
Abstract
Background
In plants, microRNAs (miRNAs) are endogenous ~22 nt RNAs that play important regulatory roles in many aspects of plant biology, including metabolism, hormone response, epigenetic control of transposable elements, and stress response. Extensive studies of miRNAs have been performed in model plants such as rice and Arabidopsis thaliana. In maize, most miRNAs and their target genes were analyzed and identified by clearly different treatments, such as response to low nitrate, salt and drought stress. However, little is known about miRNAs involved in maize ear development. The objective of this study is to identify conserved and novel miRNAs and their target genes by combined small RNA and degradome sequencing at four inflorescence developmental stages.
Results
We used deep-sequencing, miRNA microarray assays and computational methods to identify, profile, and describe conserved and non-conserved miRNAs at four ear developmental stages, which resulted in identification of 22 conserved and 21-maize-specific miRNA families together with their corresponding miRNA*. Comparison of miRNA expression in these developmental stages revealed 18 differentially expressed miRNA families. Finally, a total of 141 genes (251 transcripts) targeted by 102 small RNAs including 98 miRNAs and 4 ta-siRNAs were identified by genomic-scale high-throughput sequencing of miRNA cleaved mRNAs. Moreover, the differentially expressed miRNAs-mediated pathways that regulate the development of ears were discussed.
Conclusions
This study confirmed 22 conserved miRNA families and discovered 26 novel miRNAs in maize. Moreover, we identified 141 target genes of known and new miRNAs and ta-siRNAs. Of these, 72 genes (117 transcripts) targeted by 62 differentially expressed miRNAs may attribute to the development of maize ears. Identification and characterization of these important classes of regulatory genes in maize may improve our understanding of molecular mechanisms controlling ear development.
Similar content being viewed by others
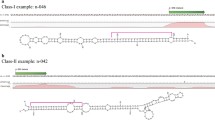


Background
Maize is one of the most productive crops worldwide, and is widely used as a model plant in genetics research [1]. Maize produces two distinct inflorescences, commonly referred to as the tassel and the ear. In this respect, it differs from other grasses such as rice and wheat. The tassel arises from the apex of the mature plant, while ears originate from axillary bud apices [2]. One obvious difference in morphology between the two inflorescences is the presence (tassel) or absence (ear) of a variable number of long branches originating at the base. In previous studies, the wide range of natural variation among different inbred lines was used to identify quantitative trait loci (QTL) underlying a variety of phenotypes by association mapping [3, 4]. Several genes associated with maize ear development have been identified in genetic and molecular studies [2]. However, knowledge about maize ear development is still limited, and most of the genes involved in this process are still unknown.
In plants, small RNA-guided post-transcriptional regulatory mechanisms play important roles in many aspects of plant biology, including metabolism [5], hormone responses [6], epigenetic control of transposable elements [7], and responses to biotic stress [8] and abiotic stress [9]. The two main types of small RNAs are microRNAs (miRNAs) and small interfering RNAs (siRNAs) [10]. Over recent decades, many miRNA families have been discovered in plants, and have been shown to regulate more aspects of plant biology than siRNAs [5, 11, 12]. Published reports as well as publicly accessible miRNA datasets (miRBase, version 20, http://www.mirbase.org/) [13], mainly based on model plants, suggest that miRNAs in plants are complex and abundant [14–17]. Therefore, identification of miRNAs and their targets in diverse species has been a major focus in recent years.
So far, conserved miRNAs in maize have been identified by sequence homology analyses [18–20], and new miRNA sequences have been identified by traditional [21–23] or high-throughput [17, 24–30] sequencing methods. These miRNA sequences can be found in miRBase databases [13]. Functional analysis has been carried out for only a few maize miRNAs, mainly in their role in flower development [21, 31–36].
There are three main objectives of this study. The first objective is to identify conserved and novel miRNAs in maize ears at four different developmental stages. The second objective is to combine publically available Arabidopsis thaliana, Oryza sativa, Sorghum bicolor, and Zea mays miRNAs data with the new Zea mays miRNAs data to generate a miRNA microarray platform to analyze the dynamics of miRNA expression. Finally, to discover the targets of conserved and non-conserved miRNAs, we aimed to identify the remnants of small RNA-directed target cleavage by sequencing the 5′ ends of uncapped RNAs using a degradome sequencing approach.
Results
Overview over small RNA library sequencing
To study the involvement of regulatory miRNAs in the complex process of ear development, we profiled miRNA accumulation during ear development in the maize inbred line B73. We constructed a maize small RNA library using mixed RNAs obtained from ears at four different developmental stages. Sequencing was conducted on the Illumina platform. We obtained more than 10.67 million raw clean reads, ranging from 18 nt to 30 nt in length. After trimming adaptor sequences and removing contaminated reads, clean reads were aligned against the Maize B73 RefGen v2 working gene set using SOAP2 software [37]. We found that 7,981,459 (3,436,342 distinct) reads matched perfectly to the maize genome, representing 74.85% of total reads (66.64% of distinct reads). Of the distinct reads, 5.22% matched with non-coding RNAs in Rfam and NCBI Genbank databases; these non-coding RNAs included snoRNAs (0.01%), snRNAs (0.03%), tRNAs (0.15%), rRNAs (1.69%), and siRNAs (3.34%) (Additional file 1). The remaining reads were then used to identify conserved and new miRNAs. The length of these small RNAs ranged from 20 nt to 24 nt. Of these, the 24 nt category was the most abundant small RNA (48.55%) (Figure 1a), followed by 22 nt (14.18%) and 21 nt (8.78%). These were consistent with the typical lengths of plant mature small RNAs reported in other studies [14, 27, 38, 39].

Size distribution of small RNAs by deep sequencing and characterizations of maize miRNA. (a) Size distribution of sequenced sRNAs in total RNA; (b) The composition of maize miRNAs of various lengths; (c) The relative nucleotide bias at each position of the miRNAs in total RNA.
