the archaeological record. In Britain, leprosy was widespread throughout the Middle Ages until its gradual and unexplained decline between the 14th and 16th centuries. The nature of this ancient endemic leprosy and its relationship to modern strains is only partly understood. Modern leprosy strains are currently divided into 5 phylogenetic groups, types 0 to 4, each with strong geographical links. Until recently, European strains, both ancient and modern, were thought to be exclusively type 3 strains. However, evidence for type 2 strains, a group normally associated with Central Asia and the Middle East, has recently been found in archaeological samples in Scandinavia and from two skeletons from the medieval leprosy hospital (or leprosarium) of St Mary Magdalen, near Winchester, England.
Results
Here we report the genotypic analysis and whole genome sequencing of two further ancient M. leprae genomes extracted from the remains of two individuals, Sk14 and Sk27, that were excavated from 10th-12th century burials at the leprosarium of St Mary Magdalen. DNA was extracted from the surfaces of bones showing osteological signs of leprosy. Known M. leprae polymorphisms were PCR amplified and Sanger sequenced, while draft genomes were generated by enriching for M. leprae DNA, and Illumina sequencing. SNP-typing and phylogenetic analysis of the draft genomes placed both of these ancient strains in the conserved type 2 group, with very few novel SNPs compared to other ancient or modern strains.
Conclusions
The genomes of the two newly sequenced M. leprae strains group firmly with other type 2F strains. Moreover, the M. leprae strain most closely related to one of the strains, Sk14, in the worldwide phylogeny is a contemporaneous ancient St Magdalen skeleton, vividly illustrating the epidemic and clonal nature of leprosy at this site. The prevalence of these type 2 strains indicates that type 2F strains, in contrast to later European and associated North American type 3 isolates, may have been the co-dominant or even the predominant genotype at this location during the 11th century.
Similar content being viewed by others


Background
Leprosy has been known since the earliest recorded times, with references in ancient texts [1] and paleopathological evidence in the archaeological record dating back at least 4000 years [2]. In Britain, evidence for leprosy has been recorded from as early as the 4th century AD [3] but is thought to have been particularly prevalent between the 11th and 14th centuries, as evidenced by a rise in the number of active leprosy hospitals known as leprosaria[4]. However, by the end of this period, leprosy was declining such that leprosaria were being abandoned or put to other uses. Firstly in southern Britain but later in regions further to the north, until by the 18th century leprosy was only recorded in the far northern isles of Shetland, the last sufferer dying in the Edinburgh Infirmary in 1798 [5]. This decline in the prevalence of leprosy was reflected throughout Europe and remains an intriguing feature of infectious disease history for which there is no clear reason. Although leprosy is now rare in Europe, it remains a significant disease in many parts of the world with approximately 220,000 new cases in 2011 [6]. Despite the use of multi-drug therapy against leprosy since the 1980s, the prevalence of the disease remains stubbornly high in many areas. A wider understanding of the origins and history of leprosy, including why leprosy died out in Europe, may help to develop more effective strategies for controlling the modern disease.
Leprosy genomes show an unusual degree of conservation with strains being >99.9% identical. Only 807 polymorphic sites are recorded across all known strains [7], most of which are Single Nucleotide Polymorphisms (SNPs), with some Variable Nucleotide Tandem Repeats (VNTRs) [8]. This is despite the presence of more than 1300 pseudogenes, equating to 41% of the genome [9]. A subset of these polymorphic loci were used to develop a molecular typing scheme and to generate a phylogeny for M. leprae. This predicted an ancestral strain, thought to have a genotype between that of modern type 2 and type 3 strains, originating in East Africa approximately 100,000 years ago [8]. From there, leprosy appeared to have disseminated across the world along routes of human migration and trade, with type 1 strains dominating in Southeast Asia, type 3 s in the Near East and Europe, type 3Is in Northwestern Europe and ultimately in North America and type 4 s in West Africa and later Brazil [8]. Recently, new data have challenged this interpretation, with an apparently more basal group, type 0, being identified in patients from China and New Caledonia [7].
