
Abstract
Background
The Type VI secretion system (T6SS) has been identified in several different bacteria, including the plant pathogenPantoea ananatis. Previous in silico analyses described three different T6SS loci present in the pathogenic strain of P. ananatis LMG 20103. This initial investigation has been extended to include an additional seven sequenced strains of P. ananatis together with 39 strains from different ecological niches. Comparative and phylogenetic analyses were used to investigate the distribution, evolution, intra-strain variability and operon structure of the T6SS in the sequenced strains.
Results
Three different T6SS loci were identified in P. ananatis strain LMG 20103 and designated PA T6SS 1-3. PA T6SS-1 was present in all sequenced strains of P. ananatis and in all 39 additional strains examined in this study. In addition, PA T6SS-1 included all 13 core T6SS genes required for synthesis of a functional T6SS. The plasmid-borne PA T6SS-2 also included all 13 core T6SS genes but was restricted to only 33% (15/46) of the strains examined. In addition, PA T6SS-2 was restricted to strains of P. ananatis isolated from symptomatic plant material. This finding raises the possibility of an association between PA T6SS-2 and either pathogenicity or host specificity. The third cluster PA T6SS-3 was present in all strains analyzed in this study but lacked 11 of the 13 core T6SS genes suggesting it may not encoded a functional T6SS. Inter-strain variability was also associated with hcp and vgrG islands, which are associated with the T6SS and encode a variable number of proteins usually of unknown function. These proteins may play a role in the fitness of different strains in a variety of ecological niches or as candidate T6SS effectors. Phylogenetic analysis indicated that PA T6SS-1 and PA T6SS-2 are evolutionarily distinct.
Conclusion
Our analysis indicates that the three T6SSs of P. ananatis appear to have been independently acquired and may play different roles relating to pathogenicity, host range determination and/or niche adaptation. Future work will be directed toward understanding the roles that these T6SSs play in the biology of P. ananatis.
Similar content being viewed by others


Background
Pantoea ananatis is a Gram-negative, motile, facultative anaerobe belonging to the gamma Proteobacteria. This bacterium can survive and multiply in a variety of ecological niches as a saprophyte, endophyte, epiphyte and pathogen [1]. In its latter role, P. ananatis infects a wide range of economically important plants. In South Africa the pathogen causes disease on maize, onion and Eucalyptus spp. [2–5] but has also been reported to infect pineapple [6], rice [7], melon [8], sudan grass and sorghum [9, 10]. The mechanism of spread of P. ananatis between host plants is largely unknown. However, Walcot et al. [11] isolated virulent strains of P. ananatis from onion seeds, which went on to produce disease symptoms on susceptible onion plants [11]. In addition, Gitaitis et al. [12] demonstrated that tabacco thrips (Frankliniella fusca) were able to transmit P. ananatis into 52% of onion plants analysed [12]. These studies show that seeds and insect vectors are important sources of inoculation and could serve as vehicles for the spread of P. ananatis to different geographical regions. Current control measures are limited to cultivation of resistant plant cultivars, eradication of infected plant material and/or the use of biocontrol in the form of lytic phages [1, 13, 14]. Despite the wide geographical and host range of P. ananatis, there is limited information on the genetic determinants of virulence and ecological fitness of the species.
To date, seven different secretion systems have been described in bacteria; namely type I-VII [15, 16]. These secretion systems release factors that modulate the host environment to favour bacterial fitness and, in some cases, virulence. The type VI secretion system (T6SS) was first described by Pukatzki et al. [17] in Vibrio cholerae and was shown to be required for virulence against amoeba and macrophages [17]. This secretion system consists of 15-23 different proteins, which assemble to form an injectisome-like structure similar to an inverted contractile phage particle [18–20]. The T6SS has since been identified in the genome of several pathogenic but also non-pathogenic Gram-negative bacteria, suggesting that it may be involved in functions unrelated to pathogenicity [21–24]. The role of the T6SS in virulence, symbiosis, biofilm formation and stress has been documented in several bacteria [25–30]. In addition, the T6SSs of Pseudomonas aeruginosa, Vibrio cholerae, Pseudomonas fluorescens, Pseudomonas protegens, Burkholderia thailandensis and Serratia marcescens have been shown to secrete bactericidal effectors which inhibit growth of bacterial species that lack the cognate immunity protein [31–40]. Similarly, the T6SSs of Pseudomonas syringae pv. tomato DC3000, Acinetobacter baumannii, Acinetobacter baylyi, Vibrio parahaemolyticus and Citrobacter rodentium have also been to play a role in inter-bacterial competition [41–44]. Some bacteria encode more than one evolutionarily distinct T6SS in their genome [23]. Multiple T6SS gene clusters found in a given bacterial genomes are believed to have been acquired by independent horizontal gene transfer events, possibly to play different roles in the biology of different bacteria [23, 24, 45].
This study focused on the T6SS of the pathogen Pantoea ananatis. To date, the genome sequences for eight strains of P. ananatis are available, representing both pathogenic and non-pathogenic strains. Type II, type III and type IV secretion systems are well documented virulence determinants of several human and plant pathogens [15, 16], although genome mining showed that they were all absent from the genome of P. ananatis strain LMG 20103 [1, 46]. These findings raise the possibility that the T6SS of P. ananatis could play a role in either virulence or ecological fitness of the species. Previous comparative studies identified three distinct T6SS loci on the genome of a single Eucalyptus pathogenic P. ananatis strain LMG 20103 [46–48]. Thus to better understand the distribution of the T6SS in this versatile pathogen we carried out an in-depth comparative analysis of the T6SS in eight sequenced strains, of which three were recently sequenced as part of this study. To this end, we analyzed the gene content, sequence similarity, synteny, operon structure, and possible evolutionary history of each T6SS loci. PCR and dot blot hybridisations were used to study the distribution of the T6SS in 39 additional strains from a variety of niches. This study represents the first detailed intra-species comparative analysis of the T6SS in P. ananatis.
Results and discussion
In silico identification of T6SS gene clusters in P. ananatis
The 13 conserved gene components of the T6SS from P. ananatis strain LMG 20103 [48] were used as a bait to identify the T6SS in all sequenced strains of P. ananatis. BLASTN and BLASTP searches were done using all eight currently available genomes of P. ananatis, which included the LPP-1 megaplasmid found in all sequenced strains [49]. In this manuscript we have used the standard T6SS gene nomenclature proposed by Shalom et al. [50]. Based on this nomenclature, the conserved T6SS genes were designated tss A-M/ (Type Six Secretion A-M), while the accessory or non-conserved T6SS genes were designated tag A-P (Type Six Associated Genes A-P) [50].