Computational identification of genuine miRNAs during maize ear development
To date, research on identifying conserved and novel miRNAs has used several standard methods and databases, including Rfam, GenBank, and miRBase. Because of their low expression levels and sequence depths, it is always difficult to predict miRNAs. Hence, we used a strict strategy with eight steps to predict and identify known and novel miRNAs based on the characteristic features of miRNAs specifically processed by Dicer-like proteins from canonical stem-loop regions of longer RNA precursors [40, 41]. We used an integrated strategy combining high-throughput sequencing with bioinformatics analyses to identify miRNAs meeting all reported previously criteria (see “Methods”) [42].
As shown in the schematic diagram of the strategy (Additional file 2), our computational analysis generated 508 loci folded within typical stem-loop structures (Additional file 3). After excluding 38 loci that overlapped with protein-coding gene exons, 76 loci overlapping transposable elements and other repetitive elements, and 9 loci with free energy lower than −20 kcal/mol (Additional file 4), the remaining 385 loci were considered to be candidate miRNA genes. We used miRAlign to identify paralogs or orthologs of these 385 candidate miRNA genes by comparing their sequences with those of known miRNAs, as described previously [43]. From this analysis, we detected 99 known miRNA genes encoding 96 mature miRNAs and three miRNA star (miRNA*) (Additional file 4). We also detected 64 novel miRNA* sequences (Additional file 5).
In plants, it is difficult to identify new miRNAs, even when they have the characteristic hairpin feature, because of abundant inverted repeats that can also fold into dysfunctional hairpins [44, 45]. Thus, we used additional strategies that were not based on phylogenetic conservation to identify non-conserved pre-miRNAs. We used MiPred (http://www.bioinf.seu.edu.cn/miRNA/) to distinguish pre-miRNAs from other similar segments in the maize genome. Among the remaining 286 candidate pre-miRNA-like hairpins, 52 were classified as pseudo-pre-miRNAs and 198 were not pre-miRNA-like hairpins. The other 36 loci, which encoded 26 non-redundant mature miRNAs (Table 1), were identified as maize-specific miRNA genes. Of these 26 miRNAs, 25 belonged to new families that have not been reported in plants (Additional file 4, Additional file 5). Here, we have designated them in the form of their zma-miR-specific-number, e.g., zma-miRs2. When several maize-specific miRNAs belonged to the same family, we named them in a similar manner to that used to name known mature miRNAs [15] (e.g. zma-miRs6a and zma-miRs6b). All of the new miRNA precursors had regular stem-loop structures. We also detected four miRNA* (Additional file 3, Additional file 4, Additional file 5, Additional file 6), providing further evidence for the existence of this class of miRNAs in maize.
Characterization of newly identified miRNAs in maize
As expected, approximately all 22 the conserved miRNA families in the small RNA library were identified in this study. However, we detected miRNA* sequences of zma-miR171h/k and zma-miR408b instead of their corresponding mature miRNA sequences (Additional file 5). We also identified five mature miRNAs (zma-miR160e, zma-miR166e/f, zma-miR169g, and zma-miR172e) previously predicted by similarity searches [18, 20] and unexpectedly found their corresponding miRNA* sequences (zma-miR160e*, zma-miR166e*/f*, and zma-miR169g*), which were not available in miRBase. Besides the known miRNAs, we also identified 26 new miRNA candidates (Table 1, Additional file 6), and nine (zma-miRs1a/b, zma-miRs2, zma-miRs4, zma-miRs7, zma-miRs9, zma-miRs12, zma-miRs14, and zma-miRs17) were previously reported [27] (Additional file 5). The sequence of miRs4 was similar to that of members of the miR169 family (Additional file 7), indicating that miRs4 may be a member of that family. Most of the new miRNAs could only be produced from one locus. However, zma-miRs6b and four other new miRNA genes (zma-miRs5, zma-miRs6a, zma-miRs8, and zma-miRs23) could be produced from two or more loci (Additional file 4). Among the newly identified miRNAs, 21-nt miRNAs were the most abundant category (52.6%) (Figure 1b). Analysis of the nucleotide sequences of these miRNAs revealed that uridine (U) was the most common nucleotide at the 5′ end (>80%) (Figure 1c), and the 10th and 11th nucleotides, which match to the cleavage site of targets, were usually adenine (A). Also, U was the most common nucleotide at positions 21 and 22 in these miRNAs.
Next, we conducted microarray assays to analyze expressions of the known and newly identified miRNAs during maize ear development. We detected transcripts of all of the conserved miRNAs and 20 out of 26 (76%) maize-specific miRNAs in the microarray experiment. Those that were undetected either had a low affinity to the chip probes or very low transcript levels (sequencing frequency <50) (Additional file 8). These results suggest that Solexa sequencing is a more specific and efficient tool than the miRNA microarray assay for identifying mature miRNAs. In our study, we detected six miRNA families (miR408/482/827/397/398/2118) in the microarray assay that were not present in the Solexa sequencing data (Additional file 8, Additional file 9). These miRNAs need to be further validated.
Although we identified 122 miRNAs (96 conserved + 26 non-conserved) and 64 miRNA*s, they showed a diverse range of abundance, and only a few miRNA families dominated in the miRNA library and microarray assay data. The six most abundantly expressed miRNA families were miR166, miR168, miR167, miR156, miR159, and miRs6. There were extremely low frequencies of miR395, miR399, miR2275, miRs12, and miRs19, possibly because these families are expressed in a tissue-specific manner. Most of the miRNA*s showed very low transcript levels (sequencing frequency <30), much lower than those of their homoplastic miRNAs, consistent with previous findings [12]. The transcript level of zma-miR408b was lower than that of zma-miR408b*, and the mature product from the 3′ arm of the hairpin suggested that the 3′ arm may be functional.