In an effort to build a more comprehensive and accurate history for leprosy that better informs our understanding of its origins and macroecology, we and others have been studying DNA from ancient M. leprae infections. Leprosy is unusual in that multibacillary infection causes pathological changes of the bone, such that ancient infections can be retrospectively identified from skeletal remains. Using SNP typing and whole genome sequencing of DNA from such lesions, we have recently been able to re-assess the genetic history of leprosy and demonstrate that 1000 years ago European leprosy was caused by strains that group with type 2 and type 3 strains [7, 10] and that a most recent common ancestor of all extant and ancient M. leprae existed only 1400 to 2700 years ago [7].
In this study we extend this work to gain further insights into the molecular epidemiology of European medieval leprosy. By sequencing the genomes of two further M. leprae strains from individuals interred at the leprosarium of St Mary Magdalen, Winchester, England, during the second half of the 11th century we can begin to understand in more detail the epidemiology of M. leprae at this site [11].
Results
Archaeology and osteology
Excavations at St Mary Magdalen, Winchester, are amongst the most extensive of any British leprosy hospital to date [11, 12]. The hospital is one of the earliest such institutions known in Britain and, as such, provides insights into both early institutional care and the nature and status of such communities at a relatively early date. Such hospitals may well have been a model for succeeding charitable institutions of social care. Thirty-eight burials have been excavated from the site, of which 33 (87%) show osteological signs of leprosy, a much higher percentage than observed in other British material. Of these, five M. leprae strains have been SNP genotyped (Sk2, Sk7, Sk8, and Sk14, and Sk19) [10] and 2 whole genomes (Sk2 and Sk8) sequenced [7].
The current study focuses on typing a further burial, Sk27, and generating whole genome sequences of the M. leprae from both Sk14 and Sk27. Both were excavated from the earlier and more northern of two cemeteries at the site (together with skeletons Sk7, Sk8, Sk9 and Sk19 [10]). Based upon comparative and diagnostic material this cemetery was thought to have been in use primarily in the decades immediately following the Norman Conquest, between 1070 AD and 1100 AD. This interpretation was confirmed by 14C dating, with dates of between 940 cal AD and 1160 cal AD [11]. Sk14 and Sk27 were 14C dated to between 955–1033 cal AD (WK 28629) and 1020–1162 cal AD (SUERC-39676), respectively.
The remains of Sk14 are those of an adolescent, probably male, displaying clear pathological features of leprosy: circumferential wasting of the foot phalanges, loss of bone of the distal ends of the distal foot phalanges, porosity of the shafts of the metatarsals and porous, woven and compact bone on the shafts of the fibulae, thinning of the palate and nasal bones and rounding of the margins of the nasal aperture [13]. In contrast, Sk27 was a middle-aged adult, with only minimal evidence of characteristic leprosy lesions. Indeed, Sk27 was originally identified as a possible control, non-lepromatous skeleton and only on closer examination was porosity and resorption of bone noted on the dorsal surface of the distal ends of the distal foot phalanges, particularly those for the first metatarsal. The grave of Sk27 was also notable for containing a scallop shell (Figure 1), indicating that the remains are likely those of a pilgrim who had completed the Way of St James to the Cathedral of Santiago de Compostela in Galicia, Spain [11].

The burial of Sk27 showing the associated scallop shell.
Confirmation that skeleton Sk27 had been infected with M. leprae
Samples of bone from Sk27 were scraped from the rhino-maxillary area under clean and controlled conditions using disposable gloves, scalpels and tubes. A control sample was taken from around the vomer (the thin bone that separates the nostrils) of a skeleton, Sk12, which showed no osteological evidence of M. leprae. The vomer is a site that often has a high M. leprae burden in lepromatous leprosy, and so is a good site from which to isolate M. leprae DNA. The presence of M. leprae DNA in the Sk14 extract was previously confirmed by real-time PCR using primers for the multicopy RLEP and the single copy locus, ML1795 (encoding the 18 kDa antigen), both of which gave single bands of the expected size (111 bp and 114 bp, respectively) [13]. The presence of M. leprae DNA in extracts from SK27 was similarly confirmed by PCR. PCR for the multicopy M. tuberculosis loci IS1081 gave no products (79 bp expected) for either sample, indicating an absence of tuberculosis. The control sample Sk12 gave no products with primers for loci of either M. leprae or M. tuberculosis, indicating that contamination of the samples from extraneous sources or cross-contamination between the samples was negligible.