Homologs of the T6SS genes were clustered in two distinct genomic regions in all sequenced strains of P. ananatis. These regions were designated PA T6SS-1 and PA T6SS-2 (Pantoea ananatis T6SS 1, 2). The PA T6SS-1 gene cluster was located on the genome of all eight sequenced strains of P. ananatis while PA T6SS-2 gene cluster was located on a ~30 kb region of the LPP-1 megaplasmid of strains AJ13355, LMG 20103 and PA-4. This 30 kb region was missing from the LPP-1 plasmid of P. ananatis strains LMG 2665T, LMG 5342, BD442, B1-9 and PA-13. We also found that PA T6SS-1 and PA T6SS-2 gene clusters contained all 13 core gene components of the T6SS [23, 24]. In addition, we also identified a 9 kb region containing homologs of icmF and dotU in all eight sequenced strains of P. ananatis. This gene cluster, was designated T6SS-3 and was found to be missing the remaining 11 core gene components of the T6SS. Whether or not PA T6SS-3 gene cluster encodes a functional T6SS or represents a truncated T4SS is currently unknown. However, this putative PA T6SS-3 gene cluster was included in this study for purposes of comparative analysis. The overall genetic organisation of each T6SS of P. ananatis is presented in Figure 1. The list of all core and accessory T6SS gene components found in the T6SS gene clusters of P. ananatis, including their putative functions and COG classification is presented in Table 1.

Genetic organization of the different type VI secretion gene clusters (T6SS) in Pantoea ananatis (PA). Genes are indicated by arrows and the direction of the arrows represents the direction of transcription of the gene related to the rest of the genome. We used the type VI secretion system gene nomenclature of Shalom et al. [50]. Conserved core gene components of the T6SS (tssA-M) are indicated in red while non-conserved genes associated with the T6SS of limited bacteria (tagA-P) are indicated in different colors. The tag genes found in the T6SS gene clusters of other bacteria are indicated in the key. The figure is not drawn to scale, PA T6SS-1 and PA T6SS-2 are both over 30 kb and contain up to 20 genes each, while PA T6SS-3 (9 kb) contains 8 genes in all sequenced strains.
Operon structure of the T6SS
The T6SS encoded by most bacteria is organized in discreet transcriptional units or operons [51, 52], suggesting coordinated expression [23, 24]. We therefore, investigated the organization of conserved genes in the P. ananatis T6SS. The core genes of PA T6SS-1 were clustered in three highly conserved operons; group 1 (tssJ-tssK-dotU-icmF) group 2 (tssB-tssC-hcp) and group 3 (tssE-tssF-tssG-tssH). PA T6SS-2 showed a considerable level of gene shuffling compared to PA T6SS-1, with gene order being highly variable between each of the different groupings. The consensus grouping in PA T6SS-2 included tssB-tssC-tssK-dotU and tssF-tssG-tssJ-tssE, while tssH-vgrG and icmF-tssA were stand-alone operons linked to non-conserved T6SS genes. The start and stop codons of all 9 genes located in PA T6SS-3 gene cluster overlapped with each other, suggesting that PA T6SS-3 represents a single transcriptional unit. These different operon structures suggested the independent acquisition of the T6SS clusters, each of which may play a different role in the biology of P. ananatis.
Distribution of the PA T6SS in other strains of P. ananatis
PCR and dot blot analysis
To determine the prevalence of the three PA T6SS clusters [1–3] among P. ananatis strains, we analyzed the distribution of each T6SS cluster in 46 different strains of the pathogen. BlastP analysis showed that the gene products of PA T6SS-1 and 2 shared less than 50% amino acid similarity (Additional file 1: Table S1). This low sequence similarity allowed specific PCR primers to be designed within conserved regions located on the DNA sequence of the targeted genes. For primer design, the targeted T6SS gene homologs from eight sequenced strains of P. ananatis were aligned in BioEdit using ClustalW multiple alignment program and PCR primers designed within the conserved DNA regions located on these genes. BlastN analysis showed that the conserved regions used for primer design were not present on the DNA sequences of T6SS gene homologs found in other clusters. We, therefore, saw no cross reactivity during the PCR amplification. The list of primers and strains of P. ananatis used for PCR analysis is provided in Additional file 2: Table S2 and Additional file 3: Table S3, respectively.
PCR amplification showed that the T6SS-1 and T6SS-3 homologs were present in all tested strains of P. ananatis. PCR amplification using PA T6SS-2 gene specific primers identified homologs in only 15 of the 46 strains of P. ananatis (33%) tested (Additional file 4: Table S4). The distribution of PA T6SS-2 was further confirmed by dot blot hybridization using probes for tssA, tssE, tssJ and tssK (results not shown). Our results showed that PA T6SS-2 was present in strains of P. ananatis isolated from symptomatic maize, onion, pineapple fruit and Eucalyptus spp. However, not all strains of P. ananatis isolated from the same host plant contained PA T6SS-2. The cluster was only found in 3 of the 6 strains of P. ananatis (50%) isolated from either maize, onion, pineapple and Eucalyptus spp., while the remaining 3 strains, isolated from the same host but different plants, did not have the cluster. In addition, PA T6SS-2 was absent in all strains of P. ananatis isolated from symptomatic wheat (2 isolates), honeydew melon (4 isolates), rice (7 isolates) and sugarcane (1 isolate). These results suggest that PA T6SS-2 could be a host range or virulence determinant of P. ananatis. Future studies will undertake pathogenicity and cross inoculation trials to validate this correlation. In addition, the cluster was also found in P. ananatis strains AJ13355 and Yomogi-101 which have been shown not to cause disease on a range of host plants ([53, 54], and data not shown), suggesting that PA T6SS-2 could be involved in other processed related to the ecological fitness of the species.
Comparative analysis of T6SS gene clusters from different strains of P. ananatis
Homologous PA T6SSs encoded by different strains of P. ananatis were highly conserved in terms of sequence similarity, gene content and operon structure (Figure 2 and Additional file 5: Figure S1). A detailed description of the genes found in individual T6SSs encoded by all sequenced strains of P. ananatis analyzed in this study are provided in (Additional file 6: Table S5-S12 and Additional file 7: Table S13-S15).

Comparison of the Pantoea ananatis type VI secretion systems 1 and 2 (T6SS-1 and T6SS-2). All 13 conserved core gene components of the T6SS are indicated in red while non-conserved genes associated with the T6SS of limited bacteria are indicated in different colors. The letters in the figure represent conserved T6SS genes based on the nomenclature of Shalom et al. [50]. PA T6SS-1 was found in all eight sequenced strains analyzed, while PA T6SS-2 was restricted to AJ13355 (saprophyte), LMG 20103 (pathogen of Eucalyptus spp) and PA-4 (onion pathogen). ‡ represents genes found in two unassembled contigs in PA T6SS-1.