Expression profiles of known and newly identified miRNAs
To analyze miRNA expression during maize ear development, we analyzed the miRNA expression profiles of ear samples collected at four different developmental stages using microarray assays. Conserved mature miRNAs are generally conserved among plant species and are stably expressed in diverse tissues. However, when microarray technology is used to analyze expression, members of the same miRNA family with 1–3 nt sequence differences need to be normalized for further analyses because hybridization can occur between members of the same miRNA family across different species [46]. Hence, a total of 53 miRNAs, about 8.4% (53/632) of the probes on the microarray, were identified as putative differentially expressed miRNAs (P = 0.01) (Table 2). Of these, 45 miRNAs aligned with 59 members of 21 maize miRNA families, while the others corresponded to members of miRNA families from three other plant species, including rice (osa-miR156/162/164/168/396/529) Arabidopsis (ath-miR156/164/167) and sorghum (sbi-miR396). The results shown in Additional file 10: Figure S3 indicated that the differentially expressed miRNAs may be specially regulated in diverse pathways during ear development. A sample of 12 expressed miRNAs was randomly selected for validation by stem-loop qRT-PCR. The trends in the expression of these miRNAs detected by microarray experiments were consistent (7 miRNAs) or partially (5 miRNAs) consistent with those determined in stem-loop qRT-PCR analyses (Figure 2).

Expression of 12 miRNAs in developmental ears in maize. The expression pattern of 12 miRNAs detected by microarray experiment (a) and stem-loop quantitative Real-Time PCR (qRT-PCR) (b). Stages 1, 2, 3, and 4 represent stage I, stage II, stage III, and stage IV, respectively. *indicated the significant difference in adjacent two developmental stages (one-way ANOVA and Fisher′s LSD, p < 0.01, n = 4). The blue, pink, green, and purple bars in graph b depicted the stem-loop qRT-PCR relative expression level ± standard error of three replicates for each miRNA in the four developmental stages of maize ears, respectively.
Target prediction of conserved and non-conserved miRNAs by degradome sequencing
To identify small RNA targets at a global level in maize, we used the recently developed degradome library sequencing technology [47, 48]. We generated four libraries from maize ears at different developmental stages (I-IV) as described above. High-throughput sequencing yielded 13,638,690 (1,706,149 distinct), 18,257,616 (2,887,536 distinct), 9,477,595 (682,164 distinct), and 8,393,209 (2,490,241 distinct) 20-nt sequences representing the 5′ ends of uncapped, poly-adenylated RNAs for stages I to IV, respectively. The total number of signatures matching to the genome was 10,596,420 for stage I, 14,571,419 for stage II, 7,415,394 for stage III, and 6,524,350 for stage IV. The number of distinct sequences in the four libraries matching to the genome was 1,123,608 (66%) for stage I, 1,995,882 (69%) for stage II, 423,065 (62%) for stage III, and 1,746,858 (70%) for stage IV. The number of signatures that matched to only one location in the genome was relatively high: 825,904 (74%) for stage I, 1,521,543 (76%) for stage II, 317,671 (75%) for stage III, and 1,318,724 (70%) for stage IV, suggesting that 20-nt signatures are sufficient to identify their origin in the maize genome. Of these, 973,186 (87%), 1,816,631 (91%), 382,792 (90%) and 1,580,297 (90%) distinct signatures for stage I, II, III, and IV, respectively, could be mapped to annotated maize gene models (B73 RefGen_v2). A small proportion of the distinct signatures (0.8%, 0.7%, 0.9%, and 0.7% for stage I, II, III, and IV, respectively) could also be mapped to the maize chloroplast or mitochondrial genomes. The number of distinct signatures matching to rRNAs, tRNAs, small nucleolar RNAs (snRNAs) or small nuclear RNAs was 10,101 (0.9%) for stage I, 9,596 (0.5%) for stage II, 4,521 (1.1%) for stage III, and 11,572 (0.7%) for stage IV. These were removed before subsequent analyses (Additional file 10). Similarly, we removed the sequences matching to repeats/transposons that were revealed by searches against the repeat database (http://www.girinst.org/server/RepBase/). Interestingly, a significant proportion of distinct signatures from the four libraries matched to introns and intergenic regions, similar to the findings of previous transcript profiling analyses [47, 48].
Based on previous studies, a characteristic scenario of miRNA-guided slicing is that the cleavage takes place precisely between the 10th and 11th nt from the 5′ end of miRNA in the complementary region of the target transcript [47–51]. We used CleaveLand pipeline [52] to identify sliced miRNA targets in the maize transcriptome. Various sequenced tags were plotted on each of the target transcripts (Additional file 11). The cleaved target transcripts were categorized into five classes (categories 0, 1, 2, 3, and 4) as reported previously for Arabidopsis[47], grapevine [49], rice [51], and soybean [50].
For conserved miRNAs and ta-siRNAs, 120 target genes (234 transcripts) were identified in ears at the four stages of development (Table 3, Figure 3 Additional file 12). Reads associated with most of these miRNA targets were over-represented (Additional file 11). However, only 15% of the miRNA targets (19 out of 127) were identified in all four stages. The targets were classified into categories 0–4 based on the abundance of degradome tags indicative of miRNA-mediated cleavage. In stage I, II, III, and IV, there were 5, 19, 7, and 20 targets classified as category 0 (where miRNA-guided cleavage remnants were the most abundantly recovered species) [53]. There were 5, 2, 20, and 3 targets in stage I, II, III, and IV, respectively, classified as category 1 (where the abundance of cleavage products was equal to the maximum, and there was more than one maximum position on the transcript). In stage I, II, III, and IV, there were 22, 28, 27, and 20 targets classified as category 2 (where the abundance of cleavage sequences was less than the maximum but greater than the median for the transcript). In stage I, II, III, and IV there were 10, 7, 13 and 5 target transcripts classified as category 3 (where the total abundance of degradome sequences at the cleavage site was equal to or less than the median for the transcript). All other transcripts were classified as category 4 (where only one raw read matched the 5′ end of a slicing remnant). Only 4, 8, 0, and 9 targets in stage I, II, III, and IV, respectively, were in category 4. Among the identified targets, category 2 was the most abundant category among the four degradome libraries (Tables 3, 4 and Additional file 11).

Summary of cleaved miRNA target categories found with degradome analyses in four developmental stages of maize ears.