Whole genome sequencing of M. lepraegenomes from skeletons SK14 and Sk27
DNA from skeletons Sk14 and Sk27 was amplified, enriched for M. leprae sequences using microarrays, and sequenced as described previously for Sk2 and Sk8 [7]. Details of the read depths, their alignment to the M. leprae TN reference genome and percentage genome coverage are given in Table 1. After quality controls less than 1% of the sequence reads aligned with human DNA, even in the control sample Sk12, only 1.8% of reads aligned to human DNA.
The control sample, Sk12, was characterized by very low coverage of the M. leprae genome, punctuated by short regions with high coverage and high diversity. This is consistent with a sample containing little or no M. leprae sequence, but with some carry-over of DNA from environmental organisms. This again confirms that there was little or no cross-contamination between samples or from modern M. leprae DNA during the sample preparation. Many of the reads that were retrieved from Sk12 had similarity to environmental organisms that contain genomic regions that are likely to be conserved across mycobacteriaceae and beyond. Such regions apparently have sufficient similarity to the microarray probes to hybridize, particularly in the absence of competing M. leprae DNA, but are sufficiently dissimilar to generate the high diversity observed. Regions with high coverage included approximately 50 genes such as rpoB, tuf, fusA and certain smaller regions within genes that often represent conserved motifs, for example within ABC transporters. Base calls from regions with high coverage in the control sample were not considered reliable and were discounted from the data.
Sanger sequencing of targeted SNPs (Table 2) was in agreement with Illumina sequencing so validating both typing and sequencing methods. Analysis of VNTR genotypes was not possible for either Sk14 or Sk27 as insufficient numbers of reads spanned the ML0058c (21–3) region, while ML2344-ML2345 (AGA)20 and ML2172-ML2173 (GTA)9 had high coverage in the Sk12, the control sample, and so were deemed unreliable.
Analysis of the ancient M. lepraegenomes
We have previously used PCR and Sanger sequencing of a subset of SNPs [8] to genotype the M. leprae from Sk14 as type 2F [10]. Using identical methods, we also identified Sk27 as a type 2F strain (Table 2).
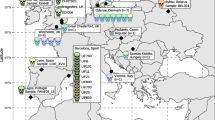
Independent analysis of the sequence reads at the University of Tübingen and the University of Surrey (Table 1), generated differing values for coverage and read depth. However, both centres reconstructed very similar genomes with only a single SNP discrepancy (not included in subsequent analysis) between the comparable datasets. Comparison of the Sk14 and Sk27 M. leprae genomes with other modern and ancient genomes identified the presence of very few novel polymorphs (Additional file 1: Table S1). Sk14 contained 3 novel indels, all of which were in pseudogenes, and 3 novel SNPs, only one of which was non-synonymous, encoding a change of Gly163 to valine in hypothetical protein ML1637. Sk27 had a single novel synonymous SNP and no identifiable novel indels. Phylogenetic analysis generated trees comparable to those of Schuenemann et al.[7] and placed Sk14 and Sk27 in the highly conserved type 2F branch of M. leprae (Figure 2 and Additional file 2: Figure S1). It is notable that the strain most closely related to Sk14 is another isolate from Winchester, Sk8, described in our previous studies [7, 10].

Phylogenetic relationships between modern and ancient M. leprae. A maximum likelihood tree describing the relationship between Sk14, Sk27 and other ancient (in bold) and modern M. leprae genomes [7]. Locations and dates (14C estimates or year of isolation) are given in brackets. Groupings are derived from Schuenemann et al. [7]. The position of Sk27 is poorly defined but is clearly within group 2 and closely related to other ancient genomes.
Discussion
The history of leprosy in Europe is dominated by a drawn-out epidemic that may have lasted a millennium, but peaked between the 11th and 14th centuries before declining during the following centuries. In other parts of the world the incidence of leprosy has remained high despite the administration of multi-drug chemotherapy. Understanding the origins and macroecology of leprosy worldwide, and specifically why it declined in Europe, may help in the development of improved control strategies against the endemic worldwide leprosy of today.