-
1)
PA T6SS-1
The genetic architecture of PA T6SS-1 was shown to be conserved amongst all sequenced strains of P. ananatis. The tssD (hcp) and tssI (vgrG) genes found in this cluster encoded Hcp and VgrG proteins that do not have C-terminal extensions as found in “evolved” VgrG and Hcp proteins [45, 55, 56]. The C-terminal extension of some evolved VgrG proteins, such as VgrG1 of V. cholerae and Aeromonas hydrophila, have been associated with actin cross-linking and actin ADP ribosylation activity in mammalian host cells, respectively [55, 56]. P. ananatis strains PA-4, BD442, B1-9 and LMG 20103 had a single vgrG gene, while strains AJ13355, PA-13, LMG 5342 and LMG 2665T had an additional copy of vgrG. These vgrG genes appear to encode VgrG proteins with different domain architectures, characterized by the presence or absence of a C-terminal Beta-N-acetylglucosaminidase domain (Figure 3). This C-terminal domain is associated with lysozymes belonging to the glycoside hydrolase family 73 (PF01832) [57, 58]. It is possible, therefore, that the different VgrG proteins encoded by each vgrG gene are mobilized to the T6SS baseplate under different physiological conditions or play different roles either as effectors, structural elements or both.
Figure 3 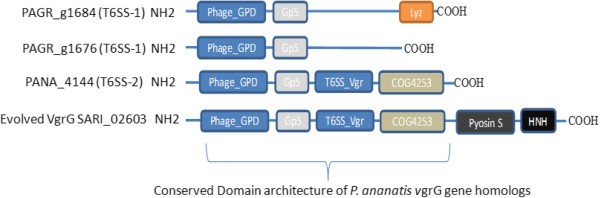
Homolog of vgrG genes found in the type VI secretion system gene clusters of sequenced strains of Pantoea ananatis . Domains are represented in different colors. P. ananatis strain PA-13 has two structurally different vgrG genes (PAGR_g1684 and PAGR_g1676) within T6SS-1. Lyz = lysozyme / Beta-N-acetylglucosaminidase domain is found in PAGR_g1684 which is missing from the vgrG homolog (PAGR_g1676). PANA_4144 found in T6SS-2 of strain LMG 20103 has a domain architecture similar to part of SARI_02603 of Salmonella enterica subspecies arizonae.
Regions associated with hcp and vgrG contain genes that encode a variable number of accessory and hypothetical proteins that account for strain specific differences. The first variable region in PA T6SS-1 is located between the major rhsD element and vgrG. Genes found in this region encode mostly hypothetical proteins and proteins with either a PAAR (proline-alanine-alanine-arginine) repeat or pentapeptide_4 domains. PAAR repeat proteins of bacteria have categorised into different classes (Class 1-7) based on their domain architectures [58]. PAAR repeat proteins of several bacteria have effector domains on the N or C-terminal [58]. Some of these effector domains include: transthyretin, lipase, nuclease, deaminase, and ADP-ribosyl transferase [58]. The genes products of PA-13 PAGR_g1683 and LMG 5342 PANA5342_1748 belong to the Class 1 PAAR domain architecture proteins and share 100% sequence similarity (Figure 4). In addition, PAGR_g1683 and PANA5342_1748 have no N or C terminal extensions and showed structural homolog to the PAAR repeat protein of V. cholerae (4jiv_D–Hhpred score 105.4). A recent study showed that the PAAR repeat proteins of E. coli and V. cholerae bind to the Gp5-VgrG complex by means of non-covalent interactions [59]. In addition, PAAR repeat proteins of V. cholerae and Acinetobacter baylyi were shown to be bactericidal effectors associated with T6SS-mediated killing of E. coli[59]. These findings have led to the speculation that, PAAR repeat proteins carrying different effector domain located on either their N or C-terminal extensions may also bind to the VgrG spike and mediate secretion of these effectors by the T6SS [59, 60]. It is also speculated that PAAR repeat proteins may form non-covalent interactions with different effectors, thereby recruiting them to the T6SS spike complex [59]. It is, therefore, possible that the PAAR repeat proteins encoded by genes located in the vgrG island of P. ananatis T6SS-1 gene cluster may either be T6SS effectors associated with inter-bacterial competition or may mediate secretion of other effectors.
Figure 4 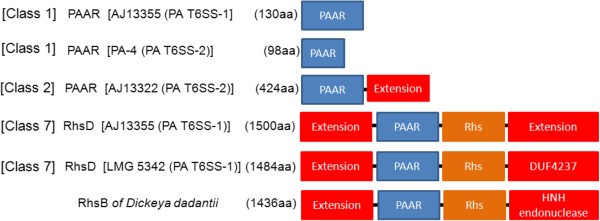
Domain architecture of PAAR repeat proteins associated of Pantoea ananatis type VI secretion system 1 and 2 (T6SS-1 and T6SS-2). We used the nomenclature of Shneider et al. [59], which categorized PAAR proteins into different classes [1–7] based on domain architecture. The different domains and extensions associates with P. ananatis PAAR proteins are indicated by different colors. No putative effector domains were associated with N or C-terminal extensions located in most PAAR repeat proteins of P. ananatis. However, the RhsD protein of LMG 5342, LMG 2665T and B1-9 showed extensive structural homology to the insecticidal YenC2 toxins of Yersinia entomophaga. The domain architecture of P. ananatis RhsD protein is similar to the RhsB endonuclease toxin of Dickeya dadantii.
PAAR-repeat domains are also found in the rhsD gene of PA T6SS-1. A recent study, aimed at identifying polymorphic toxins in bacterial genomes using comparative analysis, sequence and structural analysis, identified RhsD as putative T6SS effectors of P. ananatis, based on the presence of PAAR repeats on this protein [59]. Similarly, the RhsD protein of Serratia marcescens was recently identified in a T6SS secretome analysis, suggesting that this protein is secreted by the T6SS [40]. Bacteria Rhs proteins have been associated with different phenotypes such as social motility, inflammasome-mediated cell death, virulence in mice, insecticidal toxin production, polysaccharide transport and bacteriocin production [61–69]. In addition, the rhsA and rhsB genes of Dickeya dadantii encode endonuclease toxins which have been associated with contact-dependent killing of other bacteria species [70, 71]. Toxin producing strains of D. dadantii also express the cognate immunity factors from the rhsI gene located downstream of the rhsA and rhsB genes [70, 71]. The rhsD locus of P. ananatis has a genetic organization similar to that of D. dadantii consisting of the rhsD gene which is followed by the rhsI homolog (Figure 5). In addition, P. ananatis rhsD genes have a conserved N-terminal domain and a variable C-terminal domain which is characteristic of several bacteria Rhs toxins (Additional file 8: Figure S2) [72, 73]. We identified a DUF4237 domain of unknown function on the C-terminal extensions of RhsD proteins of P. ananatis strains B1-9, LMG 2665T and LMG 5342, which is missing from the remaining strains. Furthermore, the RhsD proteins of strains B1-9, LMG 2665T and LMG 5342 showed extensive structural homology to the insecticidal YenC2 toxin of Yersinia entomophaga (4igl_B–Hhpred score 574, 559.5 and 573.4, respectively) [65, 74]. These findings suggest that the rhsD loci associated with the T6SS-1 gene clusters of P. ananatis may encode different toxin/immunity factors which play different roles as either bactericidal or insecticidal toxins. Alternatively, RhsD proteins of P. ananatis may mediate secretion of other T6SS effectors which are non-covalently associated with either their N or C-terminal extension, as predicted for other PAAR proteins [59].