We identified target genes for almost all of the 22 conserved miRNA families. The conserved miRNAs were able to target various numbers of genes, ranging from 1 to 18. Among the conserved miRNA families, zma-miR156 and zma-miR529 had the highest number of gene targets. zma-miR156 targeted 13 unique genes including SPL genes and zma-miR529 targeted 18 unique genes including ZCN19 (a possible maize FT ortholog) (Table 3), indicating that these two families might play key roles in ear development [31, 54]. Most of the conserved miRNAs targeted multiple gene loci. Their gene targets were members of different families of transcription factors, such as SBP-box transcription factor, AUXIN RESPONSE FACTOR (ARF), TCP, MYB, bZIP, AP2, and GRAS. We also identified 57 new target genes of conserved miRNAs in maize (Table 3). Among the 127 miRNA target genes, 67 (53%) had been predicted previously [17], 56 (44%) cross-validated with other degradome libraries prepared from plants under different stress conditions [25, 28–30], and 8 have been validated by 5′-RACE and/or genetic experiments [25, 28, 30, 32, 33, 36]. The targets of conserved miRNAs were highly abundant in the four sequenced target libraries, and were often classified as category 0, 1, or 2 targets (Table 3, Additional file 11). For instance, miR169 targeted seven different CCAAT-binding transcription factors in the four stages (category 0 or 2) with very high abundance, but it also guided the slicing of three other non-conserved targets with very low abundance. Interestingly, some target transcripts were regulated by pairs of miRNAs: both miR156 and miR529 targeted five members of the same SBP family, and the miR159/319 pair regulated three MYB domain transcription factors. This result suggested that there is complex regulation of these genes by these miRNA pairs, consistent with the findings of a previous study [49] (Table 3).
Out of 26 non-conserved zma-miRNAs including 21 new miRNAs with four corresponding miRNA*, we identified targets for only seven miRNAs (miR2118, miR2775, miRs4, miRs9, miRs14, miRs15 and miRs17). We used absolute numbers to plot the cleavages on target mRNAs; this was referred to as a target plot (t-plot) by German et al.[48]. Except for miRs4, the targets mostly belonged to category 2 or 4 with very low abundance, which differed from the targets of conserved miRNAs (Tables 3, 4). Four identified targets of miRs4 (category 0 or 2) were the same as those of miR169, providing further evidence that miRs4 is a member of the miR169 family.
GO analysis of targets regulated by differentially expressed miRNAs
In our study, we predicted 72 genes (117 transcripts) for 62 differentially expressed miRNAs from 11 miRNA families. More than 90% of these miRNAs had putative functions (Tables 2, 3 and Additional file 13). 73% of these differentially expressed miRNA families played an important role in post-transcriptional regulation by targeting mRNAs encoding transcription factors in SBP, ARF, GRAS, and AP2 families (Tables 2, 3 and Additional file 13). GO analysis revealed that the target genes were involved in a wide spectrum of regulatory functions and biological processes including regulation of transcription, DNA binding, response to hormone stimulus, nucleic acid metabolic process, gene expression, cellular macromolecule synthesis, and cellular nitrogen compound metabolism. This was consistent with the fact that the small RNA and the degradome libraries for miRNA sequence analysis were constructed from developing maize ears (Additional file 14). In addition, a specific feature of our study was that we found more genes in families involved in metabolic process, biological regulation, cellular biosynthetic process, and nucleic acid binding function at the later stages of maize ear development (Table 2, Additional file 14). The accumulation of dry matter such as starch and saccharides is the main event in ear development, and a large number of target genes may participate in this pathway. The differentially expressed miRNAs may regulate expression of these target genes to control ear development and biomass yield in maize.
Discussion
Small RNAs play important roles in gene regulation in plants [12]. In this study, we have annotated miRNA genes based on the complete assembly of the maize genome. In total, 98 known miRNAs and 26 new miRNAs were identified in maize ears by deep sequencing. This confirmed previous results reported by Zhang et al. [17]. These newly identified miRNAs may belong to lineage-specific families, and showed little or no expression at the miRNA level. We identified 62 miRNAs as differentially expressed miRNAs by microarray assays.
The recently reported high-throughput experimental approach [47, 48] allowed us to create a detailed miRNA: target interaction atlas for maize. In the current work, we identified a total of 131 genes (245 transcripts) targeted by 102 small RNAs including 98 miRNAs and 4 ta-siRNAs (Tables 3, 4 and Additional file 12). Among the 131 genes, 54 were cross-validated in other degradome libraries [25, 28–30], by 5′-RACE, and/or by genetic experiments [17, 25, 30, 32, 36], showing that degradome sequencing is a powerful tool for identifying targets regulated by miRNAs. Surprisingly, most highly conserved miRNAs were detectable in maize ears at all four developmental stages (Additional file 8), but sliced targets were not detected at all stages (Additional file 12). It is possible that the differentially expressed miRNAs regulate both the spatial pattern and the level of target mRNA expression, as previously demonstrated in some cases [55, 56]. It is equally possible that this represents a limitation of degradome sequencing. Results can be affected by many unpredictable factors such as ligation efficiency, PCR bias, etc. There were 127 target genes of 22 conserved miRNA families. Among the target genes, 72.4% encoded transcription factors (Table 3). These targets were not only conserved families, such as SBP, MYB, ARF, bZIP (basic-leucine Zipper), NAC, GRAS, AP2, and TCP transcription factor gene families, but also non-conserved genes encoding metallophosphoesterase, DICER-LIKE1, No Apical Meristem (NAM) proteins, and PHD finger proteins. The conserved targets may participate in maize ear development. We also identified 13 genes targeted by non-conserved miRNAs. One ARF gene and three DNA-binding transcription factor genes cleaved by ta-siRNAs were also identified (Table 4). The conserved miRNAs silenced more targets than did maize-specific miRNAs. It is possible that conserved miRNAs play a crucial role in post-transcriptional regulation in different plant species [12]. However, maize-specific miRNAs may function only to regulate gene expression during gramineae- or maize-specific biological processes. Although conserved miRNAs mainly regulate genes encoding transcription factors, maize-specific miRNAs are considered to be young miRNAs that have evolved recently, and are often expressed at lower levels than conserved miRNAs [39, 57].