Ancient DNA can be uniquely useful in constructing and assessing models of pathogen evolution [14–16] and macroecological patterns of infection. This is particularly so for M. leprae, whose extremely limited genotypic diversity has facilitated the development of models describing the evolution of modern strains and their historic worldwide dissemination. To more fully inform these models and to capture the diversity of M. leprae we have begun a bioarchaeology program of research to sequence and reconstruct the genomes of ancient M. leprae that infected medieval people at the site of the leprosarium of St Mary Magdalen, at Winchester, in southern Britain [7]. Here we continue our characterization of these M. leprae strains and present two new 1000 year old mycobacterial genomes, which in combination with previous samples, allow us to describe, in detail, the historical epidemiology of leprosy at this location across the centuries.
Rather than reveal a greater diversity of mycobacterial strains, the genomes that we present are striking in their similarity to modern M. leprae, providing further confirmation that the medieval European leprosy epidemic involved strains almost identical to those that today infect people in other parts of the world. It could be argued that this lack of diversity is an artifact of enriching the ancient DNA for M. leprae sequence using microarrays based upon modern M. leprae genomes, potentially allowing genomic re-arrangements or insertions to go undetected. However, such a scenario would seem unlikely. Firstly, these samples fall within the limits of diversity observed in the de novo assembled genomes, both ancient and modern [7–9], none of which have alternative genome architectures. Secondly, we would expect any such changes in genome architecture to be observable in the data as apparent indels at the ends of sequence reads, each representing the boundaries of any such re-arrangement. No such indels were identified in the data. This leaves us to conclude that these ancient strains were indeed highly similar to modern strains and, therefore, the diminishing incidence of leprosy in Europe was not caused by the evolution of M. leprae to a distinct genotypes that was less virulent or less transmissible. A more probable explanation is that changing social conditions reduced the rate of transmission and that concomitant epidemics of tuberculosis and Yersinia pestis (plague) resulted in high rates of mortality in leprosy infected individuals, effectively reducing the reservoir of transmissible infection [17] and hastening the selection of resistant host genotypes in the human population. A further factor may have been an increase in immunological resistance to leprosy in the population associated with exposure to the increasing levels of tuberculosis [8, 18].
The genomes of the M. leprae from skeletons Sk14 and Sk27 group firmly with other type 2F strains. Moreover, the most closely related M. leprae strain in the worldwide phylogeny (Figure 2) to Sk14 is another ancient St Magdalen skeleton, Sk8, described in our previous studies [7, 10]. Both individuals were contemporaneous children or adolescents, illustrating vividly the epidemic and conserved nature of leprosy at this site.
Until recently, type 2 M. leprae had only been identified in Asia and the Near East. It had been assumed that these type 2 strains gave rise to type 3 strains and moved with human migration to become the archetypal strains in Western Europe [8]. However, we and others have shown the presence of type 2 strains in Europe [10, 19] in the 11th and 12th centuries, alongside the more typical type 3K, 3M and 3I strains [8, 20] (Table 3). It is now clear that, at least at Winchester, type 2F strains, rather than being unusual, may well have been a co-dominant or even the predominant strain during the 11th century. The lack of type 2F strains and presence of type 3I strains in later burials both here at Winchester, and elsewhere in Europe [21] may be indicative of the shift away from type 2F strains towards the type 3 strains of leprosy typically seen in later European and North American (thought to be epidemiologically related to European strains [7, 8]) strains. It is difficult, from these data, to determine whether the type 2F strains identified at St Mary Magdalen were truly endemic to the area, or whether they were a relatively new introduction, possibly associated with trade or pilgrimage, evidence for which is graphically illustrated by the presence of a scallop shell in the grave of Sk27. Whatever their origins, the apparent displacement of type 2F strains by type 3 strains in later archaeological and contemporary samples is unlikely to have been due to a selective pressure as there are very few genetic differences between strains, and those that are present are not predicted to encode phenotypic changes. An alternative model describing a shift towards a type 3 European M. leprae population driven by allelic drift associated with a waning European leprosy epidemic would seem more plausible.
Conclusion
Through the study of bioarchaeology and specifically ancient DNA, we are gaining a greater understanding of the medieval European leprosy epidemic. By concentrating on a single well-characterized site typical of British leprosaria, St Mary Magdalen near Winchester, we have gained a detailed and almost personal insight into the wider epidemic and how it changed across the centuries. Our findings reveal that at this site during the 11th century, leprosy strains were typically part of a closely related group of type 2F strains, not the type 3 strains that predominate in later European samples.