Figure 5 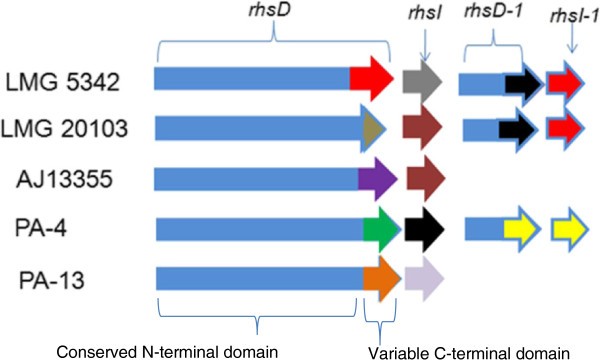
Genetic organization of the different rhs loci associated with the type VI secretion system 1 gene cluster (T6SS-1) of Pantoea ananatis . Representative strains of P. ananatis with distinct rhs loci are indicated in the figure. Strains LMG 5342, B1-9 and LMG 2665T have identical rhsD/rhsI gene homologs. Similarly, strain PA-4 and BD442 also have identical rhsD/rhI gene homologs. Conserved and variable regions associated with rhsD genes are indicated in the figure. The rhsI gene is located downstream of the rhsD gene and the colors indicate the different rhI genes encoded by different strains of P. ananatis. The figure is not drawn to scale.
Homologs of tagAB and tagB, which encode pentapeptide repeat proteins were also identified in the vgrG islands of P. ananatis strains PA-13, AJ13355 and LMG 5342. In strain PA 13, the pentapeptide repeat proteins are encoded by PAGR_g1680 and PAGR_g1681 which correspond to tagAB and tagB genes, respectively. Sequence analysis performed on PAGR_g1680 and PAGR_g1681 predicted no signal sequence and no transmembrane helices [75, 76]. PsortB analysis predicted the gene product of PAGR_g1680 to be a secreted effector while the sub-cellular localization of PAGR_g1681 protein is unknown [77]. Homologs of tagAB and tagB genes have also been in identified in the T6SS gene clusters of Serratia marcescens, Azotobacter vinelandii, Burkolderia bronchiseptica, including the T6SS-5 gene clusters of B. mallei, B. pseudomallei and B. thailandensis, however, these genes have not been functionally characterised and the biological function of their gene products is currently unknown [30, 38, 50].
The second variable region found in P. ananatis T6SS-1 gene cluster is located between hcp and tagH and contains a gene encoding a protein with a peptidoglycan binding domain (PG_binding). In strain LMG 5342, the protein is encoded by PANA5342_1731 and showed structural homology to the lytic transglycosylase enzyme of Pseudomonas aeruginosa bacteriophage phiKZ (3bkh_A Hhpred–score 146) while the corresponding homologs found in strains LMG 20103, PA-4, B1-9 and AJ13355 showed structural homology to the pesticin toxin of Yersinia pestis (4aqm_A Hhpred–score 290.2, 283.4, 290.2 and 283.4, respectively). Pesticin and lytic transglycosylase enzymes are bactericidal effectors which degrade peptidoglycan by cleaving the β-1,4 glycosidic bond between N-acetylmuramic acid and the N-acetylglucosamine moieties [78–82]. These findings suggest that genes found in the hcp island of P. ananatis T6SS-1 cluster may encode bactericidal effectors which are functionally related to the Type VI secretion glycoside hydrolase effectors 1-3 (Tge1-3) [37, 83]. Genes encoding proteins with the PG_binding domain were not identified in the T6SS-1 hcp island of P. ananatis strain LMG 2665T. However, the gene product of LMG 2665T N454_00628 showed weak structural homology to Colicin S4 of Escherichia coli (3few_X–Hhpred score 55.8). Colicin S4 is a pore-forming bacteriocin which kills bacteria species not expressing the immunity factor [84, 85]. It is, therefore, possible that the gene product of LMG 2665T N454_00628 may be a bacteriocidal effector which plays a role in inter-bacterial competition. We did not identify homologs of Type VI amidase 1-4 (Tae 1-4), Type VI lipase effectors 1-5 (Tle 1-5), Small secreted proteins 1-6 (Ssp 1-6) and Type VI secretion exported 1, 2 (Tse 1, 2) in the T6SS-1 gene cluster of P. ananatis, based on sequence and structural analysis [34, 36, 39, 40].
-
2)
PA T6SS-2
The genetic architecture of PA T6SS-2 is highly conserved in all strains that harbor the cluster. PA T6SS-2 was found to contain a single vgrG gene that encodes a VgrG protein with an additional C-terminal DUF2345 (COG4253) domain of unknown function. Conserved Domain architecture analysis showed that this domain was conserved in the vgrG genes of several different bacteria. All strains of P. ananatis that harbor this cluster encode two copies of the tssA gene within the cluster showing 22% amino acid similarity. Duplication of the tssA gene has also been reported in the T6SS gene clusters of Vibrio cholerae, Escherichia coli O157:H7, Yersinia pseudotuberculosis and Salmonella enterica serotype Gallinarum [21, 45, 50]. The reason for this duplication is unknown, as is the exact role that TssA plays as an essential T6SS structural protein.
Two variable regions were identified in PA T6SS-2, which contained genes encoding non-conserved T6SS components and hypothetical proteins. The first variable region is located between tssA and tssF and encodes several strain specific hypothetical proteins including proteins with a PAAR_motif. The genes products of LMG 20103 PANA_4136, AJ13355 PAJ_p0154 and PA-4 N455_00706 contain 424 amino acids, sharing 99% amino acid similarity and belong to the Class 2 PAAR domain architecture proteins. Furthermore, these PAAR proteins all contained identical C-terminal extensions with no putative effector domains. The gene products of PANA_4136, PAJ_p0154 and PA-4 N455_00706 may either be T6SS effectors or mediate secretion of other effectors bound to their C-terminal extensions [59, 60]. The second variable region in PA T6SS-2 gene cluster is located between icmF and vgrG. This region in LMG20103 contains four genes (PANA_4140 to 4143) encoding hypothetical proteins with homologs present in strain AJ13355 but is missing from strain PA-4. PANA_4143 encodes a protein with a peptidase_M23 domain. This protein is a putative endopeptidase and is predicted to be a T6SS effector based on its high structural homology to the secreted chitinase G of Streptomyces coelicolor[48, 86]. The corresponding variable region in P. ananatis strain PA-4 contains a gene with an esterase_lipase domain and belongs to the family lecithin: cholesterol acyltransferase (PF02450). This protein is predicted to be involved in extracellular metabolism of plasma lipoproteins, including cholesterol [87]. Genes with an esterase_lipase domain have been found in the vicinity of vgrG genes outside the major T6SS of Pseudomonas spp. In P. aeroginosa these genes form part of the “cargo” genes predicted to have been acquired by recent horizontal gene transfer [88]. In addition, the N455_00710 gene which is located in the vgrG island of strain PA-4 T6SS-2 cluster, encodes a Class 1 PAAR domain architecture protein containing 98 amino acids with no N or C-terminal extensions. This PAAR protein may play a role as a T6SS effector associated with inter-bacterial competition [59, 60]. Homologs of functionally characterized T6SS effectors such as type VI lipase effectors 1-5, type VI amidase 1-4, type VI secretion glycoside hydrolase effectors, Small secreted proteins 1-6 and Type VI secretion exported 1-2, were not identified in P. ananatis T6SS-2 gene cluster, using sequence analysis and structural homology search tools.