Previous studies showed that miR156 and miR172 function throughout flower development from the earliest stages (floral induction, stage I) to very late stages (floral organ cell-type specification, stage IV) [31–34]. miR156a-l probably targets several SPL genes during the juvenile-to-adult phase transition in maize (Figure 4a, Tables 2 and 3), and is postulated to indirectly activate miR172 via SPL[31]. miR172 has been shown to negatively regulate GL15 (Table 3), which promotes maintenance of the juvenile state [31]. The levels of miR156 and miR172 are conflicting during phase transition (Figure 4b). Meanwhile, miR172e likely controls IDS1 and SID1, which are responsible for maize spikelet sex determination and meristem cell fate [32–34], by both translational repression and mRNA degradation (Table 3; Figure 4a). Beyond miR156 and miR172, miR164 targets genes encoding NAM proteins, and may be involved in regulating ear development (Table 3), similar to how miR164 is postulated to regulate NAC-domain targets in Arabidopsis [58]. Although most miRNA families appear to target a single class of targets, the miR159/319 family regulates both MYB and TCP transcription factors, which may control petal morphogenesis as previously reported [59].

miR156 and miR172 in maize flower development (Adapted from Poethig (2009). (a) The development of the maize ear (female inflorescence) and the juvenile phase regulated by miR156 and miR172. In the ear, the inflorescence meristem (IM, stage I) directly gives rise to spikelet pair meristems (SPM, stage II). SPMs give rise to two spikelet meristems (SM, stage III), which subsequently form two floral meristems (FM, stage IV). miR172 and its targets IDS1 and SID1 function to influence the SM to FM conversion. IDS1 is also a negative regulator of its homolog SID1. Both IDS1 and SID1 are positive regulators of the SM to FM transition. (b) The expression pattern of zma-miR156a and zma-miR172e. * indicated the significant difference in adjacent two developmental stages (one-way ANOVA and Fisher’s LSD, p < 0.01, n = 4).
Some miRNAs have been shown to be involved the signaling pathway that mediates responses to the phytohormone auxin. For example, miR167 targets four AUXIN RESPONSE FACTOR (ARF) genes, and miR160 targets six ARF genes. In addition to the miRNAs mentioned above, one miRNA family (miR162) targets a gene central to miRNA genesis; the differentially expressed zma-miR162 targets DICER-LIKE1 (DCL1), a homolog of DCL1 in Arabidopsis that is required for miRNA accumulation [60]. In summary, genome-wide identification of all targets provided useful information to explore the functions of miRNAs in maize.
Conclusions
In this study, we have confirmed the expression of conserved, known non-conserved and new maize miRNAs using high-throughput approaches to better understand the role of miRNAs in developmental maize ears. Besides, we have identified 131 target genes of both known and new miRNAs and ta-siRNAs using recently developed tools for the global identification of miRNA targets. Specifically, 72 genes (117 transcripts) targeted by 62 differentially expressed miRNAs from 11 miRNA families may play important roles in ear development in maize. Maize represents a model for cultivated crop plants. As these characters are quite different for other model plants (e.g. Arabidopsis and Medicago), we expect to discover new roles of miRNAs in post-transcriptional regulation. We also provided some evidence of the important function of miRNAs in regulating developmental process. Identification and characterization of this important class of regulatory genes in maize may improve our understanding of molecular mechanism controlling maize ear development.
Methods
Plant materials and RNA isolation
Seed of maize inbred lineB73 was first sterilized and germinated in an incubator, then grown in a controlled environment at 28°C/21°C (day/night) under a 16-h day/8-h night photoperiod with a relative humidity of 70%. Ear development can be divided into four stages: the growth point elongation phase, spikelet differentiation phase, the floret primordium differentiation phase and floret organ differentiation phase. Plant materials (ears) were collected as described previously [48]. Briefly, ears were manually collected at the four developmental stages according to the plant features (number of visible leaves, leaf age index, and number of unfolded and folded leaves) combined with microscopic observation. All the samples were harvested and immediately frozen in liquid nitrogen and stored at −80°C. The total RNA from each sample was then isolated using Trizol (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions.
Small RNA library preparation and sequencing
The total RNAs were pooled for each of four developmental stages for Solexa sequencing. After small RNA cloning, the sequencing procedures were conducted as described previously [61]. In brief, sequencing was performed as follows: approximate 100 ug of total RNA was purified by polyacrylamide gel electrophoresis (PAGE), to enrich for molecules in the range of 18–30nt, and ligated with adapters to the 5′ and 3′ terminals of the RNA. Then, small RNA molecules were used as templates for cDNA synthesis. In total, 18 PCR cycles and agarose gels were used for amplification and fragments of around 90 nt including both small RNA and adaptors, separately. The purified DNA was used Solexa sequence analysis performed by the Illumina platform. Digital-quality data were generated from the image files produced by the sequencer. After quality control using common pipeline, clean reads were directly used for further bioinformatics analysis.
Degradome library construction
Small cDNA libraries using the sliced ends of poly adenylated transcripts from maize ears of four developmental stages were constructed according to previous reports [47, 48]. By using the Oligotex kit (Qiagen, location), 200 μg of total RNA was used for extracting poly (A) RNA, which were ligated with an RNA adapter consisting of a MmeI recognition site in its 3′ end. After ligation, first-strand cDNA was generated using oligod (T) and the PCR product was amplified using five PCR cycles. The PCR product was purified and digested with MmeI. The digested PCR product was then ligated to a double-stranded DNA oligonucleotide with degenerate nucleotides at the 5′-or 3′-ends. The ligation product was further gel purified and amplified using 10 PCR cycles. The final PCR product was purified and sequenced using Illumina’s sequencing by synthesis (SBS) sequencing technology.