Methods
Bone retrieval and methods
The skeletons were excavated between 2009 and 2011 by a team from the University of Winchester. The burials were recovered as articulated remains from single graves and associated grave fills were subject to 100% sampling. Pathological conditions were recorded and photographed in detail and diagnosed with reference to appropriate sources [11, 12]. All necessary permits were obtained for the described field studies, including a licence (−0070) to exhume and retain human remains, provided by the Ministry of Justice, 102 Petty France, London, SW1H 9AJ.
Bone sampling and DNA extraction
Samples of bone were taken from the rhino-maxillary region of skeletons Sk14 and Sk27 to maximize the chances of recovering M. leprae DNA. Measures were taken to minimize the opportunities for cross-contamination between cases and, use of modern DNA as a positive control was avoided completely [10]. The vomer region of a skeleton, Sk12, with no signs of leprosy was sampled as a negative control and was treated identically to the other samples throughout. The bones were ground to a fine powder and DNA extracted using the NucliSens™ extraction kit (bioMériux Limited, Boxtel, The Netherlands) as described previously [13] in laboratories physically separate from rooms in which leprosy DNA has been previously amplified.
PCR amplification of specific loci
Real-time PCR assays were carried out on a Mx3005P qPCR system (Agilent Technologies, Wokingham, UK). Methods for the PCR amplification of the multi-copy RLEP locus, the single copy locus coding for the 18 kDa antigen (ML1795), and for the M. tuberculosis specific multi-copy loci IS1081 locus are all described by Taylor, 2013 [10].
Whole genome amplification, enrichment and analysis
DNA was extracted, enriched and sequenced either at the University of Surrey alone, for Sk27, or independently at both the University of Surrey and University of Tübingen for Sk14. Both protocols are fully described by Schuenemann et al. [7]. Both involved removing uracil residues with USER enzyme (New England Biolabs, Hitchin, UK), repairing the DNA, ligating linkers to the ends, and PCR amplifying before enriching for M. leprae sequences using microarrays (once at the University of Tübingen and twice at the University of Surrey). Samples were pair-end sequenced with double indices on HiSeq2000 or MiSeq machines.
At the University of Surrey, reads were quality controlled and aligned to the M leprae TN genome [9] using Bowtie2 [24]. Duplicate reads were removed with MarkDuplicate script [25] and SNPs assigned. The criteria for inclusion in the SNP/indel list were that polymorphs had to be outside of annotated repeat regions, to have a read depth of more than 5 reads in the sample, but less than 5 reads in the control sample Sk12, have a QUAL score of > =50 (not required for longer indels) and appear in more than 80% of reads. Indels had an additional requirement that all qualifying reads had to span the indel.
At the University of Tübingen the reads were first quality controlled using FastQC, followed by adapter clipping, merging of corresponding paired-end reads and, finally, quality trimming of the resulting reads. The reads were mapped against the M. leprae TN genome as a reference using the Burrows-Wheeler Aligner (BWA). Mapped reads were subject to duplicate removal using Samtools’ rmdup method. The Genome Analysis Toolkit (GATK) was then used to generate a mapping assembly and to call SNPs for each strain. The TN reference base was called if the quality score was at least 30 and the position was covered by at least 5 reads. A variant position (SNP) was called if the position was covered by at least 5 reads and the fraction of mapped reads containing the SNP was at least 90%.
To be included in phylogenetic analysis, SNPs had to either be identified at both centers, or identified at only one center, with no contradictory data from the other. Phylogenetic analysis were carried using MEGA5 [26] using Maximum Likelihood (using the Tamura-Nei model), Maximum Parsimony (using Max-mini Branch-&-bound algorithm) and Neighbour-Joining (using the number of differences method) methods.
Availability of supporting data section
All raw read files have been submitted to the related trace archive of the National Center for Biotechnology Information Sequence Read Archive entry PRJNA200950. Alignments and phylogenetic trees have been submitted to TreeBASE with study ID 15537.
References
Jacob JT, Franco-Paredes C: The stigmatization of leprosy in India and its impact on future approaches to elimination and control. PLoS Neglect Trop Dis. 2008, 2: e113-10.1371/journal.pntd.0000113.
Robbins G, Tripathy VM, Misra VN, Mohanty RK, Shinde VS, Gray KM, Schug MD: Ancient skeletal evidence for leprosy in India (2000 B.C.). PLoS One. 2009, 4: e5669-10.1371/journal.pone.0005669.