-
3)
PA T6SS-3
Comparative analysis of PA T6SS-3 showed that there was no variability of this cluster between the different strains of P. ananatis. The genetic architecture, gene order and gene content of PA T6SS-3 was conserved in all sequenced strains of P. ananatis. Interestingly, all genes found in PA T6SS-3 showed high sequence similarity to genes found in Pantoea sp. At-9b, Pantoea sp. aB-valens, P. vagans C9-1, P. agglomerans E325 and Erwinia billingiae Eb661. These Pantoea and Erwinia spp. have a homologous cluster highly similar to PA T6SS-3 in terms of gene content and operon structure [48]. The high conservation of this cluster suggests a strong selective pressure to maintain the gene content and order, although its specific role is unknown. The fact that the cluster is missing 11 core gene components of the T6SS suggests that this system does not encode a functional T6SS, although this is yet to be confirmed.



“Orphan” Hcp and VgrG proteins
When the T6SS genes hcp and vgrG are present outside the T6SS they are often referred to as “orphan” vgrG and hcp genes. Our analysis did not identify “orphan” vgrG genes in any of the sequenced genomes of P. ananatis. However, we identified three different “orphan” hcp genes in P. ananatis strain PA-13 (PAGR_g1583, PAGR_g1584 and PAGR_g3636). These “orphan” hcp genes were designated hcp-1, hcp-2 and hcp-3 to correspond to PAGR_g1583, PAGR_g1584 and PAGR_g3636, respectively. Genes hcp-1 and hcp-2 are adjacent to each other and have homologs present in all sequenced strains of P. ananatis, while hcp-3 is restricted to P. ananatis PA-13. Multiple alignments of the amino acid sequences of representative P. ananatis Hcp proteins showed that Hcp-3 protein is highly divergent from Hcp-1, Hcp-2 and T6SS-associated Hcp proteins (Additional file 9: Figure S3). All “orphan” hcp genes in P. ananatis are found in the vicinity of genes encoding hypothetical proteins and a putative endoribonuclease SymE, which is part of an SOS inducible toxin/antitoxin system [89]. It remains to be determined whether “orphan” hcp genes of P. ananatis are association with the major T6SSs, as either T6SS effectors or structural proteins, or whether the conserved association between symE and “orphan” hcp genes is important in other aspects of the biology of P. ananatis.
Phylogenetic analysis of the T6SS
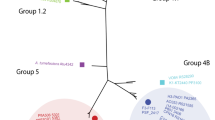
Phylogenetic analysis was used to infer the evolutionary history of the T6SS using the Maximum Likelihood Method base on the Le and Gascuel (LG) + G + F amino acid substitution model, as determined by ProtTest [90–92]. Representative bacteria from the different T6SS phylogenetic groups A-D were included in the analysis [23, 45] (Additional file 10: Table S16). The analysis showed that PA T6SS-1 belonged to phylogenetic Group A, while PA T6SS-2 belonged to Group C (Figure 6). PA T6SS-1 was phylogenetically closest to T6SS loci 1 of Pantoea sp At-9b (an insect endophyte), while PA T6SS-2 was closest to T6SS loci 2 of Erwinia amylovora CFBP1430 (a plant pathogen). Our phylogenetic analysis resembled the analysis performed by Bingle et al. [23] which showed that phylogenetic Group A to D contained pathogenic and non-pathogenic bacteria associated with different ecological niches [23]. Similarly, functionally characterized T6SS with a known role in virulence or inter-bacterial competition were represented in the different Groups. For example, the H1-T6SS of P. aeruginosa which is known secreted Tse1 to 3 antimicrobial effectors was found in Group A while the T6SSs of Vibrio cholerae and Peudomonas. syringae pv tomato with a similar role in inter-bacterial competition belonged to Group D [31, 32, 41]. In addition, all four phylogenetic Groups contained bacteria T6SSs which have been associated with virulence. Together, these finding suggest that bacteria T6SSs found in phylogenentic Group A to D are evolutionarily distinct and play different roles in pathogenic and non-pathogenic bacteria [23, 24].

Evolutionary relationships of the different type VI secretion systems of Pantoea ananatis using concatenated TssB and TssC amino acid sequences. Phylogenetic analysis were conducted in MEGA6 [90]. The amino acid substitution model was determined by ProtTest [91]. The evolutionary history was inferred using the Maximum Likelihood method based on the Le and Gascuel (LG) + G + F model [92]. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The TssB/C homologs used in this study are indicated next to the name of the bacteria. Blue octagons represent T6SS phylogenetic Groups A-D proposed by Bingle et al. [23]. Representative position of PA T6SS-1 and PA T6SS-2 are shown in blue and green rectangles, respectively. Details of all bacteria used in are given in (Additional file 10: Table S16).
Our phylogenetic tree also showed that P. ananatis T6SS-1 and T6SS-2 clustered away from the T6SS of several important plant pathogens such as Xanthomonas citri, X. campestris, X. oryzae, Pseudomonas syringae pv. syringae, Pseudomonas syringae pv. phaseolicola, Pectobacterium atrosepticum SCRI1043, Pectobacterium carotovorum subsp. carotovorum and Pectobacterium carotovorum subsp. brasiliensis. Functional studies have shown that the T6SS gene clusters of P. atrosepticum and P. syringae are only partially required for disease development. The T6SS mutants of these bacteria were either only slightly reduced in the ability to cause disease or caused disease symptoms on susceptible host plants similar to the wild type strains [41, 93–96]. These findings, together with our T6SS phylogenetic groupings, suggests that 1) the T6SS of different plant pathogens were acquired from unrelated bacteria or distantly-related ancestors, 2) the T6SS clusters found in different phytopathogens may play different roles depending on the host plant or ecological niche, and 3) T6SS-1 and T6SS-2 may play an important role in the virulence of P. ananatis in susceptible host plants.