MiRNA microarray assays
MiRNA microarray assays of different developmental stages were performed by LC Sciences (Houston, TX, USA). The custom μparaflo™ microfluidic chip contained 632 unique plant miRNAs of release version 18, representing 1,187 miRNAs (including miRNA*) from 4 plant species (http://microrna.sanger.ac.uk/) [13], and 26 additional unique miRNAs of maize identified by Solexa sequencing, representing 26 novel miRNAs. Each chip contained four repetitions of each probe. In total, the 1,215 miRNAs were composed of 224, 496, 148 and 347 miRNAs from Arabidopsis thaliana, Oryza sativa, Sorghum bicolor and Zea mays, separately. RNA labeling, microarray hybridization, array scanning, and data’s analysis were performed essentially as previously described [46].
Bioinformatics analysis of sequencing data
Both small RNA reads and degradome reads were generated by Illumina Genome Analyzer II. As for the small RNA library, the data were processed and analyzed as previously described by Wang et al.[27] and Zhang et al.[17]. In brief, unique reads ranging from18-25 nt were collected and mapped to the maize genome (B73 RefGen_v2 (release 5b. 60 in February 2011)) reference sequences [62] by SOAP2 [37]. After removing sequences matching non-coding rRNAs, tRNAs, snRNAs and snoRNAs in the Rfam and NCBI Genbank databases, the matched Solexa reads that were extracted 250 nt of the sequence flanking the genomic sequences were used for RNA secondary structure prediction, which was performed by mFold 3.5 [63] and analyzed by MIREAP (https://sourceforge.net/projects/mireap/) to identify new candidates using default settings. The candidate miRNA list was further trimmed based on the criteria as described [17, 42]. Based on the hairpin structure of the pre-miRNA, the corresponding miRNA star sequence was also identified.
Degradome reads were filtered using custom Perl script. The remaining distinct 20–21 nt sequences that perfectly matched maize contigs were collected for further analysis. The 15 nt upstream and 5′ end of the reads that mapped to maize contigs were extracted to generate 30-sequence tags, which were used to align to newly identified miRNAs and miRBase (Release19.0, August, 2012) using the Cleave and pipeline [52]. Alignments were collected as candidate targets if they fulfilled the criteria as described before [50].
GO functional enrichment analysis of all candidate targets during different developmental stages was carried out using Blast2GO (version 2.3.5, http://www.blast2go.org/) and GO annotations were performed using AgriGO (http://bioinfo.cau.edu.cn/agriGO/). KEGG pathway analyses of differentially expressed genes were performed using Cytoscape software (version 2.6.2) (http://www.cytoscape.org/) with the ClueGO plugin (http://www.ici.upmc.fr/cluego/cluegoDownload.shtml) [64].
Stem-loop quantitative real-time PCR (qRT-PCR) analysis
Validations of 13 randomly selected mature miRNAs were carried out by stem-loop reverse transcription-PCR (RT–PCR) (Additional file 15). Total RNA (200 ng) was used to initiate the reverse transcription reaction. Primers for the stem–loop RT-PCR were designed using methods as described by Chen et al.[65] and Varkonyi-Gasic et al.[66]. The stem-loop RT-PCR was using the Applied Bio systems 7500 Real-Time PCR System (Applied Bio systems, Foster City, CA). All primers were listed in Additional file 12: Table S8. All reactions were run in triplicate. 5S rRNAs was used as the internal control for stem–loop RT-PCR [67].
Data access
The RNA sequencing data have been deposited in the NCBI under the accession number GSE47837.
References
Bennetzen JL, Hake SC: Its Biology. Handbook of Maize. 2009, New York: Springer
Vollbrecht E, Schmidt R: Development of the Inflorescences. Handbook of Maize: Its Biology. Edited by: Bennetzen JL, Hake SC. 2009, New York: Springer, 13-40.
Upadyayula N, da Silva HS, Bohn MO, Rocheford TR: Genetic and QTL analysis of maize tassel and ear inflorescence architecture. Theor Appl Genet. 2006, 112 (4): 592-606.
Zhao W, Canaran P, Jurkuta R, Fulton T, Glaubitz J, Buckler E, Doebley J, Gaut B, Goodman M, Holland J, et al: Panzea: a database and resource for molecular and functional diversity in the maize genome. Nucleic Acids Res. 2006, 34 (Database issue): D752-D757.
Nag A, Jack T: Sculpting the flower; the role of microRNAs in flower development. Curr Top Dev Biol. 2010, 91: 349-378.
Liu Q, Chen YQ: Insights into the mechanism of plant development: interactions of miRNAs pathway with phytohormone response. Biochem Biophys Res Commun. 2009, 384 (1): 1-5.
Lisch D: How important are transposons for plant evolution?. Nat Rev Genet. 2013, 14 (1): 49-61.
Voinnet O: Post-transcriptional RNA silencing in plant-microbe interactions: a touch of robustness and versatility. Curr Opin Plant Biol. 2008, 11 (4): 464-470.
Sunkar R, Chinnusamy V, Zhu J, Zhu JK: Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci. 2007, 12 (7): 301-309.
Carthew RW, Sontheimer EJ: Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009, 136 (4): 642-655.
Chuck G, Candela H, Hake S: Big impacts by small RNAs in plant development. Curr Opin Plant Biol. 2009, 12 (1): 81-86.
Jones-Rhoades MW, Bartel DP, Bartel B: MicroRNAS and their regulatory roles in plants. Annu Rev Plant Biol. 2006, 57: 19-53.
Kozomara A, Griffiths-Jones S: miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39 (Database issue): D152-D157.
Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL, et al: High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One. 2007, 2 (2): e219-
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen X, Green PJ, et al: Criteria for annotation of plant MicroRNAs. Plant Cell. 2008, 20 (12): 3186-3190.
Nobuta K, Venu RC, Lu C, Belo A, Vemaraju K, Kulkarni K, Wang W, Pillay M, Green PJ, Wang GL, et al: An expression atlas of rice mRNAs and small RNAs. Nat Biotechnol. 2007, 25 (4): 473-477.
Zhang L, Chia JM, Kumari S, Stein JC, Liu Z, Narechania A, Maher CA, Guill K, McMullen MD, Ware D: A genome-wide characterization of microRNA genes in maize. PLoS Genet. 2009, 5 (11): e1000716-
Maher C, Timmermans M, Stein L, Ware D: 2004 IEEE Computational Systems Bioinformatics Conference (CSB’04). Identifying MicroRNAs in Plant Genomes. 2004, 718-723.
Zhang B, Pan X, Anderson TA: Identification of 188 conserved maize microRNAs and their targets. FEBS Lett. 2006, 580 (15): 3753-3762.
Zhang BH, Pan XP, Wang QL, Cobb GP, Anderson TA: Identification and characterization of new plant microRNAs using EST analysis. Cell Res. 2005, 15 (5): 336-360.
Juarez MT, Kui JS, Thomas J, Heller BA, Timmermans MC: microRNA-mediated repression of rolled leaf1 specifies maize leaf polarity. Nature. 2004, 428 (6978): 84-88.
Mica E, Gianfranceschi L, Pe ME: Characterization of five microRNA families in maize. J Exp Bot. 2006, 57 (11): 2601-2612.
Zhang Z, Lin H, Shen Y, Gao J, Xiang K, Liu L, Ding H, Yuan G, Lan H, Zhou S, et al: Cloning and characterization of miRNAs from maize seedling roots under low phosphorus stress. Mol Biol Rep. 2012, 39 (8): 8137-8146.
Ding D, Wang Y, Han M, Fu Z, Li W, Liu Z, Hu Y, Tang J: MicroRNA transcriptomic analysis of heterosis during maize seed germination. PLoS One. 2012, 7 (6): e39578-
Jiao Y, Song W, Zhang M, Lai J: Identification of novel maize miRNAs by measuring the precision of precursor processing. BMC Plant biology. 2011, 11: 141-
Shen Y, Jiang Z, Lu S, Lin H, Gao S, Peng H, Yuan G, Liu L, Zhang Z, Zhao M, et al: Combined small RNA and degradome sequencing reveals microRNA regulation during immature maize embryo dedifferentiation. Biochem Biophys Res Commun. 2013, 441 (2): 425-430.
Wang L, Liu H, Li D, Chen H: Identification and characterization of maize microRNAs involved in the very early stage of seed germination. BMC Genom. 2011, 12: 154-
Zhai L, Liu Z, Zou X, Jiang Y, Qiu F, Zheng Y, Zhang Z: Genome-wide identification and analysis of microRNA responding to long-term waterlogging in crown roots of maize seedlings. Physiol Plant. 2013, 147 (2): 181-193.
Zhao M, Tai H, Sun S, Zhang F, Xu Y, Li WX: Cloning and characterization of maize miRNAs involved in responses to nitrogen deficiency. PLoS One. 2012, 7 (1): e29669-
Zhao Y, Xu Z, Mo Q, Zou C, Li W, Xu Y, Xie C: Combined small RNA and degradome sequencing reveals novel miRNAs and their targets in response to low nitrate availability in maize. Ann Bot. 2013, 112 (3): 633-642.
Chuck G, Cigan AM, Saeteurn K, Hake S: The heterochronic maize mutant Corngrass1 results from overexpression of a tandem microRNA. Nat Genet. 2007, 39 (4): 544-549.
Chuck G, Meeley R, Hake S: Floral meristem initiation and meristem cell fate are regulated by the maize AP2 genes ids1 and sid1. Development. 2008, 135 (18): 3013-3019.
Chuck G, Meeley R, Irish E, Sakai H, Hake S: The maize tasselseed4 microRNA controls sex determination and meristem cell fate by targeting Tasselseed6/indeterminate spikelet1. Nat Genet. 2007, 39 (12): 1517-1521.
Chuck G, Meeley RB, Hake S: The control of maize spikelet meristem fate by the APETALA2-like gene indeterminate spikelet1. Genes Dev. 1998, 12 (8): 1145-1154.
Irish EE: Experimental analysis of tassel development in the maize mutant tassel seed 6. Plant Physiol. 1997, 114 (3): 817-825.
Lauter N, Kampani A, Carlson S, Goebel M, Moose SP: microRNA172 down-regulates glossy15 to promote vegetative phase change in maize. Proc Natl Acad Sci U S A. 2005, 102 (26): 9412-9417.
Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J: SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009, 25 (15): 1966-1967.
Moxon S, Jing R, Szittya G, Schwach F, Rusholme Pilcher RL, Moulton V, Dalmay T: Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome Res. 2008, 18 (10): 1602-1609.
Rajagopalan R, Vaucheret H, Trejo J, Bartel DP: A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006, 20 (24): 3407-3425.
Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, Dreyfuss G, Eddy SR, Griffiths-Jones S, Marshall M, et al: A uniform system for microRNA annotation. RNA. 2003, 9 (3): 277-279.
Berezikov E, Cuppen E, Plasterk RH: Approaches to microRNA discovery. Nat Genet. 2006, 38 (Suppl): S2-S7.
Kutter C, Schob H, Stadler M, Meins F, Si-Ammour A: MicroRNA-mediated regulation of stomatal development in Arabidopsis. Plant Cell. 2007, 19 (8): 2417-2429.
Wang X, Zhang J, Li F, Gu J, He T, Zhang X, Li Y: MicroRNA identification based on sequence and structure alignment. Bioinformatics. 2005, 21 (18): 3610-3614.
Jiang P, Wu H, Wang W, Ma W, Sun X, Lu Z: MiPred: classification of real and pseudo microRNA precursors using random forest prediction model with combined features. Nucleic Acids Res. 2007, 35 (Web Server issue): W339-W344.
Ng KL, Mishra SK: De novo SVM classification of precursor microRNAs from genomic pseudo hairpins using global and intrinsic folding measures. Bioinformatics. 2007, 23 (11): 1321-1330.
Ding D, Zhang L, Wang H, Liu Z, Zhang Z, Zheng Y: Differential expression of miRNAs in response to salt stress in maize roots. Ann Bot. 2009, 103 (1): 29-38.
Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ: Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol. 2008, 18 (10): 758-762.
German MA, Pillay M, Jeong DH, Hetawal A, Luo S, Janardhanan P, Kannan V, Rymarquis LA, Nobuta K, German R, et al: Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat Biotechnol. 2008, 26 (8): 941-946.
Pantaleo V, Szittya G, Moxon S, Miozzi L, Moulton V, Dalmay T, Burgyan J: Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010, 62 (6): 960-976.
Song QX, Liu YF, Hu XY, Zhang WK, Ma B, Chen SY, Zhang JS: Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol. 2011, 11: 5-
Zhou M, Gu L, Li P, Song X, Wei L, Chen Z, Cao X: Degradome sequencing reveals endogenous small RNA targets in rice (Oryza sativa L. ssp. indica). Front Biol. 2010, 5 (1): 67-90.
Addo-Quaye C, Miller W, Axtell MJ: CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics. 2009, 25 (1): 130-131.
Ma Z, Coruh C, Axtell MJ: Arabidopsis lyrata small RNAs: transient MIRNA and small interfering RNA loci within the Arabidopsis genus. Plant Cell. 2010, 22 (4): 1090-1103.
Colasanti J, Muszynski M: The Maize Floral Transition. In: Handbook of Maize. Its Biology. Edited by: Bennetzen J, Hake S. 2009, New York: Springer, 41-55.
Nagpal P, Ellis CM, Weber H, Ploense SE, Barkawi LS, Guilfoyle TJ, Hagen G, Alonso JM, Cohen JD, Farmer EE, et al: Auxin response factors ARF6 and ARF8 promote jasmonic acid production and flower maturation. Development. 2005, 132 (18): 4107-4118.
Wu MF, Tian Q, Reed JW: Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression, and regulates both female and male reproduction. Development. 2006, 133 (21): 4211-4218.
Allen E, Xie Z, Gustafson AM, Sung GH, Spatafora JW, Carrington JC: Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat Genet. 2004, 36 (12): 1282-1290.
Mallory AC, Dugas DV, Bartel DP, Bartel B: MicroRNA regulation of NAC-domain targets is required for proper formation and separation of adjacent embryonic, vegetative, and floral organs. Curr Biol. 2004, 14 (12): 1035-1046.
Nag A, King S, Jack T: miR319a targeting of TCP4 is critical for petal growth and development in Arabidopsis. Proc Natl Acad Sci U S A. 2009, 106 (52): 22534-22539.
Park W, Li J, Song R, Messing J, Chen X: CARPEL FACTORY, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr Biol. 2002, 12 (17): 1484-1495.
Lu C, Meyers BC, Green PJ: Construction of small RNA cDNA libraries for deep sequencing. Methods. 2007, 43 (2): 110-117.
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al: The B73 maize genome: complexity, diversity, and dynamics. Science. 2009, 326 (5956): 1112-1115.
Zuker M: Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31 (13): 3406-3415.
Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J: ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009, 25 (8): 1091-1093.
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al: Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33 (20): e179-
Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP: Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant methods. 2007, 3: 12-
Lang Q, Jin C, Lai L, Feng J, Chen S, Chen J: Tobacco microRNAs prediction and their expression infected with Cucumber mosaic virus and Potato virus X. Mol Biol Rep. 2011, 38 (3): 1523-1531.
Acknowledgements
The work was supported by grants from the National Natural Science Foundation of China (31271740), the National Hi-Tech program of China (2012AA10307), and the Major Project of China on New varieties of GMO Cultivation (2011ZX08003-003).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
HL, GP, TL and ZZ designed the study. HL, CQ, ZC, TZ, XY, SC, HZ, XH, YS, YZ, and MX performed the analyses. HL, CQ, TZ, XY, TL, ZZ, and GP drafted the manuscript. All of the authors critically revised and provided final approval of this manuscript.
Hongjun Liu, Cheng Qin, Zhe Chen contributed equally to this work.
Electronic supplementary material
12864_2013_5669_MOESM2_ESM.pdf
Additional file 2: Figure S1: Step-by-step schematic representation of the strategy for maize miRNAs discovery and validation. (PDF 36 KB)
12864_2013_5669_MOESM6_ESM.xlsx
Additional file 6: Table S5: The detailed precursor sequences and exact genome locations of all maize miRNAs. (XLSX 20 KB)
12864_2013_5669_MOESM7_ESM.pdf
Additional file 7: Figure S2: The sequence conservation of mature miRNAs between members of known miR169 family and miRs4. (PDF 54 KB)
12864_2013_5669_MOESM9_ESM.pdf
Additional file 9: Figure S3: Clustering of differently expressed miRNA members during the process of ear development in maize. M-1, M-2, M-3 and M-4 represent stage I, stage II, stage III and stage IV, respectively. (PDF 183 KB)
12864_2013_5669_MOESM11_ESM.pdf
Additional file 11: Figure S4: T-plots for some miRNA and ta-siRNA targets in four developmental stages of maize ears. (PDF 1 MB)
12864_2013_5669_MOESM12_ESM.xlsx
Additional file 12: Table S8: Cleaved miRNA and ta-siRNA targets in four developmental stages of maize ears. (XLSX 124 KB)
12864_2013_5669_MOESM13_ESM.xlsx
Additional file 13: Table S9: List of targets regulated by differentially expressed miRNAs in four developmental stages of maize ears. (XLSX 38 KB)
12864_2013_5669_MOESM14_ESM.xlsx
Additional file 14: Table S10: Functional classification of targets regulated by differentially expressed miRNAs based on Gene Ontology (GO). (XLSX 15 KB)
12864_2013_5669_MOESM15_ESM.xlsx
Additional file 15: Table S11: Primers of stem-loop qRT-PCR assay used for 12 randomly selected miRNAs in this study. (XLSX 12 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Liu, H., Qin, C., Chen, Z. et al. Identification of miRNAs and their target genes in developing maize ears by combined small RNA and degradome sequencing. BMC Genomics 15, 25 (2014). https://doi.org/10.1186/1471-2164-15-25
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-25