Reader R: New evidence for the antiquity of leprosy in early Britain. J Archaeol Sci. 1974, 1: 205-207. 10.1016/0305-4403(74)90043-0.
Rawcliffe C: Leprosy in Medieval England. 2006, Woodbridge, UK: Boydell Press
Richards P: The Medieval Leper and His Northern Heirs. 1977, Cambridge, UK: DS Brewer
World Health Organisation: Global leprosy situation, 2012. Wkly Epidemiol Rec. 2013, 34: 317-328.
Schuenemann VJ, Singh P, Mendum TA, Krause-Kyora B, Jäger G, Bos KI, Herbig A, Economou C, Benjak A, Busso P, Nebel A, Boldsen JL, Kjellström A, Wu H, Stewart GR, Taylor GM, Bauer P, Lee OY, Wu HH, Minnikin DE, Besra GS, Tucker K, Roffey S, Sow SO, Cole ST, Nieselt K, Krause J: Genome-wide comparison of medieval and modern Mycobacterium leprae. Science. 2013, 341: 179-183. 10.1126/science.1238286.
Monot M, Honoré N, Garnier T, Zidane N, Sherafi D, Paniz-Mondolfi A, Matsuoka M, Taylor GM, Donoghue HD, Bouwman A, Mays S, Watson C, Lockwood D, Khamesipour A, Dowlati Y, Jianping S, Rea TH, Vera-Cabrera L, Stefani MM, Banu S, Macdonald M, Sapkota BR, Spencer JS, Thomas J, Harshman K, Singh P, Busso P, Gattiker A, Rougemont J, Brennan PJ, et al: Comparative genomic and phylogeographic analysis of Mycobacterium leprae. Nat Genet. 2009, 41: 1282-1289. 10.1038/ng.477.
Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, Honoré N, Garnier T, Churcher C, Harris D, Mungall K, Basham D, Brown D, Chillingworth T, Connor R, Davies RM, Devlin K, Duthoy S, Feltwell T, Fraser A, Hamlin N, Holroyd S, Hornsby T, Jagels K, Lacroix C, Maclean J, Moule S, Murphy L, Oliver K, Quail MA, et al: Massive gene decay in the leprosy bacillus. Nature. 2001, 409: 1007-1011. 10.1038/35059006.
Taylor GM, Tucker K, Butler R, Pike AWG, Roffey S, Marter P, Lee OY-C, Minnikin DE, Besra GS, Singh P, Cole ST, Stewart GR: Detection and strain typing of ancient Mycobacterium leprae from a medieval leper hospital. PLoS One. 2013, 8: e62406-10.1371/journal.pone.0062406.
Roffey S, Tucker K: A contextual study of the medieval hospital and cemetery of St Mary Magdalen, Winchester, England. Int J Paleopathol. 2012, 2: 170-180. 10.1016/j.ijpp.2012.09.018.
Roffey S: Medieval leper hospitals in England: an archaeological perspective from St Mary Magdalen, Winchester. Mediev Archaeol. 2012, 56: 203-234. 10.1179/0076609712Z.0000000007.
Taylor GM, Watson CL, Bouwman AS, Lockwood DNJ, Mays SA: Variable nucleotide tandem repeat (VNTR) typing of two palaeopathological cases of lepromatous leprosy from Mediaeval England. J Archaeol Sci. 2006, 33: 1569-1579. 10.1016/j.jas.2006.02.008.
Bos KI, Schuenemann VJ, Golding GB, Burbano HA, Waglechner N, Coombes BK, McPhee JB, DeWitte SN, Meyer M, Schmedes S, Wood J, Earn DJD, Herring DA, Bauer P, Poinar HN, Krause J: A draft genome of Yersinia pestis from victims of the black death. Nature. 2011, 7370: 506-510.
Taubenberger JK, Reid AH, Lourens RM, Wang R, Jin G, Fanning TG: Characterization of the 1918 influenza virus polymerase genes. Nature. 2005, 437: 889-893. 10.1038/nature04230.
Bouwman AS, Kennedy SL, Müller R, Stephens RH, Holst M, Caffell AC, Roberts CA, Brown TA: Genotype of a historic strain of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2012, 145: 18511-18516.