To infer the evolutional history of “orphan” hcp genes (hcp-1, 2 and 3) in P. ananatis we constructed a second Maximum Likelihood tree using the General Reverse Transcriptase (rtRev) + I + G + F amino acid substitution models, as determined by ProtTest [97]. Our phylogenetic analysis also included representative T6SS-associated hcp genes and “orphan” hcp genes from other bacteria (Additional file 11: Table S17). The resulting phylogeny revealed four distinct clusters, which we have designated as hcp Cluster 1-4 to distinguish them from T6SS phylogenetic Group A-D [23] and Group I-V [24]. Our results indicate that hcp-1 and hcp-2 belong to Cluster 3, together with the T6SS-associated hcp genes found in PA-T6SS-1 (Additional file 12: Figure S4). Hcp-3 (PAGR_g3636) belongs to Cluster 2, together with T6SS-associated hcp genes present in PA-T6SS-2. Hcp-3, however, forms part of a subset of Cluster 2, and was phylogenetically close to orphan Hcp proteins from klebsiella varriicola At-22 and Erwinia amylovora ATCC BAA-2158. These hcp groupings suggest either independent acquisition of “orphan” hcp genes from different ancestors or gene duplication and rearrangement of T6SS-associated hcp genes.
Conclusion
Comparative analyses of the T6SS in the genomes of sequenced strains of P. ananatis identified three putative gene clusters PA T6SS-1, PA T6SS-2 and PA T6SS-3. The former two of these were potentially functional as they contained the 13 core genes necessary for synthesis of a functional T6SS [23, 24, 30]. PA T6SS-1 was widespread in the genome of all sequenced strains including environmental isolates, while PA T6SS-2 was plasmid borne and restricted mostly to pathogenic strains of P. ananatis isolated from certain classes of plants. This finding suggests a potential association of PA T6SS-2 with host range determination. However, the finding that PA T6SS-1 and PA T6SS-2 were present in both pathogenic and non-pathogenic strains of P. ananatis supports the concept that the T6SS may evolve to play different roles unrelated to pathogenicity, e.g. competition against other microbes, fitness and/or niche adaptation [18, 23, 27, 39]. The genetic organization and phylogenetic groupings of PA T6SS-1 and −2 further suggests that these clusters were independently acquired to play differing roles in the different strains of P. ananatis. Furthermore, the variable regions associated with hcp and vgrG genes could account for specialization of each T6SS based on the needs of the specific strain. In the future, key questions that need to be addressed include determining: 1) whether the T6SSs of P. ananatis are functionally active and what roles they play in host-pathogen interactions and fitness; 2) which in vitro and in vivo conditions activate the T6SSs; 3) the presence of different potential effectors secreted by the T6SSs of P. ananatis and their physiological relevance to fitness and host-pathogen interactions; and 4) how T6SSs are regulated in these strains.
Methods
P. ananatisT6SS data acquisition
The annotated genome sequences of different P. ananatis strains were downloaded from NCBI with the accession numbers LMG 5342 (chromosome HE617160.1, plasmid pPANA10 HE617161.1); AJ13355 (chromosome AP012032.1, plasmid AP012033.1); PA-13 (chromosome CP003085.1, plasmid PAGR_pCP003086.1); LMG 20103 (chromosome CP001875.2) and the draft genome of B1-9 (CAEJ00000000.1). The draft genomes of P. ananatis BD 442, PA-4 and LMG 2665T were sequenced and partially assembled in our laboratory. The sequences and fully annotated contigs representing the different type VI secretion system gene clusters have been deposited in GenBank for strain BD 442 (KF552073, KF552074), PA-4 (KF590029, KF590030, KF590028) and LMG 2665T (KF590026, KF590027). All eight sequenced strains of P. ananatis were obtained from different geographical regions and were isolated from different diseased plants or other environmental samples. Detailed information on the strains is presented in Additional file 3: Table S3.
In silicoidentification of T6SS cluster
Genes associated with the T6SS, including flanking regions identified in P. ananatis strain LMG 20103 by De Maayer et al. [46], were used as bait to search for T6SS homologs in all sequenced P. ananatis genomes and plasmids using BLASTN and BLASTP [98, 99]. Nucleotide sequences representing entire T6SS regions, including flanking regions, were extracted from each genome and used for de novo gene prediction using F-GenesB (http://www.Softberry.com). All predicted genes were searched against non-redundant protein databases at NCBI to identify homologs. SignalP 4.0 and TMHMM Server v.2.0 were used to predict signal peptides and trans-membrane helices [75, 76]. Protein localization and functional classifications were done using PSORTb [77], InterProScan [100], Conserved domains and CDD domain architecture-Search tool on NCBI [101]. Protein structural homology analyses were done using the HHpred [102]. Proteins were clustered based on their COG groups [103].
PCR and dot blot hybridisations
A minimum of four primer pairs were designed per T6SS cluster to amplifying locus specific genes. For primer design, the nucleotide sequences for each gene of interest were extracted from all eight sequenced strains of P. ananatis. BLASTP analysis showed that T6SS gene products from homologous T6SS cluster found in all sequenced strains of P. ananatis were over 98% similar [Additional file 6: Table S9-S12 and Additional file 7: Table S15]. In addition, BLASTP analysis showed that there was less than 70% similarity between gene products found in the different T6SS clusters (Additional file 1: Table S1). The low sequence similarity between gene homologs from in each T6SS cluster, therefore, allowed specific primers to be designed within conserved T6SS gene regions. Importantly, PCR analysis showed that the primers were specific and did not amplify T6SS homologs found in other T6SS gene clusters found in the genome sequence of P. ananatis. Genomic DNA was isolated from 46 different P. ananatis strains using the Quick-gDNA™MiniPrepkit (ZYMO RESEARCH, USA) following the manufacturer’s instructions. PCR amplification using SuperTherm DNA polymerase (Southern Cross Biotechnology, RSA) was performed with a VeritiR Thermal Cycler (Applied Biosystems, USA). DNA sequencing was done using the ABI PRISM3100 Genetic Analyzer (Applied Biosystems) at the DNA Sequencing Facility (University of Pretoria-RSA). Colony hybridisation was used to validate the PCR results. Probes for hybridisation were labeled with Digoxigenin-11-dUTP using DIG PCR labeling Kit (Roche Applied Science, USA) according to the manufacturer’s instructions. Membrane hybridization, washing and detection were done using DIG DNA labeling and detection kit (Roche Applied Science, USA) as directed by the manufacturer.
Phylogenetic analyses
Two phylogenetic analyses were carried out, one using concatenated amino acid sequences of TssB and TssC and the second using amino acid sequences of Hcp and “orphan” Hcp. The TssBC analysis showed the phylogenetic grouping of the different T6SSs of P. ananatis, while a further phylogenetic grouping examined the relationship between T6SS-associated Hcp and orphan Hcp proteins. Amino acid sequences of TssB, TssC, Hcp and “orphan” Hcp proteins representing bacteria from T6SS phylogenetic Groups A-D were downloaded from NCBI [23, 24]. Amino acid sequences were aligned by ClustalW and phylogenetic analysis conducted in MEGA6 [90]. The amino acid substitution models were determined by ProtTest and applied to this study [91]. The evolutionary history of TssB/C and Hcp proteins were inferred by using the Maximum Likelihood method based on 1) the Le and Gascuel (LG) + G + F amino acid substitution model [92] for TssB/C proteins and 2) the General Reverse Transcriptase (rtRev) + I + G + F amino acid substitution model [97] for Hcp proteins.
Availability of supporting data
The data sets supporting the results of this article are included within the article and its additional files. Alignments and Phylogenetic trees which support the findings presented in this research article are available online in the Dryad Digital Repository (doi: 10.5061/dryad.vd7k7).
References
Coutinho TA, Venter SN: Pantoea ananatis: an unconventional plant pathogen. Mol Plant Pathol. 2009, 10: 325-335. 10.1111/j.1364-3703.2009.00542.x.
Coutinho TA, Preisig O, Mergaert J, Cnockaert MC, Riedel KH, Swings J, Wingfield MJ: Bacterial blight and dieback of Eucalyptus species, hybrids and clones in South Africa. Plant Dis. 2002, 86: 20-25. 10.1094/PDIS.2002.86.1.20.
Goszczynska T, Botha WJ, Venter SN, Coutinho TA: Isolation and identification of the causal agent of brown stalk rot, a new disease of maize in South Africa. Plant Dis. 2007, 91: 711-718. 10.1094/PDIS-91-6-0711.
Goszczynska T, Moloto VM, Venter SN, Coutinho TA: Isolation and identification of Pantoea ananatis from onion seed in South Africa. Seed Sci Technol. 2006, 34: 655-668.
Goszczynska T, Venter SN, Coutinho TA: PA 20, a semi-selective medium for isolation and enumeration of Pantoea ananatis. J Microbiol Methods. 2006, 64: 225-231. 10.1016/j.mimet.2005.05.004.
Serrano FB: Bacterial fruitlet brown-rot of pineapple in the Philippines. Phil J Sci. 1928, 36: 271-305.
Cother EJ, Reinke R, Mckenzie C, Lanoiselet VM, Noble DH: An unusual stem necrosis of rice caused by Pantoea ananas and the first record of this pathogen on rice in Australia. Australas Plant Pathol. 2004, 33: 495-503. 10.1071/AP04053.
Wells JM, Sheng WS, Ceponis MJ, Chen TA: Isolation and characterization of strains of Erwinia ananas from honeydew melons. Phytopathol. 1987, 77: 511-514. 10.1094/Phyto-77-511.
Azad HR, Holmes GJ, Cooksey DA: A new leaf blotch disease of sudangrass caused by Pantoea ananas and Pantoea stewartii. Plant Dis. 2000, 84: 973-979. 10.1094/PDIS.2000.84.9.973.
Cota LVD, Costa RV, Silva DD, Parreira DF, Lana UGP, Casela CR: First report of pathogenicity of Pantoea ananatis in sorghum (Sorghum bicolor) in Brazil. Australas Plant Dis Notes. 2010, 5: 120-122. 10.1071/DN10044.
Walcott RR, Gitaitis RD, Castro AC, Sanders FH, Diaz-Perez JC: Natural infestation of onion seed by Pantoea ananatis, causal agent of center rot. Plant Dis. 2002, 86: 106-111. 10.1094/PDIS.2002.86.2.106.
Gitaitis RD, Walcott RR, Wells ML, Diaz Perez JC, Sanders FH: Transmission of Pantoea ananatis, causal agent of center rot of onion, by tobacco thrips, Frankliniella fusca. Plant Dis. 2003, 87: 675-678. 10.1094/PDIS.2003.87.6.675.
Azegami K: Suppressive effect of bacteriophage on bacterial palea browning of rice caused by Pantoea ananatis. J Gen Plant Pathol. 2013, 79: 145-154. 10.1007/s10327-013-0435-9.
Paccola-Meirelles LD, Meirelles WF, Parentoni SN, Marriel IE, Ferreira AS, Casela CR: Reaction of maize inbred lines to a bacterium Panotea ananas, isolated from Phaeosphaeria leaf spot lesion. Crop Breed Appl Biotechnol. 2002, 2: 587-590. 10.12702/1984-7033.v02n04a12.
Economou A, Christie PJ, Fernandez RC, Palmer T, Plano GV, Pugsley AP: Secretion by numbers: protein traffic in prokaryotes. Mol Microbiol. 2006, 62: 308-319. 10.1111/j.1365-2958.2006.05377.x.
Tseng TT, Tyler BM, Setubal JC: Protein secretion systems in bacterial-host associations, and their description in the gene ontology. BMC Microbiol. 2009, 9 (Suppl1): S2-
Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ: Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci U S A. 2006, 103: 1528-1533. 10.1073/pnas.0510322103.
Filloux A, Hachani A, Bleves S: The bacterial type VI secretion machine: yet another player for protein transport across membranes. Microbiol. 2008, 154: 1570-1583. 10.1099/mic.0.2008/016840-0.
Leiman PG, Basler M, Ramagopal UA, Bonanno JB, Sauder JM, Pukatzki S, Burley SK, Almo SC, Mekalanos JJ: Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc Natl Acad Sci U S A. 2009, 106: 4154-4159. 10.1073/pnas.0813360106.
Pell LG, Kanelis V, Donaldson LW, Howell PL, Davidson AR: The phage λ major tail protein structure reveals a common evolution for long-tailed phages and the type VI bacterial secretion system. Proc Natl Acad Sci U S A. 2009, 106: 4160-4165. 10.1073/pnas.0900044106.
Shrivastava S, Mande SS: Identification and functional characterization of gene components of type VI secretion system in bacterial genomes. PLoS ONE. 2008, 3: e2955-10.1371/journal.pone.0002955.
Das S, Chaudhuri K: Identification of a unique IAHP (IcmF Associated Homologous Proteins) cluster in Vibrio cholera and other Proteobacteria through in silico analysis. In Silico Biol. 2003, 3: 287-300.
Bingle LEH, Bailey CE, Pallen MJ: Type VI secretion: a beginner’s guide. Curr Opin Microbiol. 2008, 11: 3-8. 10.1016/j.mib.2008.01.006.
Boyer FG, Berthod FJ, Vandenbrouck Y, Attree I: Dissecting the bacterial type VI secretion system by a genome wide in silico analysis: what can be learned from available microbial genomic resources?. BMC Genomics. 2009, 10: 104-10.1186/1471-2164-10-104.
Bladergroen MR, Badelt K, Spaink HP: Infection-blocking genes of a symbiotic Rhizobium leguminosarum strain that are involved in temperature-dependent protein secretion. MPMI. 2003, 16: 53-64. 10.1094/MPMI.2003.16.1.53.
Aschtgen MS, Bernard CS, De Bentzmann S, Lloubès R, Cascales E: SciN is an outer membrane lipoprotein required for type VI secretion in enteroaggregative Escherichia coli. J Bacteriol. 2008, 190: 7523-7531. 10.1128/JB.00945-08.
Weber B, Hasic M, Chen C, Wai SN, Milton DL: Type VI secretion modulates quorum sensing and stress response in Vibrio anguillarum. Environ Microbiol. 2009, 11: 3018-3028. 10.1111/j.1462-2920.2009.02005.x.
Zheng J, Leung KY: Dissection of a type VI secretion system in Edwardsiella tarda. Mol Microbiol. 2007, 66: 1192-1206. 10.1111/j.1365-2958.2007.05993.x.
Mougous JD, Cuff ME, Raunser S, Shen A, Zhou M, Gifford CA, Goodman AL, Joachimiak G, Ordoñez CL, Lory S, Walz T, Joachimiak A, Mekalanos JJ: A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science. 2006, 312: 1526-1530. 10.1126/science.1128393.
Schwarz S, West TE, Boyer F, Chiang WC, Carl MA, Hood RD, Rohmer L, Tolker-Nielson T, Skerrett SJ, Mougous JD: Burkholderia type VI secretion systems have distinct roles in eukaryotic and bacterial cell interactions. PLoS Pathog. 2010, 6: e1001068-10.1371/journal.ppat.1001068.
Hood RD, Singh P, Hsu F, Guvener T, Carl MA, Trinidad RR, Silverman JM, Ohlson BB, Hick KG, Plemel RL, Li M, Schwarz S, Wang WY, Merz AJ, Goodlett DR, Mougous JD: A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe. 2010, 7: 25-37. 10.1016/j.chom.2009.12.007.
MacIntyre DL, Miyata ST, Kitaoka M, Pukatzki S: The Vibrio cholerae type VI secretion system displays antimicrobial properties. Proc Natl Acad Sci U S A. 2010, 107: 19520e-19524e. 10.1073/pnas.1012931107.
Miyata ST, Kitaoka M, Brooks TM, McAuley SB, Pukatzki S: Vibrio cholerae requires the type VI secretion system virulence factor VasX to kill Dictyostelium discoideum. Infect Immun. 2011, 79: 2941-2949. 10.1128/IAI.01266-10.
Russell AB, Singh P, Brittnacher M, Bui NK, Hood RD, Carl MA, Agnello DM, Schwarz S, Goodlett DR, Vollmer W, Mougous JD: A widespread bacterial type VI secretion effector superfamily identified using a heuristic approach. Cell Host Microbe. 2012, 11: 538-549. 10.1016/j.chom.2012.04.007.
Dong TG, Ho BT, Yoder-Himes DR, Mekalanos JJ: Identification of T6SS dependent effector and immunity proteins by Tn-seq in Vibrio cholerae. Proc Natl Acad Sci U S A. 2013, 110: 2623-2628. 10.1073/pnas.1222783110.
Russell AB, LeRoux M, Hathazi K, Agnello DM, Ishikawa T, Wiggins PA, Wai SN, Mougous JD: Diverse type VI secretion phospholipases are functionally plastic antibacterial effectors. Nature. 2013, 496: 508-512. 10.1038/nature12074.
Whitney JC, Chou S, Russel AB, Biboy J, Gardiner TE, Ferrin MA, Brittnacher M, Vollmer W, Mougous JD: Identification, structure, and function of a novel type VI secretion peptidoglycan glycoside hydrolase effector-immunity pair. J Biol Chem. 2013, 288: 26616-26624. 10.1074/jbc.M113.488320.
Murdoch SL, Trunk K, English G, Fritsch MJ, Pourkarimi E, Coulthurst SJ: The opportunistic pathogen Serratia marcescens utilizes type VI secretion to target bacterial competitors. J Bacteriol. 2011, 193: 6057-6069. 10.1128/JB.05671-11.
English G, Trunk K, Rao VA, Srikannathasan V, Hunter WN, Coulthurst SJ: New secreted toxin and immunity proteins encoded within the Type VI secretion system gene cluster of Serratia marcescens. Mol Microbiol. 2012, 86: 921-936. 10.1111/mmi.12028.
Fritsch MJ, Trunk K, AlcoforadoDiniz J, Guo M, Trost M, Coulthurst SJ: Proteomic identification of novel secreted anti-bacterial toxins of the Serratia marcescens type VI secretion system. Mol Cell Proteomics. 2013, 12: 2735-2749. 10.1074/mcp.M113.030502.
Haapalainen M, Mosorin H, Dorati F, Wu R-F, Roine E, Taira S, Nissinen R, Laura Mattinen L, Jackson R, Pirhonen M, Lind N-C: Hcp2, a secreted protein of the phytopathogen Pseudomonas syringae pv. tomato DC3000, is required for fitness for competition against bacteria and yeasts. J Bacteriol. 2012, 194: 4810-4822. 10.1128/JB.00611-12.
Carruthers MD, Nicholson PA, Tracy EN, Munson RS: Acinetobacter baumannii utilizes a type VI secretion system for bacterial competition. PLoS ONE. 2013, 8: e59388-10.1371/journal.pone.0059388.
Salomon D, Gonzalez H, Updegraff BL, Orth K: Vibrio parahaemolyticus type VI secretion system 1 is activated in marine conditions to target bacteria, and is differentially regulated from system 2. PLoS ONE. 2013, 8: e61086-10.1371/journal.pone.0061086.
Gueguen E, Cascales E: Promoter swapping unveils the role of the Citrobacter rodentium CTS1 type VI secretion system in interbacterial competition. Appl Environ Microbiol. 2013, 79: 32-38. 10.1128/AEM.02504-12.
Blondel CJ, Jimenez JC, Contreras I, Santiviago CA: Comparative genomic analysis uncovers 3 novel loci encoding type six secretion systems differentially distributed in Salmonella serotypes. BMC Genomics. 2009, 10: 1-17. 10.1186/1471-2164-10-1.
De Maayer P, Chan WY, Venter SN, Toth IK, Birch PRJ, Joubert F, Coutinho TA: Genome sequence of Pantoea ananatis LMG20103, the causative agent of Eucalyptus blight and dieback. J Bacteriol. 2010, 192: 2936-2937. 10.1128/JB.00060-10.
Sarris PF, Trantas EA, Skandalis N, Tampakaki AP, Kapanidou M, Kokkinidis M, Panopoulos NJ: Phytobacterial type VI secretion system: Gene distribution, phylogeny, structure and biological functions. “Plant Pathology Book”. Edited by: Cumagun CJ. 2010, InTech, 53-84. doi:10.5772/33235 ISBN 978–953–51–0489–6, 3
De Maayer P, Venter SV, Kamber T, Duffy B, Coutinho TA, Smits TH: Comparative genomics of the type VI secretion systems of Pantoea and Erwinia species reveals the presence of putative effector islands that may be translocated by the VgrG and Hcp proteins. BMC Genomics. 2011, 12: 576-10.1186/1471-2164-12-576.
De Maayer P, Chan WY, Blom J, Venter SN, Duffy B, Smith THM, Coutinho TA: The large universal Pantoea plasmid LPP-1 plays a major role in biological and ecological diversification. BMC Genomics. 2012, 13: 625-10.1186/1471-2164-13-625.
Shalom G, Shaw JG, Thomas MS: In vivo expression technology identifies a type VI secretion system locus in Burkholderia pseudomallei that is induced upon invasion of macrophages. Microbiol. 2007, 153: 2689-2699. 10.1099/mic.0.2007/006585-0.
Dandekar T, