
Abstract
Background
Alzheimer’s disease (AD) is one of the leading genetically complex and heterogeneous disorder that is influenced by both genetic and environmental factors. The underlying risk factors remain largely unclear for this heterogeneous disorder. In recent years, high throughput methodologies, such as genome-wide linkage analysis (GWL), genome-wide association (GWA) studies, and genome-wide expression profiling (GWE), have led to the identification of several candidate genes associated with AD. However, due to lack of consistency within their findings, an integrative approach is warranted. Here, we have designed a rank based gene prioritization approach involving convergent analysis of multi-dimensional data and protein-protein interaction (PPI) network modelling.
Results
Our approach employs integration of three different AD datasets- GWL,GWA and GWE to identify overlapping candidate genes ranked using a novel cumulative rank score (SR) based method followed by prioritization using clusters derived from PPI network. SR for each gene is calculated by addition of rank assigned to individual gene based on either p value or score in three datasets. This analysis yielded 108 plausible AD genes. Network modelling by creating PPI using proteins encoded by these genes and their direct interactors resulted in a layered network of 640 proteins. Clustering of these proteins further helped us in identifying 6 significant clusters with 7 proteins (EGFR, ACTB, CDC2, IRAK1, APOE, ABCA1 and AMPH) forming the central hub nodes. Functional annotation of 108 genes revealed their role in several biological activities such as neurogenesis, regulation of MAP kinase activity, response to calcium ion, endocytosis paralleling the AD specific attributes. Finally, 3 potential biochemical biomarkers were found from the overlap of 108 AD proteins with proteins from CSF and plasma proteome. EGFR and ACTB were found to be the two most significant AD risk genes.
Conclusions
With the assumption that common genetic signals obtained from different methodological platforms might serve as robust AD risk markers than candidates identified using single dimension approach, here we demonstrated an integrated genomic convergence approach for disease candidate gene prioritization from heterogeneous data sources linked to AD.
Similar content being viewed by others


Background
Alzheimer’s disease (AD) is a gradually progressive neurodegenerative disease, characterized by cognitive impairment in elderly. Genetics is known to play a major role in its development with studies showing both gene-gene and gene-environment interactions as risk factors [1, 2]. The number of people afflicted with AD is estimated to be more than 24 million worldwide, and the heritability is estimated to be 60-80% [3–5]. Over the last decade, several high throughput experimental approaches involving genome-wide linkage (GWL) scans, genome-wide association (GWA) studies, and genome-wide expression (GWE) profiling, have been extensively utilized to identify the underlying genetic risk factors. Linkage studies were instrumental in the initial identification of four genes (APP, PSEN1, PSEN2 and APOE) associated with AD [6]. Later, several other loci spanning many genes were discovered in AD using GWL scans. However, linkage studies in sporadic or late onset AD (LOAD) suffers from limitations of low resolution of results, lack of availability of large multigeneration families and inclusion of phenocopies [7].
With the advent of high throughput genotyping platforms in recent years, several GWA studies were carried out using population based case–control designs which resulted in the identification of additional AD risk genes [7, 8]. However, these studies require very large sample size specifically to detect genetic variant with small attributable risk. Additionally, case control studies are prone to issues of population stratification and population admixture. In recent years, a limited number of global gene expression profiling studies have been conducted using post-mortem AD brain tissues [9, 10]. These studies have led to identification of genes related to multiple cellular pathways known to be involved in AD pathogenesis and progression. However, the major drawback of such studies includes limited access to brain samples from AD subjects as well as age matched controls. Further, variable RNA quality due to post-mortem delay and the difficulty in establishing temporal and regional specificity of gene expression changes adds up to the limitations [11]. Although different genetic based approaches have led to the accumulation of massive amounts of data, however, due to differential limitations of each study, limited success has been achieved in identifying common underlying genetic markers related to AD progression and pathogenesis. This warrants designing of novel approaches complementing the existing ones for disease gene discovery.
In recent years, integrative approaches combining multiple data sources have been widely used to identify susceptible genes in complex disorders such as AD [12, 13], epilepsy [14], type 2 diabetes [15, 16], prostate cancer [17], depression [18], schizophrenia [19] and Parkinson’s disease (PD) [20]. Such approaches may help imbibe disease specific biological knowledge that may not be available from one dimensional approaches. Further, network modelling of gene-gene and protein-protein interactions (PPI) provides a relatively new integrative approach to understand complex disease and identify disease-related genes [21, 22]. For instance, candidate genes in complex disorders, such as AD [23–27], obstructive sleep apnea [28], heart failure [29], cancer [30] and cardiorenal syndrome [31], have already been explored extensively using PPI based approach. Thus, a convergent analysis approach involving multi-dimensional datasets combined with network or pathway analysis might serve as a comprehensive approach for disease candidate gene prioritization.
In this study, we aimed to develop a system biology approach based on genomic convergence of genetic data from multiple high-dimensional genome-wide studies and network modelling of protein-protein interactions to prioritize candidate genes linked to AD. We identified 108 common overlapping genes from integrated analysis of three datasets - GWL [8, 32, 33], GWA [34] and GWE [[35, 36]; GSE5281] and ranked them using our ranked based scoring method. We identified direct protein interactors of 108 candidate genes and then created a layered PPI network comprising of 640 nodes based on subcellular localization of proteins. Finally, we performed Markov Cluster algorithm (MCL) based clustering using clusterMaker and functional enrichment analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID) to identify functional modules and significant Gene Ontology (GO) annotation clusters, respectively [37–39]. Hence, integrating AD linkage, genetic association, and gene expression data followed by network modelling of PPI resulted in a list of evidence-based candidate genes for future experimental validation and related pathways for better understanding of underlying AD patho-physiology. This multi-dimensional evidence-based approach can be applied to other complex disorders having publically available high throughput data.
Results
The objective of this study was to identify potential candidate genes involved in AD development and progression by an integrative genomic convergence approach involving rank based scoring method. The datasets, for integrative analysis, were retrieved from AlzGene database (GWL), I-GAP (International Genomics of Alzheimer’s Project) study (GWA) and NCBI Gene Expression Omnibus (GEO) database: GSE5281 (GWE). The common overlapping genes occurring in all the three datasets were identified and ranked by cumulative rank score obtained by addition of gene ranks based on either p values or scores. The final 108 overlapping genes were used for ‘GO analysis’ and to create a layered PPI network comprising 640 nodes and 2214 edges. These identified putative proteins were then used to identify functionally important clusters and common biomarkers among plasma/serum and CSF proteome. The entire work flow is depicted in Figure 1.

Flow chart describing the entire work flow. Integrated data analysis was performed on three genome wide datasets to identify overlapping 108 AD putative candidate genes which were ranked by using cumulative rank based scoring method. These genes were further used to create a PPI and identify overlapping proteins among 108 and proteins from CSF and plasma proteome. PPI was then used to create a layered network based on the subcellular localization information of 640 genes, to identify clusters using MCL algorithm and to retrieve functional annotation using DAVID web tool.
Putative AD linked candidate genes from integrative analysis
For GWL data analysis, genomic linkage regions linked to AD were retrieved from AlzGene database with LOD scores ≥ 2.0 or p value <0.05 (1p31.1-q31.1, 3q12.3-q25.31, 6p21.1-q15, 7pter-q21.11, 8p22-p21.1, 9q21.31-q32, 10p14-q24, 17q24.3-qter, 19p13.3-qter) and used for further analysis. Among these 9 linkage regions, 7 were included from meta-analysis of five independent genome scans carried out by Butler et al. [32], using genome search meta-analysis (GSMA) approach and 2 regions from Hamshree et al. [33] that combined three large samples to give a total of 723 affected relative pairs (ARPs) and analyzed using multipoint, model-free ARP linkage analysis approach. A total of 2976 genes were retrieved using UCSC genome browser [40] from these linkage regions and genes were ranked according to their score obtained from GeneWanderer web server [41].
Further, for the GWA dataset, 19,532 single nucleotide polymorphisms (SNPs) with p value <0.0001 [34] were selected. These SNPs were mapped to their corresponding genes using NCBI Variation Reporter, SCAN (SNP and CNV Annotation) database [42] and SPOT web tool [43]. This led us to the identification of 1,686 genes which were ranked based on weighted p value obtained though genomic information network prioritization and scoring method implemented in SPOT [43]. For replication analysis, we used another GWA dataset from Boada et al. [44] which included genotyped and imputed SNPs (1,098,485) from 7 reported GWA studies comprising ~8082 cases and ~12040 controls for stage I meta-analysis. With this cohort used in stage I analysis with P < 0.001, 1202 SNPs were obtained. When candidate genes identified in the main and replication datasets were compared, we found a concordance of 35.4% (see Additional file 1).
For GWE data analysis, the GSE5281 dataset was selected and analyzed using GEO2R tool accessed from GEO web server [45]. In our study, expression data from six brain regions – entorhinal cortex (EC), hippocampus (HIP), posterior cingulate cortex (PC), middle temporal gyrus (MTG), superior frontal gyrus (SFG) and primary visual cortex (VCX), were used for analysis. The genes with adjusted p value < 0.05 and fold change ≥ 2.0 for upregulated genes and ≤ 0.5 for down regulated genes were selected from each region and then merged. This analysis resulted in 7961 genes which were ranked by their corresponding adjusted p values. For replication analysis, we used another GWE dataset - GSE15222 that comprised expression data from post-mortem brain cortical regions of 176 late-onset AD cases and 188 controls [46]. A concordance of 58.2% was found between GSE5281 and GSE15222 datasets after analysis (see Additional file 1).
The intersection of all the three datasets resulted in the final set of 108 putative candidate genes (Figure 2) and their individual ranks were added to get score. Based on this rank score the genes were re-ranked with gene having the lower cumulative rank score getting the higher rank. The top 10 genes are listed in Table 1 and the list of 108 genes is provided in Additional file 2.

Venn diagram of putative overlapping AD candidate genes among different genome wide datasets. The venn diagram represents the genes in the three individual datasets and overlapping 108 putative AD target genes identified by integrated analysis of the three datasets.
As all the six brain regions are found to be associated with AD pathology with different degree of involvement depending upon disease severity, we analysed expression profile data of each region separately and obtained candidate genes specific in each brain region. We identified 25, 16, 40, 38, 27 and 1 candidate genes specific in EC, HIP, PC, MTG, SFG and VCX brain regions, respectively, from overlap with GWA and GWL repertoires (see Additional files 3 and 4).
Protein-protein interaction network, layering and network analysis
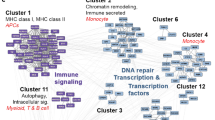
Identification of proteins that interact directly with proteins encoded by identified 108 target genes might help elucidate the molecular mechanism underlying AD patho-physiology. Thus, in the present study, we created a PPI network from the 108 candidate genes using APID2NET plugin in Cytoscape [47, 48] comprising 640 nodes and 2214 edges. Then, a layered network based on the sub-cellular localization information of 640 proteins using “Cerebral” plugin [49] in Cytoscape was obtained from the PPI network. The layered network is depicted in Figure 3. Further, another cytoscape plugin “clusterMaker” [37] was used on the PPI to create clusters using MCL clustering algorithm [50]. This resulted in the identification of 6 important clusters with 7 proteins (EGFR, ACTB, CDC2, IRAK1, APOE, ABCA1 and AMPH) forming the central hub nodes (Figure 4a-f). All 63 clusters obtained from MCL clustering are provided in Additional file 5.

Layered Protein-Protein Interaction network (PPI) of 108 proteins. A layered network based on the subcellular localization of 640 proteins in the PPI was created. The nodes representing functionally important genes were highlighted in the layered network using colour codes - green (genes forming hub nodes in clusters (7), occurring in top 15 of ranked genes (108) and also present in putative biomarker dataset (38)); cyan (genes forming hub nodes in clusters, occurring in 108 AD genes and also present in putative biomarker dataset); yellow (genes occurring both in cluster hub and in 108 ranked genes); pink (genes forming hub nodes in clusters and occurring in top 15 of ranked genes); blue (remaining 59 from 108 list); red (38 biomarkers from AD, CSF and plasma overlap); grey (remaining proteins from 640 candidates).

Important clusters obtained from clustering of 640 proteins using MCL algorithm in clusterMaker. (a-f) Biologically significant gene clusters were identified from PPI using MCL algorithm. The nodes representing functionally important genes were coloured in the pattern described for the layered network.
Functional annotation analysis by GO terms
We performed functional GO enrichment analysis of the 108 AD candidate genes, using functional annotation clustering tool implemented in DAVID [38, 39], to identify association of candidate genes with different ‘GO terms’. The significantly over represented ‘GO terms’, identified neurogenesis (p = 0.0032) as the top cluster, followed by regulation of neurogenesis (p = 0.0062). The other significantly over represented biological processes included peptidyl tyrosine phosphorylation (p = 0.0041), cytoplasmic membrane-bounded vesicles (p = 0.006), regulation of MAP kinase activity (p = 0.0005), kinase activity (p = 0.0081), purinergic nucleotide receptor activity, G-protein coupled (p = 0.0153), neuron development (p = 0.0098), response to calcium ion (p = 0.0067), sensory perception of light stimulus (p = 0.0041), endocytosis (p = 0.0192) (Figure 5). This analysis was also repeated for 640 candidate genes (Additional file 6).

Clustering of GO terms: significantly over represented top 11 functionally annotated clusters from biological process, cellular component and molecular function of 108 proteins.
AD putative biochemical biomarkers
In this study, we also looked for the identification of cerebrospinal fluid (CSF) and plasma based AD specific biomarker and found 3 common proteins (APOE, EGFR, ACTB) among 108 AD proteins and proteins from CSF and plasma proteome (Figure 6) and 38 common proteins among 640 putative AD proteins and proteins from CSF and plasma proteome (Additional file 7), which might serve as potential biochemical biomarkers for early detection of AD cases in future.

Putative AD specific biomarkers. The venn diagram depicts overlap among putative 108 AD proteins, proteins from CSF and plasma proteome.
Validation of Rank based approach by using PD datasets
For validation of our rank based gene prioritization approach, we selected PD, another common complex neurodegenerative disorder that involves the deposition of α-synuclein as intracellular Lewy bodies leading to progressive degeneration of dopaminergic neurons within multiple brain regions. It clinically manifests as both a movement disorder, characterized by tremor, rigidity, bradykinesia and postural instability and a distinct form of cognitive impairment, characterized by visuospatial impairment and fluctuations in mental state [51, 52]. We applied our rank based method to identify overlapping genes in three PD datasets – GWL, GWA, and GWE. We retrieved GWL dataset from PDgene database (http://www.pdgene.org/) [53]. It included genetic loci showing evidence for linkage in the meta-analysis of five GWL scans comprising 862 families with 1384 affected subjects using the GSMA method by Rosenberger et al. [54].
For GWA dataset, we retrieved SNPs with pre-computed p values from a NCBI dbGaP database with study accession: phs000089.v3.p2 (http://www.ncbi.nlm.nih.gov/gap) [55]. This dataset comprises PD cases drawn from population of North American Caucasians, and neurologically normal controls from the population which are banked in the National Institute of Neurological Disorders and Stroke (NINDS Repository) collection for a stage I genome wide analysis. Initially, genome-wide, SNP genotyping of these samples was carried out in 267 PD subjects and 270 controls, and later extended to include genotyping in 939 PD cases and 802 controls. This collection was included in the first stage study by Fung et al. [56], and the expanded study by Simon-Sanchez et al. [57, 58]. A total of 7,943 SNPs (stage I) were selected for further analysis, with p value < 0.01, from raw data comprising total of 453,217 SNPs.
For GWE dataset, we selected the gene expression data from NCBI GEO database (GSE20295) (http://www.ncbi.nlm.nih.gov/geo) [59] for analysis. It contained gene expression profiling data in post-mortem tissue of three brain regions (the substantia nigra, putamen, and Brodmann’s area 9) from matched groups of 15 neuropathologically confirmed PD and 15 controls with no history of major brain illness.
The analysis of three PD datasets using rank based scoring method led us to the identification of 59 putative target genes from the overlap of 1528 genes from GWL, 2882 genes from GWA and 2923 genes from GWE which could have significant association with PD development and progression (Figure 7a). The entire list of 59 genes is provided in Additional file 8. The comparison of 108 AD and 59 PD putative candidate genes resulted in only 2 common genes (ABCA1 and LPAR1) between the two groups (Figure 7b).

Prioritized putative PD candidate genes and overlap with prioritized AD candidate genes. (a) The venn diagram represents the genes in the three individual datasets and overlapping 59 putative PD target genes identified by integrated analysis of the three datasets. (b) The venn diagram represents the overlapping genes among AD and PD putative target genes.
Discussion
AD is a complex polygenic disorder with lack of understanding of natural course of the disorder and absence of reliable biomarkers that can predict disease onset and progression. Although, genome-wide studies, such as genetic linkage, association and expression, have allowed unbiased identification of candidate genes and pathways associated with AD development and progression, susceptibility loci or genes for AD with clinical significance have not yet been reported. This can be attributed to certain limitations associated with these methods. For instance, linkage studies require large, multi-generational pedigrees within which both affected and unaffected individuals are required for testing and even in such cases, this approach yields only regions of linkage and not the causative gene [60–62].This suggests that only a fraction of the genes, significant in these analyses, are causal genes. On the other hand, GWA studies often lack statistical power to detect SNPs with small effect size and therefore cannot detect all causal genes [63]. Further, in case of gene expression studies, identified genes are expected to contain a mix of causal and the differentially expressed genes because of the ripple effect of the causal genes [64]. The huge amount of AD specific genetic data accumulated in the past decade also indicates involvement of multiple pathways wherein each gene confers only a modest risk. Therefore, integration of datasets from multiple disciplines may lead to identification of candidate genes from different pathways and may provide an opportunity to uncover the biological functions and molecular mechanisms underlying AD through PPI network and GO analysis.
In this work, we presented a framework for integrated analysis of multi-dimensional datasets by using a rank based scoring method. First, we retrieved and analysed data from three datasets –GWL, GWA and GWE, based on the assumption that genes identified by all the three experimental technique might be significantly involved in AD pathology. Then, we used a ranked based method in which overlapping genes were first identified in all the three datasets and then each gene was assigned cumulative rank score (SR), based on addition of corresponding rank in individual datasets. The genes in each datasets were ranked based on either p values or scores. Finally, the genes were ranked based on their SR with gene having lower SR getting the higher rank. This analysis led us to the identification of 108 ranked genes from the overlap of 2729 genes from GWL, 1686 genes from GWA and 7961 genes from GWE which might serve as putative target genes having significant association with AD development and progression.
A majority of top ranked putative candidate genes have been found to be significantly associated with molecular mechanism and pathways related to AD development and progression and may serve as critical candidates for predicting AD risk. For instance, first ranked RPN1 gene encodes for a type I integral membrane protein, ribophorin that have been shown to directly interact with opioid receptors (OR). Overexpression of RPN1 is reported to enhance cell surface expression of δOR and μOR but not that of κOR [65]. Significant reductions in μOR binding are observed in the subiculum and HIP regions of brain from AD cases as compared to controls. Further, binding of δOR is also found to be decreased in the amygdala and putamen of AD brains [66]. In addition, δOR have been associated with increased processing of amyloid beta (Aβ) precursor protein (APP) by BACE1 and γ-secretase, but not that of Notch, N-cadherin or APLP1. Moreover, knockdown or blocking of δOR in AD mouse model decreases secretase activities and abolishes Aβ pathology and Aβ-dependent behavioral abnormalities [67]. Second ranked gene, RGS4, encodes for regulator of G protein signalling 4 protein, is reported to be involved in neuronal calcium dependent signaling, a cellular process related to both AD and aging [68]. In parietal cortex of AD subjects, 53% and 40% lower levels of RGS4 and Gq/11 proteins is found as compared to age-matched controls. Further, it was proposed that alteration of dynamic equilibrium between the cytosolic and membrane levels of RGS4 and Gq/11 may lead to the regional differences in the coupling of muscarinic M1 receptors in AD which in turn may lead to variable response to currently available cholingeric treatment strategies [69]. HIP1 gene encodes for Huntingtin interacting protein 1 (HIP1) that is predominantly expressed in brain and is proposed as a novel brain tumor marker that interacts with EGFR [70]. In a published genome-wide study of aging, rs17149227 (p value < 10-5) close to HIP1 gene, is found to be associated for time to death from meta-analysis of 9 cohorts [71]. Mills et al. (2005) proposed that transcriptional deregulation of HIP1 may play a significant role in the pathogenesis of neurodegenerative diseases [72].
A recently found strong LOAD candidate is PTK2B/CAKB/FAK2/PYK2 gene that encodes for a cytoplasmic protein tyrosine kinase, which is highly expressed in the CNS, particularly in HIP [73]. Aβ fibrils has been shown to induce THP-1 cells resulting in the stimulation of PYK2 tyrosine phosphorylation as a consequence of Lyn and Syk activation, intracellular calcium release, and PKC stimulation [74]. Activation of CAKb/Pyk2 is required for inducing long-term potentiation (LTP) in CA1 HIP neurons which may depend upon downstream activation of Src to upregulate N-methyl-D-aspartate-type (NMDA) glutamate receptors [75–77]. Further, in the case of AD, the immunoreactivity for c-Jun is found to be elevated in diseased brain [78, 79] and interestingly, PYK2 represents a stress sensitive mediator of c-Jun N-terminal kinase (JNK) signaling pathways.
ICA1 encodes for 69 kDa islet cell autoantigen, a BAR (Bin/amphiphysin/Rvs)-domain-containing protein with highest expression levels in brain, pancreas, and stomach mucosa [80]. It is identified as the major binding partner of protein PICK1 (protein interacts with C-kinase 1). ICA1 regulates AMPA receptor trafficking, an important mechanism underlying synaptic plasticity, by forming heteromeric ICA69-PICK1 complexes and preventing formation of PICK1- PICK1 homomeric complexes [81]. Spitzenberger et al. demonstrated that mutation of ICA69 homologue gene ric-19 in C. elegans leads to impairment of acetylcholine release at neuromuscular junctions suggesting role of ICA69 in neuroendocrine secretion [82]. AMPH1 gene encodes for protein amphiphysin I, an important regulator for synaptic vesicle endocytosis (SVE) when massive amounts of Ca2+ flow into presynaptic terminals, a phenomenon observed in AD [83]. In AMPH1 knockout mice, decreased synaptic vesicle recycling efficiency and cognitive deficits has been observed [84]. In a recent study, AMPH1 level is found to be reduced in AD brain regions known to accumulate aggregates of hyperphosphorylated tau proteins [85]. Further, stimulated neurons are also shown to abnormally accumulate amphiphysin, at the membrane during Aβ treatment [86].
Interestingly, ATP5H/KCTD2 locus is reported as the major candidate gene associated with AD pathogenesis in the study by Boada et al. [44] that is used in this study as the replication dataset. ATP5H gene encodes for mitochondrial ATP synthase that plays an important function in mitochondrial energy production and neuronal hyperpolarization during cellular stress conditions, such as hypoxia or glucose deprivation [44]. EGFR gene encodes for epidermal growth factor receptor protein, a cell surface protein that binds to epidermal growth factor. It has been put forward as a preferred target for treating amyloid beta induced memory loss in a recent study by Wang L et al. [87]. Interestingly, it has come up as one of the most significant candidate in our study occurring in top 10 ranked genes among 108 candidates, as central hub node in cluster and in the overlap of AD protein and proteins from plasma and CSF proteome. Increased expression of EGFR is observed in fibroblasts deficient in PS/gamma-secretase activity or APP expression [88]. Further, studies also indicate role of PS1 in trafficking and turnover of EGFR as well as perturbed endosomal-lysosomal trafficking in cell cycle control and Alzheimer disease and suggest potential pathogenic effects of elevated EGFR [89]. In a recent study, altered EGFR transcript levels are reported among APOE4 (high risk) when compared to APOE3 (low risk) genotype groups [90].
A major candidate gene for LOAD due to its role in cholesterol transport and metabolism is ABCA1 gene that encodes for ATP-binding cassette transporter A1, a membrane-associated protein. Increased expression of ABCA1 is highly correlated with severity of dementia in AD HIP [91]. Further, ABCA1 has been shown in mouse models of AD to enable the clearance of Aβ from the brain, through its role in the apolipoprotein (APOE) lipidation in the CNS [92–95]. In APP transgenic mice, ABCA1 deficiency increased Aβ deposition in the brain paralleled by decreased levels of ApoE [96]. In addition, ABCA1 is also found to be up-regulated in primary mouse cortical neurons and cultured astrocytes in response to oligomeric Aβ42 [97, 98]. Recent studies pointed out that ABCA1 mediates the beneficial effects of the liver X receptor (LXR) agonist GW3965 on object recognition memory and amyloid burden in APP/PS1mice [99, 100]. Based on strong evidence the LXR-ABCA1-APOE regulatory axis is now considered a promising therapeutic target in AD [101]. However, a meta-analysis report of 13 studies involving a total of 12,248 subjects failed to find association of common SNPs in ABCA1 with AD risk [102]. In contrast, Lupton et al. in a very recent study sequenced all ABCA1 coding regions in 311 LOAD cases and 360 control individuals of Greek ethnicity and observed significantly higher proportion of rare non-synonymous variants in control individuals compared to AD cases. These findings suggest that high throughput sequencing may identify rare variants that are left undetected by GWAS [92]. ACTB gene encodes for protein β-actin. It is found to have the worst candidate with reliable expression among a set of suitable endogenous reference genes (ERG) in human post-mortem brain when used for the expression analysis of potential candidate genes associated with AD [103]. ACTB was found to be upregulated by 10.2 folds in AD cerebral cortex compared with age-matched control brain [104]. Further, immunoprecipitation of proteins from AD and control brain showed oxidative modification of β-actin in the AD brain [105]. In addition, β-Secretase-cleaved APP is shown to accumulate at actin inclusions in neurons induced by stress or Aβ [106]. Several recent studies also indicate that abnormalities of actin cytoskeleton may play a critical role in AD pathology by mediating synaptic degeneration [107, 108].
We aimed to identify direct protein interactors of proteins encoded by identified 108 candidate genes by PPI network modelling with an assumption that they might provide important biological information related to molecular mechanisms underlying AD development and progression. PPI network was obtained by using APID2NET plugin in Cytoscape and included 640 protein nodes and 2214 edges. It was converted to a layered network based on subcellular localization information. We observed that majority of the proteins were localized in cytoplasm followed by nucleus. Further, we applied MCL clustering algorithm to identify functional modules with proteins forming hub nodes (EGFR, ACTB, CDC2, IRAK2, APOE, ABCA1 and AMPH) which might serve as important candidates related to AD [50]. For instance, CDC2 [109, 110], IRAK2 [111] have been reported in recently published studies with suggestive role in AD pathogenesis. GO analysis was also carried out using 108 genes to identify biological processes, molecular functions and cellular components. Top 11 annotation clusters with enrichment score > 1.3 included genes involved in diverse biological processes related to AD such as neurogenesis (DFNB31, PTK2B, RET, DLL3, APOE, CRX, ACTB, NRP1, LMX1A, PIP5K1C, ZNF488), regulation of neurogenesis (DLL3, APOE, CRX, NRP1, LMX1A, ZNF488), peptidyl tyrosine phosphorylation (TYK2, PTK2B, DDR2, SYK), cytoplasmic membrane-bounded vesicles (PLA2G4A, ABCA1, HIP1, AMPH, HGF, SFTPD, EGFR, ICA69, ATP8B3, RPN1), regulation of MAP kinase activity (PTK2B, LPAR1, APOE, RGS4, HGF, EGFR, SYK), kinase activity (PTK2B, TYK2, NME8, DDR2, NRP1, PIP5K1C, EGFR, RET, PAK4, IPMK, POLR2E, ADK, SYK), purinergic nucleotide receptor activity, G-protein coupled (SUCNR1, P2RY12, P2RY14), neuron development (DFNB31, PTK2B, RET, ACTB, NRP1, LMX1A, PIP5K1C, EGFR), response to calcium ion (PLA2G4A, PTK2B, ACTB, EGFR), sensory perception of light stimulus (DFNB31, RGS16, PCDH15, CRX, RIMS1, ELOVL4, OPN5), endocytosis (ABCA1, APOE, HIP1, AMPH, ELMO1, SFTPD).
In addition, potential CSF and plasma/serum based biomarkers were identified from the overlap of 108 and 640 AD proteins separately with proteins from CSF and plasma proteome. This resulted in the identification of 3 proteins and 38 proteins as potential biochemical biomarkers for AD among 108 and 640 identified protein datasets, respectively. Among these proteins, the CSF or plasma level of, APOE [112–120], EGFR [121] proteins have been reported to be altered in previous AD studies.
For validation of our approach, we have applied our rank based scoring method to identify PD candidate genes using three (GWL, GWA and GWE) datasets and then we compared PD candidate genes with those identified in analysis of AD datasets to check the robustness of our approach. We failed to find significant overlap in genes between AD and PD dataset in our study, which is further substantiated by a recent meta-analysis carried out by Moskvina et al. that combined the AD and PD GWA studies and failed to identify any significant evidence to support a common genetic risk between AD and PD [122]. Further, the author failed to find loci that associate with increased risk of causing both PD and AD. In addition, it is proposed that the pathological overlap among AD and PD proteins may occur at a later stage during disease progression suggesting interaction of genes from downstream cascade with susceptibility genes that increase the risk of each disease [122]. Few studies investigated simultaneous co-occurrence of AD and PD in families but yielded inconsistent results. In general, studies have reported either no risk of AD in the relatives of subjects with PD or an increased risk of AD in younger subjects with PD or those with cognitive impairment [123–125].
The recent association of several genes identified in our study to AD provides an immediate support of our work and prioritization of such candidates clearly indicates the efficiency and importance of our method. Our approach provides a list of AD candidate genes that are promising for further analysis by exploration of biological functions. The other most common candidate gene prioritization approaches use single-dimentional data-source and are based on direct PPI of the genes that are being studied. However, currently only ~10% of all human PPI have been described which is a major drawback of these approaches [126]. Here, we have tried to address these issues by using multi-dimensional data and exploiting the clustering of PPI network for identification of functional modules. Still, the limitations of our study include constraints in the gene annotation in the selected linkage regions and the availability of raw genome-wide data. Owing to these limitations, it is possible that a few putative candidate genes may have been missed out in this study during the screening process. Further, extensive experimental validation of candidate genes from our analysis is warranted in future.
Conclusion
To achieve better identification of complex disease associated genes, it is imperative to use integrative approach with disease specific methodologies. We performed integrated analysis of three different datasets – GWL,GWA and GWE and developed a rank based scoring method which resulted in the identification of 108 putative AD candidate genes. Further, network analysis led to a PPI with 640 nodes and clustering of this network resulted in 6 significant clusters with 7 genes forming central hub nodes. Finally, 3 biochemical biomarkers were also identified from the overlapping genes between 108 AD proteins and proteins in CSF-plasma proteome. EGFR and ACTB were found to be the two most significant AD risk genes ranked 8 and 10 among 108 genes respectively, present as central hub nodes in respective clusters and also as potential biochemical biomarker. We believe that our findings would provide a wealth of information for future experimental and clinical validation in AD pathogenesis and therapeutics.
Methods
Genetic linkage data retrieval and processing
We used linkage regions from AlzGene database which were based on the results of meta-analyses [32] and combined analysis [33] of previously published genome-wide linkage (GWL) data. In our study, linkage regions with LOD scores ≥ 2.0 or p value <0.05 linked to AD were selected for further analysis. The chromosomal coordinates for each linkage region were retrieved using UCSC genome browser. These were then used to extract genes from GeneWanderer web server [41] which provides a method for prioritization of candidate genes by using four different ranking strategies (random walk, diffusion kernel, shortest path and direct interaction) on a PPI network. We used random walk since it has been showed to outperform the others [41, 127, 128].
Genome wide association data retrieval and processing
We used SNPs with pre-computed p values from a recently published GWA study performed under the International Genomics of Alzheimer’s Project (I-GAP) banner [34]. The data are available for download from the following link: http://www.pasteur-lille.fr/en/recherche/u744/Igap_stage1.zip. The study performed meta-analysis on genotyped and imputed data (7,055,881 SNPs) from 4 previously published GWAS [ADGC, CHARGE, EADI, GERD consortium datasets] comprising 17,008 cases and 37,154 controls (stage 1). A total of 19,532 SNPs were found to be associated with AD risk and having p value < 1 × 10-3 after stage 1 meta-analysis. For replication analysis, we have used another GWA dataset from Boada et al. [44] that included genotyped and imputed SNPs (1,098,485) from 7 reported GWAS (Antúnez et al. [129], TGEN [130], ADNI [131], genADA [132], NIA [133], Pfizer [134], GERAD [135]) comprising ~8082 cases and ~12040 controls for stage I meta-analysis. With this cohort used in stage I analysis with P < 0.001, 1202 SNPs were obtained. These data are available as Supplementary Table S4 in the study by Baoda et al. [44].
The SNPs were mapped to genes using NCBI variation reporter tool, SCAN database [42] and SPOT tool [43]. SNPs, which remained unmapped, were excluded from further analysis. SPOT tool implements the Genomic Information Network prioritization method and provides a prioritization score that represents an order of magnitude change in p value from a test for association. SPOT score takes into account SNPs functional properties (including nonsense, frameshift, missense and 5’ and 3’-UTR designations), impact of an amino acid substitution on the properties of the protein product from PolyPhen server [136, 137], evolutionary conserved regions from ECRbase [138], all possible LD proxies - SNPs with r2 over a predefined threshold in a specific HapMap sample [139].
Gene expression data retrieval and processing
We retrieved the gene expression data from NCBI GEO (GSE5281) database (http://www.ncbi.nlm.nih.gov/geo) [38]. It contained expression data from six functionally and anatomically distinct regions in human brains, including EC, HIP, MTG, PC, SFG and VCX. The data included 161 samples and each brain region contains AD cases versus normal controls. GEO2R web application, available at http://www.ncbi.nlm.nih.gov/geo/geo2r/, was used for R-based analysis of GEO data [38]. The numbers of samples in each region of control/affected cases were 13/10 of EC, 13/10 of HIP, 12/16 of MTG, 13/9 of PC, 11/23 of SFG and 12/19 of VCX. In our study data from all 6 regions were analysed separately and then merged. For replication analysis, we used another GWE dataset - GSE15222 that comprised expression data from post-mortem brain cortical regions of 176 late-onset AD cases and 188 controls. On the GEO2R web interface, after the GSE5281 series were specified, a table populated with sample characteristics appears. The AD and control sample groups were designated to compare for each brain region separately. Default analysis setting with Benjamini & Hochberg (False discovery rate) for p value adjustments was used.
Probe sets that were not associated with known genes were removed from further analysis. If multiple probe sets represented the same gene and they showed same direction of expression, the probe set with the highest variance was used. If the direction of expression for multiple probe set was different then they were excluded from further analysis. The genes with adjusted p value < 0.05 and fold change ≥ 2.0 for upregulated genes and ≤ 0.5 for downregulated genes were selected. The genes from the six brain regions were merged and duplicates were removed.
Filtering and scoring of genes from data sets
The genes in all the three datasets were assigned HGNC (HUGO Gene Nomenclature Committee) ids separately [140]. The pseudogene, hypothetical, loci, non-coding RNA, non-protein coding genes, non-functional proteins, open reading frames (orf), chromosome X (Xp; Xq), withdrawn entries, antisense RNA, microRNA, uncharacterized genes were excluded from each data set for further analysis. The genes were ranked in GWL and GWA datasets by score and weighted p value obtained through GeneWanderer [41] and SPOT web servers [43], respectively. The genes in GWE dataset were ranked by adjusted or unadjusted p value obtained after analysis with GEO2R web tool. The genes, with higher weight or lower p value, were assigned higher ranks. The genes, appearing in all the three dataset, were identified and cumulative rank score for each gene was calculated using the following equation -
where,
Based on their rank score the genes were re-ranked with one having the lower cumulative rank score getting the higher rank.
Protein-protein Interaction network, layering and network analysis
To identify the direct interacting partners of the putative genes identified in this study from integrative analysis of three different data types, we built a PPI network using plugin APID2NET in Cytoscape version 2.8.1 [48] as described by Silla et al. [141]. Briefly, the APID2NET (APID) server creates PPI network of user-provided proteins using literature-curated protein interaction information from various databases such as BIND, BioGrid, DIP, HPRD, IncAct and MINT. UniProt ids of the 108 putative AD target genes were retrieved using uniprot id mapping tool (http://www.uniprot.org) [142] and provided as input ids in APID server to build the interaction network. For creating a PPI network, we first considered only those interactions supported by at least two experimental validations in order to minimize false-positive interactions. However, for proteins lacking interacting partners validated by two experiments, the interacting partners with one experimental validation were considered resulting in another PPI network. Three Cytoscape tools viz Advance Network Analyzer [143], Cerebral [52] and clusterMaker [37] were then applied for modelling PPI network. The Advanced Network Merge was used to model a final PPI network by taking union of both the PPI networks and for removal of duplicated edges and self loops. Isolated nodes were also manually removed from the final PPI network. Protein sub-cellular localization information for 635 proteins were retrieved from uniprot database [142] and for remaining 56 genes from human protein atlas [144] which were imported as node attributes in cytoscape. Then Cytoscape plugin “Cerebral” v.2.8.2 was applied to the final network to layout all nodes according to their sub-cellular localization such as plasma membrane, cytoplasm, nucleus, golgi apparatus, extracellular matrix, endoplasmic reticulum (ER), lysosome and mitochondria. Further, Markov Cluster algorithm (MCL) [50, 145] which was implemented in the “clusterMaker” v.1.9 plugin [37] in cytoscape was used on the PPI to create clusters with the hub nodes. The MCL algorithm has been used specifically to cluster simple graphs and weighted graphs by calculating successive powers of the associated adjacency matrix also called as Markov matrices which capture the mathematical concept of random walks on a graph [50].
GO annotation analysis
To assess the identified candidate genes in the context of GO, the DAVID functional annotation tool (version 6.7) [38, 39] was used. The functional annotation clustering of significantly over-represented GO term: cellular compartment (CC), molecular function (MF) and biological process (BP) was retrieved by using options GOTERM_CC_ALL, GOTERM_MF_ALL and GOTERM_BP_ALL. The default setting parameters and multiple corrections by the Benjamini method were used to determine the significant enrichment score of 1.3 [38, 39].
AD putative biochemical biomarkers analysis
To identify putative biochemical biomarker associated with AD, CSF and plasma proteins were retrieved from Sys-BodyFluid Database [146]. The 108 target genes were mapped to their corresponding uniprot ids using ID mapping tool available at http://www.uniprot.org[142]. The venn diagram of the overlapping proteins in all the three datasets (GWL,GWA and GWE) was created using GeneVenn tool [147] by taking intersection among these data sets.
Abbreviations
- AD:
-
Alzheimer’s disease
- GWE:
-
Genome Wide Expression
- GWL:
-
Genome Wide Linkage
- GWA:
-
Genome Wide Association
- APP:
-
Amyloid beta (A4) precursor protein
- PSEN1:
-
Presenilin 1
- PSEN2:
-
Presenilin 2
- CDC2:
-
Cyclin-dependent kinase 1
- IRAK1:
-
Interleukin-1 receptor-associated kinase 1
- CSF:
-
Cerebrospinal fluid
- LOAD:
-
Late onset Alzheimer’s disease
- PPI:
-
Protein-Protein Interaction
- SNP:
-
Single nucleotide polymorphism
- MCL:
-
Markov Clustering algorithm
- GO:
-
Gene Ontology.
References
Crews L, Masliah E: Molecular mechanisms of neurodegeneration in Alzheimer's disease. Hum Mol Genet. 2010, 19 (R1): R12-R20. 10.1093/hmg/ddq160.
Ghebranious N, Mukesh B, Giampietro PF, Glurich I, Mickel SF, Waring SC, McCarty CA: A pilot study of gene/gene and gene/environment interactions in Alzheimer disease. Clin Med Res. 2011, 9 (1): 17-25. 10.3121/cmr.2010.894.
Mayeux R, Stern Y: Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012, 2 (8): doi:10.1101/cshperspect.a006239
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, Pedersen NL: Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006, 63 (2): 168-174. 10.1001/archpsyc.63.2.168.
Pedersen NL, Posner SF, Gatz M: Multiple-threshold models for genetic influences on age of onset for Alzheimer disease: findings in Swedish twins. Am J Med Genet. 2001, 105 (8): 724-728. 10.1002/ajmg.1608.
Ertekin-Taner N: Genetics of Alzheimer's disease: a centennial review. Neurol Clin. 2007, 25 (3): 611-667. 10.1016/j.ncl.2007.03.009. v,
Guerreiro RJ, Gustafson DR, Hardy J: The genetic architecture of Alzheimer's disease: beyond APP. PSENs and APOE. Neurobiol Aging. 2012, 33 (3): 437-456. 10.1016/j.neurobiolaging.2010.03.025.
Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE: Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007, 39 (1): 17-23. 10.1038/ng1934.
Wang X, Michaelis ML, Michaelis EK: Functional genomics of brain aging and Alzheimer's disease: focus on selective neuronal vulnerability. Curr Genomics. 2010, 11 (8): 618-633. 10.2174/138920210793360943.
Reddy PH, McWeeney S: Mapping cellular transcriptosomes in autopsied Alzheimer's disease subjects and relevant animal models. Neurobiol Aging. 2006, 27 (8): 1060-1077. 10.1016/j.neurobiolaging.2005.04.014.
Tsapakis EM, Basu A, Aitchison KJ: Transcriptomics and proteomics: advancing the understanding of psychiatric pharmacogenomics. Clinical Neuropsychiatry. 2004, 1 (2): 117-124.
Hallock P, Thomas MA: Integrating the Alzheimer's disease proteome and transcriptome: a comprehensive network model of a complex disease. OMICS. 2012, 16 (1–2): 37-49.
Raj T, Shulman JM, Keenan BT, Chibnik LB, Evans DA, Bennett DA, Stranger BE, De Jager PL: Alzheimer disease susceptibility loci: evidence for a protein network under natural selection. Am J Hum Genet. 2012, 90 (4): 720-726. 10.1016/j.ajhg.2012.02.022.
Jia P, Ewers JM, Zhao Z: Prioritization of epilepsy associated candidate genes by convergent analysis. PLoS One. 2011, 6 (2): e17162-10.1371/journal.pone.0017162.
Jain P, Vig S, Datta M, Jindel D, Mathur AK, Mathur SK, Sharma A: Systems biology approach reveals genome to phenome correlation in type 2 diabetes. PLoS One. 2013, 8 (1): e53522-10.1371/journal.pone.0053522.
Sharma A, Chavali S, Tabassum R, Tandon N, Bharadwaj D: Gene prioritization in Type 2 Diabetes using domain interactions and network analysis. BMC Genomics. 2010, 11: 84-10.1186/1471-2164-11-84.
Gorlov IP, Gallick GE, Gorlova OY, Amos C, Logothetis CJ: GWAS meets microarray: are the results of genome-wide association studies and gene-expression profiling consistent? Prostate cancer as an example. PLoS One. 2009, 4 (8): e6511-10.1371/journal.pone.0006511.
Jia P, Kao CF, Kuo PH, Zhao Z: A comprehensive network and pathway analysis of candidate genes in major depressive disorder. BMC Syst Biol. 2011, 5 (3): S12-
Sun J, Jia P, Fanous AH, Webb BT, van den Oord EJ, Chen X, Bukszar J, Kendler KS, Zhao Z: A multi-dimensional evidence-based candidate gene prioritization approach for complex diseases-schizophrenia as a case. Bioinformatics. 2009, 25 (19): 2595-6602. 10.1093/bioinformatics/btp428.
Maver A, Peterlin B: Positional integratomic approach in identification of genomic candidate regions for Parkinson's disease. Bioinformatics. 2011, 27 (14): 1971-1978. 10.1093/bioinformatics/btr313.
Wang X, Gulbahce N, Yu H: Network-based methods for human disease gene prediction. Brief Funct Genomics. 2011, 10 (5): 280-293. 10.1093/bfgp/elr024.
Bakir-Gungor B, Sezerman OU: A new methodology to associate SNPs with human diseases according to their pathway related context. PLoS One. 2011, 6 (10): e26277-10.1371/journal.pone.0026277.
Liang D, Han G, Feng X, Sun J, Duan Y, Lei H: Concerted perturbation observed in a hub network in Alzheimer's disease. PLoS One. 2012, 7 (7): e40498-10.1371/journal.pone.0040498.
Regan K, Wang K, Doughty E, Li H, Li J, Lee Y, Kann MG, Lussier YA: Translating Mendelian and complex inheritance of Alzheimer's disease genes for predicting unique personal genome variants. J Am Med Inform Assoc. 2012, 19 (2): 306-316. 10.1136/amiajnl-2011-000656.
Liu ZP, Wang Y, Zhang XS, Chen L: Identifying dysfunctional crosstalk of pathways in various regions of Alzheimer's disease brains. BMC Syst Biol. 2010, 4: S11-10.1186/1752-0509-4-S2-S11.
Liu ZP, Wang Y, Zhang XS, Xia W, Chen L: Detecting and analyzing differentially activated pathways in brain regions of Alzheimer's disease patients. Mol Biosyst. 2011, 7 (5): 1441-1452. 10.1039/c0mb00325e.
Goni J, Esteban FJ, de Mendizabal NV, Sepulcre J, Ardanza-Trevijano S, Agirrezabal I, Villoslada P: A computational analysis of protein-protein interaction networks in neurodegenerative diseases. BMC Syst Biol. 2008, 2: 52-10.1186/1752-0509-2-52.
Liu Y, Patel S, Nibbe R, Maxwell S, Chowdhury SA, Koyuturk M, Zhu X, Larkin EK, Buxbaum SG, Punjabi NM, Gharib SA, Redline S, Chance MR: Systems biology analyses of gene expression and genome wide association study data in obstructive sleep apnea. Pac Symp Biocomput. 2011, 14-25.
Zhu W, Yang L, Du Z: Layered functional network analysis of gene expression in human heart failure. PLoS One. 2009, 4 (7): e6288-10.1371/journal.pone.0006288.
Sun J, Zhao Z: A comparative study of cancer proteins in the human protein-protein interaction network. BMC Genomics. 2010, 11 (3): S5-
Muhlberger I, Moenks K, Bernthaler A, Jandrasits C, Mayer B, Mayer G, Oberbauer R, Perco P: Integrative bioinformatics analysis of proteins associated with the cardiorenal syndrome. Int J Nephrol. 2010, 2011: 809378-
Butler AW, Ng MY, Hamshere ML, Forabosco P, Wroe R, Al-Chalabi A, Lewis CM, Powell JF: Meta-analysis of linkage studies for Alzheimer's disease–a web resource. Neurobiol Aging. 2009, 30 (7): 1037-1047. 10.1016/j.neurobiolaging.2009.03.013.
Hamshere ML, Holmans PA, Avramopoulos D, Bassett SS, Blacker D, Bertram L, Wiener H, Rochberg N, Tanzi RE, Myers A, Wavrant-De Vrièze F, Go R, Fallin D, Lovestone S, Hardy J, Goate A, O'Donovan M, Williams J, Owen MJ: Genome-wide linkage analysis of 723 affected relative pairs with late-onset Alzheimer's disease. Hum Mol Genet. 2007, 16 (22): 2703-2712. 10.1093/hmg/ddm224.
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, Jun G, Destefano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thornton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, Lin CF, Gerrish A, Schmidt H, Kunkle B, Dunstan ML, Ruiz A, Bihoreau MT, Choi SH, Reitz C, et al: Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013, 45 (12): 1452-1458. 10.1038/ng.2802.
Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Ramsey K, Caselli RJ, Kukull WA, McKeel D, Morris JC, Hulette CM, Schmechel D, Reiman EM, Rogers J, Stephan DA: Neuronal gene expression in non-demented individuals with intermediate Alzheimer's Disease neuropathology. Neurobiol Aging. 2010, 31 (4): 549-566. 10.1016/j.neurobiolaging.2008.05.013.
Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Ramsey K, Caselli RJ, Kukull WA, McKeel D, Morris JC, Hulette CM, Schmechel D, Reiman EM, Rogers J, Stephan DA: Altered neuronal gene expression in brain regions differentially affected by Alzheimer's disease: a reference data set. Physiol Genomics. 2008, 33 (2): 240-256. 10.1152/physiolgenomics.00242.2007.
Morris JH, Apeltsin L, Newman AM, Baumbach J, Wittkop T, Su G, Bader GD, Ferrin TE: clusterMaker: a multi-algorithm clustering plugin for Cytoscape. BMC Bioinformatics. 2011, 12: 436-10.1186/1471-2105-12-436.
da Huang W, Sherman BT, Lempicki RA: Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009, 4 (1): 44-57.
da Huang W, Sherman BT, Lempicki RA: Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37 (1): 1-13. 10.1093/nar/gkn923.
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D: The human genome browser at UCSC. Genome Res. 2002, 12 (6): 996-1006. 10.1101/gr.229102. Article published online before print in May 2002.
Kohler S, Bauer S, Horn D, Robinson PN: Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet. 2008, 82 (4): 949-958. 10.1016/j.ajhg.2008.02.013.
Gamazon ER, Zhang W, Konkashbaev A, Duan S, Kistner EO, Nicolae DL, Dolan ME, Cox NJ: SCAN: SNP and copy number annotation. Bioinformatics. 2010, 26 (2): 259-262. 10.1093/bioinformatics/btp644.
Saccone SF, Bolze R, Thomas P, Quan J, Mehta G, Deelman E, Tischfield JA, Rice JP: SPOT: a web-based tool for using biological databases to prioritize SNPs after a genome-wide association study. Nucleic Acids Res. 2010, 38 (Web Server issue): W201-209.
Boada M, Antunez C, Ramirez-Lorca R, Destefano AL, Gonzalez-Perez A, Gayan J, Lopez-Arrieta J, Ikram MA, Hernandez I, Marin J, Galán JJ, Bis JC, Mauleón A, Rosende-Roca M, Moreno-Rey C, Gudnasson V, Morón FJ, Velasco J, Carrasco JM, Alegret M, Espinosa A, Vinyes G, Lafuente A, Vargas L, Fitzpatrick AL, Launer LJ, Sáez ME, Vázquez E, Becker JT, for the Alzheimer’s Disease Neuroimaging Initiative: ATP5H/KCTD2 locus is associated with Alzheimer's disease risk. Mol Psychiatry. 2013, doi:10.1038/mp.2013.86
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A: NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41 (Database issue): D991-995.
Webster JA, Gibbs JR, Clarke J, Ray M, Zhang W, Holmans P, Rohrer K, Zhao A, Marlowe L, Kaleem M, McCorquodale DS, Cuello C, Leung D, Bryden L, Nath P, Zismann VL, Joshipura K, Huentelman MJ, Hu-Lince D, Coon KD, Craig DW, Pearson JV, Heward CB, Reiman EM, Stephan D, Hardy J, Myers AJ, NACC-Neuropathology Group: Genetic control of human brain transcript expression in Alzheimer disease. Am J Hum Genet. 2009, 84 (4): 445-58. 10.1016/j.ajhg.2009.03.011.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T: Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13 (11): 2498-2504. 10.1101/gr.1239303.
Hernandez-Toro J, Prieto C, De las Rivas J: APID2NET: unified interactome graphic analyzer. Bioinformatics. 2007, 23 (18): 2495-2497. 10.1093/bioinformatics/btm373.
Barsky A, Gardy JL, Hancock RE, Munzner T: Cerebral: a Cytoscape plugin for layout of and interaction with biological networks using subcellular localization annotation. Bioinformatics. 2007, 23 (8): 1040-1042. 10.1093/bioinformatics/btm057.
Van Dongen S: Graph clustering by flow simulation. PhD Thesis. 2000, The Netherlands: University of Utrecht
Nussbaum RL, Ellis CE: Alzheimer's disease and Parkinson's disease. N Engl J Med. 2003, 348 (14): 1356-1364. 10.1056/NEJM2003ra020003.
Mayeux R: Epidemiology of neurodegeneration. Annu Rev Neurosci. 2003, 26: 81-104. 10.1146/annurev.neuro.26.043002.094919.
Lill CM, Roehr JT, McQueen MB, Kavvoura FK, Bagade S, Schjeide BM, Schjeide LM, Meissner E, Zauft U, Allen NC, Liu T, Schilling M, Anderson KJ, Beecham G, Berg D, Biernacka JM, Brice A, DeStefano AL, Do CB, Eriksson N, Factor SA, Farrer MJ, Foroud T, Gasser T, Hamza T, Hardy JA, Heutink P, Hill-Burns EM, Klein C, Latourelle JC, et al: Comprehensive research synopsis and systematic meta-analyses in Parkinson's disease genetics: the PDGene database. PLoS Genet. 2012, 8 (3): e1002548-10.1371/journal.pgen.1002548.
Rosenberger A, Sharma M, Muller-Myhsok B, Gasser T, Bickeboller H: Meta analysis of whole-genome linkage scans with data uncertainty: an application to Parkinson's disease. BMC Genet. 2007, 8: 44-
Tryka KA, Hao L, Sturcke A, Jin Y, Wang ZY, Ziyabari L, Lee M, Popova N, Sharopova N, Kimura M, Feolo M: NCBI's Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res. 2013, -
Fung HC, Scholz S, Matarin M, Simon-Sanchez J, Hernandez D, Britton A, Gibbs JR, Langefeld C, Stiegert ML, Schymick J, Okun MS, Mandel RJ, Fernandez HH, Foote KD, Rodríguez RL, Peckham E, De Vrieze FW, Gwinn-Hardy K, Hardy JA, Singleton A: Genome-wide genotyping in Parkinson's disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2006, 5 (11): 911-916. 10.1016/S1474-4422(06)70578-6.
Simon-Sanchez J, Scholz S, Fung HC, Matarin M, Hernandez D, Gibbs JR, Britton A, de Vrieze FW, Peckham E, Gwinn-Hardy K, Crawley A, Keen JC, Nash J, Borgaonkar D, Hardy J, Singleton A: Genome-wide SNP assay reveals structural genomic variation, extended homozygosity and cell-line induced alterations in normal individuals. Hum Mol Genet. 2007, 16 (1): 1-14.
Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Krüger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, et al: Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. 2009, 41 (12): 1308-1312. 10.1038/ng.487.
Zhang Y, James M, Middleton FA, Davis RL: Transcriptional analysis of multiple brain regions in Parkinson's disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. Am J Med Genet B Neuropsychiatr Genet. 2005, 137B (1): 5-16. 10.1002/ajmg.b.30195.
Bras J, Guerreiro R, Hardy J: Use of next-generation sequencing and other whole-genome strategies to dissect neurological disease. Nat Rev Neurosci. 2012, 13 (7): 453-464.
Smith AV: Genetic analysis: moving between linkage and association. Cold Spring Harb Protoc. 2012, 2012 (2): 174-182.
Dorval V, Smith PY, Delay C, Calvo E, Planel E, Zommer N, Buee L, Hebert SS: Gene network and pathway analysis of mice with conditional ablation of Dicer in post-mitotic neurons. PLoS One. 2012, 7 (8): e44060-10.1371/journal.pone.0044060.
Hudson NJ, Dalrymple BP, Reverter A: Beyond differential expression: the quest for causal mutations and effector molecules. BMC Genomics. 2012, 13: 356-10.1186/1471-2164-13-356.
Boehnke M: Limits of resolution of genetic linkage studies: implications for the positional cloning of human disease genes. Am J Hum Genet. 1994, 55 (2): 379-390.
Ge X, Loh HH, Law PY: mu-Opioid receptor cell surface expression is regulated by its direct interaction with Ribophorin I. Mol Pharmacol. 2009, 75 (6): 1307-1316. 10.1124/mol.108.054064.
Mathieu-Kia AM, Fan LQ, Kreek MJ, Simon EJ, Hiller JM: Mu-, delta- and kappa-opioid receptor populations are differentially altered in distinct areas of postmortem brains of Alzheimer's disease patients. Brain Res. 2001, 893 (1–2): 121-134.
Teng L, Zhao J, Wang F, Ma L, Pei G: A GPCR/secretase complex regulates beta- and gamma-secretase specificity for Abeta production and contributes to AD pathogenesis. Cell Res. 2010, 20 (2): 138-153. 10.1038/cr.2010.3.
Saetre P, Jazin E, Emilsson L: Age-related changes in gene expression are accelerated in Alzheimer's disease. Synapse. 2011, 65 (9): 971-974. 10.1002/syn.20933.
Muma NA, Mariyappa R, Williams K, Lee JM: Differences in regional and subcellular localization of G(q/11) and RGS4 protein levels in Alzheimer's disease: correlation with muscarinic M1 receptor binding parameters. Synapse. 2003, 47 (1): 58-65. 10.1002/syn.10153.
Bradley SV, Holland EC, Liu GY, Thomas D, Hyun TS, Ross TS: Huntingtin interacting protein 1 is a novel brain tumor marker that associates with epidermal growth factor receptor. Cancer Res. 2007, 67 (8): 3609-3615. 10.1158/0008-5472.CAN-06-4803.
Walter S, Atzmon G, Demerath EW, Garcia ME, Kaplan RC, Kumari M, Lunetta KL, Milaneschi Y, Tanaka T, Tranah GJ, Völker U, Yu L, Arnold A, Benjamin EJ, Biffar R, Buchman AS, Boerwinkle E, Couper D, De Jager PL, Evans DA, Harris TB, Hoffmann W, Hofman A, Karasik D, Kiel DP, Kocher T, Kuningas M, Launer LJ, Lohman KK, Lutsey PL, et al: A genome-wide association study of aging. Neurobiol Aging. 2011, 32 (11): 2109-e15-28
Mills IG, Gaughan L, Robson C, Ross T, McCracken S, Kelly J, Neal DE: Huntingtin interacting protein 1 modulates the transcriptional activity of nuclear hormone receptors. J Cell Biol. 2005, 170 (2): 191-200. 10.1083/jcb.200503106.
Menegon A, Burgaya F, Baudot P, Dunlap DD, Girault JA, Valtorta F: FAK + and PYK2/CAKbeta, two related tyrosine kinases highly expressed in the central nervous system: similarities and differences in the expression pattern. Eur J Neurosci. 1999, 11 (11): 3777-3788. 10.1046/j.1460-9568.1999.00798.x.
Combs CK, Johnson DE, Cannady SB, Lehman TM, Landreth GE: Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of beta-amyloid and prion proteins. J Neurosci. 1999, 19 (3): 928-939.
Huang Y, Lu W, Ali DW, Pelkey KA, Pitcher GM, Lu YM, Aoto H, Roder JC, Sasaki T, Salter MW, MacDonald JF: CAKbeta/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron. 2001, 29 (2): 485-496. 10.1016/S0896-6273(01)00220-3.
Park SY, Avraham H, Avraham S: Characterization of the tyrosine kinases RAFTK/Pyk2 and FAK in nerve growth factor-induced neuronal differentiation. J Biol Chem. 2000, 275 (26): 19768-19777. 10.1074/jbc.M909932199.
Siciliano JC, Toutant M, Derkinderen P, Sasaki T, Girault JA: Differential regulation of proline-rich tyrosine kinase 2/cell adhesion kinase beta (PYK2/CAKbeta) and pp 125(FAK) by glutamate and depolarization in rat hippocampus. J Biol Chem. 1996, 271 (46): 28942-28946. 10.1074/jbc.271.46.28942.
Cotman CW, Anderson AJ: A potential role for apoptosis in neurodegeneration and Alzheimer's disease. Mol Neurobiol. 1995, 10 (1): 19-45. 10.1007/BF02740836.
Tian D, Litvak V, Lev S: Cerebral ischemia and seizures induce tyrosine phosphorylation of PYK2 in neurons and microglial cells. J Neurosci. 2000, 20 (17): 6478-6487.
Pilon M, Peng XR, Spence AM, Plasterk RH, Dosch HM: The diabetes autoantigen ICA69 and its Caenorhabditis elegans homologue, ric-19, are conserved regulators of neuroendocrine secretion. Mol Biol Cell. 2000, 11 (10): 3277-3288. 10.1091/mbc.11.10.3277.
Cao M, Xu J, Shen C, Kam C, Huganir RL, Xia J: PICK1-ICA69 heteromeric BAR domain complex regulates synaptic targeting and surface expression of AMPA receptors. J Neurosci. 2007, 27 (47): 12945-12956. 10.1523/JNEUROSCI.2040-07.2007.
Spitzenberger F, Pietropaolo S, Verkade P, Habermann B, Lacas-Gervais S, Mziaut H, Pietropaolo M, Solimena M: Islet cell autoantigen of 69 kDa is an arfaptin-related protein associated with the Golgi complex of insulinoma INS-1 cells. J Biol Chem. 2003, 278 (28): 26166-26173. 10.1074/jbc.M213222200.
Wu Y, Matsui H, Tomizawa K: Amphiphysin I and regulation of synaptic vesicle endocytosis. Acta Med Okayama. 2009, 63 (6): 305-323.
Di Paolo G, Sankaranarayanan S, Wenk MR, Daniell L, Perucco E, Caldarone BJ, Flavell R, Picciotto MR, Ryan TA, Cremona O, De Camilli P: Decreased synaptic vesicle recycling efficiency and cognitive deficits in amphiphysin 1 knockout mice. Neuron. 2002, 33 (5): 789-804. 10.1016/S0896-6273(02)00601-3.
De Jesus-Cortes HJ, Nogueras-Ortiz CJ, Gearing M, Arnold SE, Vega IE: Amphiphysin-1 protein level changes associated with tau-mediated neurodegeneration. Neuroreport. 2012, 23 (16): 942-946. 10.1097/WNR.0b013e32835982ce.
Kelly BL, Ferreira A: Beta-amyloid disrupted synaptic vesicle endocytosis in cultured hippocampal neurons. Neuroscience. 2007, 147 (1): 60-70. 10.1016/j.neuroscience.2007.03.047.
Wang L, Chiang HC, Wu W, Liang B, Xie Z, Yao X, Ma W, Du S, Zhong Y: Epidermal growth factor receptor is a preferred target for treating amyloid-beta-induced memory loss. Proc Natl Acad Sci U S A. 2012, 109 (41): 16743-16748. 10.1073/pnas.1208011109.
Zhang YW, Wang R, Liu Q, Zhang H, Liao FF, Xu H: Presenilin/gamma-secretase-dependent processing of beta-amyloid precursor protein regulates EGF receptor expression. Proc Natl Acad Sci U S A. 2007, 104 (25): 10613-10618. 10.1073/pnas.0703903104.
Repetto E, Yoon IS, Zheng H, Kang DE: Presenilin 1 regulates epidermal growth factor receptor turnover and signaling in the endosomal-lysosomal pathway. J Biol Chem. 2007, 282 (43): 31504-31516. 10.1074/jbc.M704273200.
Conejero-Goldberg C, Hyde TM, Chen S, Dreses-Werringloer U, Herman MM, Kleinman JE, Davies P, Goldberg TE: Molecular signatures in post-mortem brain tissue of younger individuals at high risk for Alzheimer's disease as based on APOE genotype. Mol Psychiatry. 2011, 16 (8): 836-847. 10.1038/mp.2010.57.
Akram A, Schmeidler J, Katsel P, Hof PR, Haroutunian V: Increased expression of cholesterol transporter ABCA1 is highly correlated with severity of dementia in AD hippocampus. Brain Res. 2010, 1318: 167-177.
Elali A, Rivest S: The role of ABCB1 and ABCA1 in beta-amyloid clearance at the neurovascular unit in Alzheimer's disease. Front Physiol. 2013, 4: 45-
Wildsmith KR, Holley M, Savage JC, Skerrett R, Landreth GE: Evidence for impaired amyloid beta clearance in Alzheimer's disease. Alzheimers Res Ther. 2013, 5 (4): 33-10.1186/alzrt187.
Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, Jain S, Hirsch-Reinshagen V, Wellington CL, Bales KR, Paul SM, Holtzman DM: Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008, 118 (2): 671-682.
Fitz NF, Cronican AA, Saleem M, Fauq AH, Chapman R, Lefterov I, Koldamova R: Abca1 deficiency affects Alzheimer's disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J Neurosci. 2012, 32 (38): 13125-13136. 10.1523/JNEUROSCI.1937-12.2012.
Malik B, Fernandes C, Killick R, Wroe R, Usardi A, Williamson R, Kellie S, Anderton BH, Reynolds CH: Oligomeric amyloid-beta peptide affects the expression of genes involved in steroid and lipid metabolism in primary neurons. Neurochem Int. 2012, 61 (3): 321-333. 10.1016/j.neuint.2012.05.006.
Canepa E, Borghi R, Vina J, Traverso N, Gambini J, Domenicotti C, Marinari UM, Poli G, Pronzato MA, Ricciarelli R: Cholesterol and amyloid-beta: evidence for a cross-talk between astrocytes and neuronal cells. J Alzheimers Dis. 2011, 25 (4): 645-653.
Donkin JJ, Stukas S, Hirsch-Reinshagen V, Namjoshi D, Wilkinson A, May S, Chan J, Fan J, Collins J, Wellington CL: ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem. 2010, 285 (44): 34144-34154. 10.1074/jbc.M110.108100.
Fan J, Donkin J, Wellington C: Greasing the wheels of Abeta clearance in Alzheimer's disease: the role of lipids and apolipoprotein E. Biofactors. 2009, 35 (3): 239-248. 10.1002/biof.37.
Koldamova R, Fitz NF, Lefterov I: The role of ATP-binding cassette transporter A1 in Alzheimer's disease and neurodegeneration. Biochim Biophys Acta. 2010, 1801 (8): 824-830. 10.1016/j.bbalip.2010.02.010.
Wang XF, Cao YW, Feng ZZ, Fu D, Ma YS, Zhang F, Jiang XX, Shao YC: Quantitative assessment of the effect of ABCA1 gene polymorphism on the risk of Alzheimer's disease. Mol Biol Rep. 2013, 40 (2): 779-785. 10.1007/s11033-012-2115-9.
Lupton MK, Proitsi P, Lin K, Hamilton G, Daniilidou M, Tsolaki M, Powell JF: The Role of ABCA1 Gene Sequence Variants on Risk of Alzheimer's Disease. J Alzheimers Dis. 2014, 38 (4): 897-906.
Butterfield DA, Castegna A, Lauderback CM, Drake J: Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer's disease brain contribute to neuronal death. Neurobiol Aging. 2002, 23 (5): 655-664. 10.1016/S0197-4580(01)00340-2.
Ricciarelli R, d'Abramo C, Massone S, Marinari U, Pronzato M, Tabaton M: Microarray analysis in Alzheimer's disease and normal aging. IUBMB Life. 2004, 56 (6): 349-354. 10.1080/15216540412331286002.
Leduc V, Legault V, Dea D, Poirier J: Normalization of gene expression using SYBR green qPCR: a case for paraoxonase 1 and 2 in Alzheimer's disease brains. J Neurosci Methods. 2011, 200 (1): 14-19. 10.1016/j.jneumeth.2011.05.026.
Maloney MT, Minamide LS, Kinley AW, Boyle JA, Bamburg JR: Beta-secretase-cleaved amyloid precursor protein accumulates at actin inclusions induced in neurons by stress or amyloid beta: a feedforward mechanism for Alzheimer's disease. J Neurosci. 2005, 25 (49): 11313-11321. 10.1523/JNEUROSCI.3711-05.2005.
Bamburg JR, Bloom GS: Cytoskeletal pathologies of Alzheimer disease. Cell Motil Cytoskeleton. 2009, 66 (8): 635-649. 10.1002/cm.20388.
Yao J, Khan AN: Involvement of Actin Pathology in Alzheimer’s disease. Cell Dev Biol. 2013, 2: e121-doi:10.4172/2168-9296.1000e121
Tell V, Holzer M, Herrmann L, Mahmoud KA, Schächtele C, Totzke F, Hilgeroth A: Multitargeted drug development: Discovery and profiling of dihydroxy substituted 1-aza-9-oxafluorenes as lead compounds targeting Alzheimer disease relevant kinases. Bioorg Med Chem Lett. 2012, 22 (22): 6914-8. 10.1016/j.bmcl.2012.09.006.
Chang KH, Vincent F, Shah K: Deregulated Cdk5 triggers aberrant activation of cell cycle kinases and phosphatases inducing neuronal death. J Cell Sci. 2012, 125 (Pt 21): 5124-5137.
Cui JG, Li YY, Zhao Y, Bhattacharjee S, Lukiw WJ: Differential regulation of interleukin-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2 by microRNA-146a and NF-kappaB in stressed human astroglial cells and in Alzheimer disease. J Biol Chem. 2010, 285 (50): 38951-38960. 10.1074/jbc.M110.178848.
Hu WT, Holtzman DM, Fagan AM, Shaw LM, Perrin R, Arnold SE, Grossman M, Xiong C, Craig-Schapiro R, Clark CM Pickering E, Kuhn M, Chen Y, Van Deerlin VM, McCluskey L, Elman L, Karlawish J, Chen-Plotkin A, Hurtig HI, Siderowf A, Swenson F, Lee VM, Morris JC, Trojanowski JQ, Soares H, Alzheimer's Disease Neuroimaging Initiative: Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology. 2012, 79 (9): 897-905. 10.1212/WNL.0b013e318266fa70.
Song F, Poljak A, Crawford J, Kochan NA, Wen W, Cameron B, Lux O, Brodaty H, Mather K, Smythe GA, Sachdev PS: Plasma apolipoprotein levels are associated with cognitive status and decline in a community cohort of older individuals. PLoS One. 2012, 7 (6): e34078-10.1371/journal.pone.0034078.
Vuletic S, Li G, Peskind ER, Kennedy H, Marcovina SM, Leverenz JB, Petrie EC, Lee VM, Galasko D, Schellenberg GD, Albers JJ: Apolipoprotein E highly correlates with AbetaPP- and tau-related markers in human cerebrospinal fluid. J Alzheimers Dis. 2008, 15 (3): 409-417.
Fukuyama R, Mizuno T, Mori S, Yanagisawa K, Nakajima K, Fushiki S: Age-dependent decline in the apolipoprotein E level in cerebrospinal fluid from control subjects and its increase in cerebrospinal fluid from patients with Alzheimer's disease. Eur Neurol. 2000, 43 (3): 161-169. 10.1159/000008157.
Hesse C, Larsson H, Fredman P, Minthon L, Andreasen N, Davidsson P, Blennow K: Measurement of apolipoprotein E (apoE) in cerebrospinal fluid. Neurochem Res. 2000, 25 (4): 511-517. 10.1023/A:1007516210548.
Cruchaga C, Kauwe JS, Nowotny P, Bales K, Pickering EH, Mayo K, Bertelsen S, Hinrichs A, Fagan AM, Holtzman DM Morris JC, Goate AM: Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer's disease. Hum Mol Genet. 2012, 21 (20): 4558-4571. 10.1093/hmg/dds296.
Skoog I, Hesse C, Fredman P, Andreasson LA, Palmertz B, Blennow K: Apolipoprotein E in cerebrospinal fluid in 85-year-old subjects. Relation to dementia, apolipoprotein E polymorphism, cerebral atrophy, and white matter lesions. Arch Neurol. 1997, 54 (3): 267-272. 10.1001/archneur.1997.00550150029012.
Merched A, Blain H, Visvikis S, Herbeth B, Jeandel C, Siest G: Cerebrospinal fluid apolipoprotein E level is increased in late-onset Alzheimer's disease. J Neurol Sci. 1997, 145 (1): 33-39. 10.1016/S0022-510X(96)00234-1.
Lindh M, Blomberg M, Jensen M, Basun H, Lannfelt L, Engvall B, Scharnagel H, Marz W, Wahlund LO, Cowburn RF: Cerebrospinal fluid apolipoprotein E (apoE) levels in Alzheimer's disease patients are increased at follow up and show a correlation with levels of tau protein. Neurosci Lett. 1997, 229 (2): 85-88. 10.1016/S0304-3940(97)00429-1.
Doecke JD, Laws SM, Faux NG, Wilson W, Burnham SC, Lam CP, Mondal A, Bedo J, Bush AI, Brown B, De Ruyck K, Ellis KA, Fowler C, Gupta VB, Head R, Macaulay SL, Pertile K, Rowe CC, Rembach A, Rodrigues M, Rumble R, Szoeke C, Taddei K, Taddei T, Trounson B, Ames D, Masters CL, Martins RN, Alzheimer's Disease Neuroimaging Initiative; Australian Imaging Biomarker and Lifestyle Research Group: Blood-based protein biomarkers for diagnosis of Alzheimer disease. Arch Neurol. 2012, 69 (10): 1318-1325. 10.1001/archneurol.2012.1282.
Moskvina V, Harold D, Russo G, Vedernikov A, Sharma M, Saad M, Holmans P, Bras JM, Bettella F, Keller MF, Nicolaou N, Simón-Sánchez J, Gibbs JR, Schulte C, Durr A, Guerreiro R, Hernandez D, Brice A, Stefánsson H, Majamaa K, Gasser T, Heutink P, Wood N, Martinez M, Singleton AB, Nalls MA, Hardy J, Owen MJ, O'Donovan MC, Williams J, et al: Analysis of Genome-Wide Association Studies of Alzheimer Disease and of Parkinson Disease to Determine If These 2 Diseases Share a Common Genetic Risk. JAMA Neurol. 2013, 70 (10): 1268-76.
Marder K, Tang MX, Alfaro B, Mejia H, Cote L, Louis E, Stern Y, Mayeux R: Risk of Alzheimer's disease in relatives of Parkinson's disease patients with and without dementia. Neurology. 1999, 52 (4): 719-724. 10.1212/WNL.52.4.719.
Levy G, Louis ED, Mejia-Santana H, Cote L, Andrews H, Harris J, Waters C, Ford B, Frucht S, Fahn S, Ottman R, Marder K: Lack of familial aggregation of Parkinson disease and Alzheimer disease. Arch Neurol. 2004, 61 (7): 1033-1039.
Rocca WA, Bower JH, Ahlskog JE, Elbaz A, Grossardt BR, McDonnell SK, Schaid DJ, Maraganore DM: Risk of cognitive impairment or dementia in relatives of patients with Parkinson disease. Arch Neurol. 2007, 64 (10): 1458-1464. 10.1001/archneur.64.10.1458.
Hart GT, Ramani AK, Marcotte EM: How complete are current yeast and human protein-interaction networks?. Genome Biol. 2006, 7 (11): 120-10.1186/gb-2006-7-11-120.
Moreau Y, Tranchevent LC: Computational tools for prioritizing candidate genes: boosting disease gene discovery. Nat Rev Genet. 2012, 13 (8): 523-536. 10.1038/nrg3253.
Bornigen D, Tranchevent LC, Bonachela-Capdevila F, Devriendt K, De Moor B, De Causmaecker P, Moreau Y: An unbiased evaluation of gene prioritization tools. Bioinformatics. 2012, 28 (23): 3081-3088. 10.1093/bioinformatics/bts581.
Antunez C, Boada M, Gonzalez-Perez A, Gayan J, Ramirez-Lorca R, Marin J, Hernandez I, Moreno-Rey C, Moron FJ, Lopez-Arrieta J, Mauleón A, Rosende-Roca M, Noguera-Perea F, Legaz-García A, Vivancos-Moreau L, Velasco J, Carrasco JM, Alegret M, Antequera-Torres M, Manzanares S, Romo A, Blanca I, Ruiz S, Espinosa A, Castaño S, García B, Martínez-Herrada B, Vinyes G, Lafuente A, Becker JT, et al: The membrane-spanning 4-domains, subfamily A (MS4A) gene cluster contains a common variant associated with Alzheimer's disease. Genome Med. 2011, 3 (5): 33-10.1186/gm249.
Reiman EM, Webster JA, Myers AJ, Hardy J, Dunckley T, Zismann VL, Joshipura KD, Pearson JV, Hu-Lince D, Huentelman MJ, Craig DW, Coon KD, Liang WS, Herbert RH, Beach T, Rohrer KC, Zhao AS, Leung D, Bryden L, Marlowe L, Kaleem M, Mastroeni D, Grover A, Heward CB, Ravid R, Rogers J, Hutton ML, Melquist S, Petersen RC, Alexander GE, et al: GAB2 alleles modify Alzheimer's risk in APOE epsilon4 carriers. Neuron. 2007, 54 (5): 713-720. 10.1016/j.neuron.2007.05.022.
Mueller SG, Weiner MW, Thal LJ, Petersen RC, Jack CR, Jagust W, Trojanowski JQ, Toga AW, Beckett L: Ways toward an early diagnosis in Alzheimer's disease: the Alzheimer's Disease Neuroimaging Initiative (ADNI). Alzheimers Dement. 2005, 1 (1): 55-66. 10.1016/j.jalz.2005.06.003.
Li H, Wetten S, Li L, St Jean PL, Upmanyu R, Surh L, Hosford D, Barnes MR, Briley JD, Borrie M, Coletta N, Delisle R, Dhalla D, Ehm MG, Feldman HH, Fornazzari L, Gauthier S, Goodgame N, Guzman D, Hammond S, Hollingworth P, Hsiung GY, Johnson J, Kelly DD, Keren R, Kertesz A, King KS, Lovestone S, Loy-English I, Matthews PM, et al: Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol. 2008, 65 (1): 45-53.
Wijsman EM, Pankratz ND, Choi Y, Rothstein JH, Faber KM, Cheng R, Lee JH, Bird TD, Bennett DA, Diaz-Arrastia R, Goate AM, Farlow M, Ghetti B, Sweet RA, Foroud TM, Mayeux R, NIA-LOAD/NCRAD Family Study Group: Genome-wide association of familial late-onset Alzheimer's disease replicates BIN1 and CLU and nominates CUGBP2 in interaction with APOE. PLoS Genet. 2011, 7 (2): e1001308-10.1371/journal.pgen.1001308.
Hu X, Pickering E, Liu YC, Hall S, Fournier H, Katz E, Dechairo B, John S, Van Eerdewegh P, Soares H, Alzheimer's Disease Neuroimaging Initiative: Meta-analysis for genome-wide association study identifies multiple variants at the BIN1 locus associated with late-onset Alzheimer's disease. PLoS One. 2011, 6 (2): e16616-10.1371/journal.pone.0016616.
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, et al: Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009, 41 (10): 1088-1093. 10.1038/ng.440.
Ramensky V, Bork P, Sunyaev S: Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002, 30 (17): 3894-3900. 10.1093/nar/gkf493.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR: A method and server for predicting damaging missense mutations. Nat Methods. 2010, 7 (4): 248-249. 10.1038/nmeth0410-248.
Loots G, Ovcharenko I: ECRbase: database of evolutionary conserved regions, promoters, and transcription factor binding sites in vertebrate genomes. Bioinformatics. 2007, 23 (1): 122-124. 10.1093/bioinformatics/btl546.
Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S, Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q, International HapMap Consortium, et al: A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007, 449 (7164): 851-861. 10.1038/nature06258.
Gray KA, Daugherty LC, Gordon SM, Seal RL, Wright MW, Bruford EA: Genenames.org: the HGNC resources in 2013. Nucleic Acids Res. 2013, 41 (Database issue): D545-552.
Silla Y, Sundaramoorthy E, Talwar P, Sengupta S: S-linked protein homocysteinylation: identifying targets based on structural, physicochemical and protein-protein interactions of homocysteinylated proteins. Amino Acids. 2013, 44 (5): 1307-1316. 10.1007/s00726-013-1465-5.
Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Mazumder R, O'Donovan C, Redaschi N, Suzek B: The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res. 2006, 34 (Database issue): D187-191.
Assenov Y, Ramirez F, Schelhorn SE, Lengauer T, Albrecht M: Computing topological parameters of biological networks. Bioinformatics. 2008, 24 (2): 282-284. 10.1093/bioinformatics/btm554.
Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, Wernerus H, Björling L, Ponten F: Towards a knowledge-based Human Protein Atlas. Nat Biotechnol. 2010, 28 (12): 1248-1250. 10.1038/nbt1210-1248.
Brohee S, van Helden J: Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinformatics. 2006, 7: 488-10.1186/1471-2105-7-488.
Li SJ, Peng M, Li H, Liu BS, Wang C, Wu JR, Li YX, Zeng R: Sys-BodyFluid: a systematical database for human body fluid proteome research. Nucleic Acids Res. 2009, 37 (Database issue): D907-912.
Pirooznia M, Nagarajan V, Deng Y: GeneVenn - A web application for comparing gene lists using Venn diagrams. Bioinformation. 2007, 1 (10): 420-422. 10.6026/97320630001420.
Acknowledgement
We thank Prof. Samir K. Brahmachari (IGIB) for his vision and Dr. Rajesh Gokhale, Director, Institute of Genomics and Integrative Biology (CSIR) for his support. We appreciate Dr. Shantanu Sengupta (IGIB) for critical evaluation of the manuscript. Financial support from Council of Scientific and Industrial Research (CSIR) (BSC0123) is duly acknowledged. PT, YS and SG acknowledge CSIR, Govt. of India for providing their fellowships. We thank the anonymous reviewers for their helpful suggestions for improving the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
PT conceived, designed, processed, interpreted the data and wrote the manuscript. YS and SG have contributed in work design, interpretation and manuscript writing. RK has conceived and supervised the study. MG, RA and SK have contributed by critical evaluation of the study and improving the manuscript. All the authors read and approved the final manuscript.
Electronic supplementary material
12864_2013_7037_MOESM1_ESM.xlsx
Additional file 1: AD GWA and GWE replication datasets. The file contains the list of 294 genes from AD GWA replication dataset (Boada et al.) and list of 182 genes from AD GWE replication dataset (Webster et al., GEO:GSE15222). (XLSX 56 KB)
12864_2013_7037_MOESM2_ESM.xlsx
Additional file 2: Final list of AD genes from three data sets and final list of ranked 108 genes. The file contains the list of genes from three datasets, final overlapping 108 genes ranked by their cumulative rank score. (XLSX 385 KB)
12864_2013_7037_MOESM3_ESM.png
{kind=link}
Additional file 3: Venn diagrams of overlapping genes from independent analysis of genes from 6 brain region separately with GWA and GWL datasets. The file contains Venn diagrams of genes from three datasets, final overlapping 108 genes ranked by their cumulative rank score. (PNG 2 MB)
12864_2013_7037_MOESM4_ESM.xlsx
Additional file 4: List of overlapping genes from independent analysis of genes from 6 brain region separately with GWA and GWL datasets. The file contains the list of genes from three datasets, final overlapping 108 genes ranked by their cumulative rank score. (XLSX 11 KB)
12864_2013_7037_MOESM5_ESM.pdf
Additional file 5: Clusters identified from PPI using MCL algorithm implemented in clusterMaker. The file details the 69 clusters identified by MCL algorithm from the PPI containing 640 genes. (PDF 77 KB)
12864_2013_7037_MOESM6_ESM.xlsx
Additional file 6: Annotation clusters from DAVID. The file contains top 11 and 10 annotation clusters with GO analysis from DAVID for 108 and 640 genes respectively. (XLSX 22 KB)
12864_2013_7037_MOESM7_ESM.png
{kind=link}
Additional file 7: Putative AD specific biomarkers among 640 AD proteins and proteins from CSF and plasma proteome. The file contains Venn diagram showing overlap of 640 AD proteins and proteins from CSF and plasma proteome. (PNG 440 KB)
12864_2013_7037_MOESM8_ESM.xlsx
Additional file 8: Final list of PD genes from three data sets and final list of 59 candidate overlapping genes. The file contains the list of genes from three datasets, final overlapping 59 candidate genes. (XLSX 319 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Talwar, P., Silla, Y., Grover, S. et al. Genomic convergence and network analysis approach to identify candidate genes in Alzheimer's disease. BMC Genomics 15, 199 (2014). https://doi.org/10.1186/1471-2164-15-199
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-199