Donoghue HD, Marcsik A, Matheson C, Vernon K, Nuorala E, Molto JE, Greenblatt CL, Spigelman M: Co-infection of Mycobacterium tuberculosis and Mycobacterium leprae in human archaeological samples: a possible explanation for the historical decline of leprosy. Proc Biol Sci. 2005, 272: 389-394. 10.1098/rspb.2004.2966.
Manchester K: Tuberculosis and leprosy in antiquity: an interpretation. Med Hist. 1984, 28: 162-173. 10.1017/S0025727300035705.
Economou C, Kjellström A, Lidén K, Panagopoulos I: Ancient-DNA reveals an Asian type of Mycobacterium leprae in medieval Scandinavia. J Archaeol Sci. 2013, 40: 465-470. 10.1016/j.jas.2012.07.005.
Taylor GM, Donoghue HD: Multiple loci variable number tandem repeat (VNTR) analysis (MLVA) of Mycobacterium leprae isolates amplified from European archaeological human remains with lepromatous leprosy. Microbes Infect. 2011, 13: 923-929. 10.1016/j.micinf.2011.05.003.
Watson CL, Lockwood DNJ: Single nucleotide polymorphism analysis of European archaeological M. leprae DNA. PLoS One. 2009, 4: e7547-10.1371/journal.pone.0007547.
Taylor GM, Blau S, Mays S, Monot M, Lee OYC, Minnikin DE, Besra GS, Cole ST, Rutland P: Mycobacterium leprae genotype amplified from an archaeological case of lepromatous leprosy in Central Asia. J Archaeol Sci. 2009, 36: 2408-2414. 10.1016/j.jas.2009.06.026.
Suzuki K, Takigawa W, Tanigawa K, Nakamura K, Ishido Y, Kawashima A, Wu H, Akama T, Sue M, Yoshihara A, Mori S, Ishii N: Detection of Mycobacterium leprae DNA from archaeological skeletal remains in Japan using whole genome amplification and polymerase chain reaction. PLoS One. 2010, 5: e12422-10.1371/journal.pone.0012422.
Langmead B, Salzberg S: Fast gapped-read alignment with Bowtie 2. Nat Meth. 2012, 9: 357-359. 10.1038/nmeth.1923.
Picard. [http://picard.sourceforge.net]
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28: 2731-2739. 10.1093/molbev/msr121.
Acknowledgements
We are grateful to the following people for providing samples, support and advice: Richard Talbot of ARK Genomics, Christa Lanz, Alexander Herbig, and Claudia Bauer. This work was supported by the European Research Council (ERC-APGREID), the Carl Zeiss Foundation and the Wellcome Trust.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TAM planned the study, prepared DNA for NGS sequencing, analyzed the NGS data and drafted the manuscript. VJS prepared DNA for NGS sequencing; SR co-ordinated the archaeology and contributed to the manuscript; GMT planned the study, carried out DNA extractions, carried out and contributed to the manuscript; HW contributed to the analysis of the genome data; PS prepared DNA from the archaeological material, carried out some of the SNP analysis and contributed to the manuscript. KT contributed to the excavations and carried out the osteological examinations; JH contributed to the preparation of the DNA for NGS sequencing; STC contributed to the SNP analysis and the manuscript; AMK contributed to the NGS analysis; KN contributed to the NGS analysis and phylogenetics; JK contributed to the manuscript; GRS was involved in planning the study and contributed to the manuscript. All authors approved and read the final manuscript.
Electronic supplementary material
12864_2013_5937_MOESM1_ESM.xlsx
Additional file 1: Table S1: A summary of the known polymorphic loci of M. leprae as described by Schuenemann et al [7] with additional data for Sk14 and Sk27. (XLSX 137 KB)
12864_2013_5937_MOESM2_ESM.docx
Additional file 2: Figure S1: The evolutionary histories of Sk27, Sk14 and the strains described by Schuenemann et al, 2013 [7], inferred using Maximum Likelihood, Neighbour Joining and Maximum Parsimony methods. (DOCX 26 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Mendum, T.A., Schuenemann, V.J., Roffey, S. et al. Mycobacterium leprae genomes from a British medieval leprosy hospital: towards understanding an ancient epidemic. BMC Genomics 15, 270 (2014). https://doi.org/10.1186/1471-2164-15-270
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-270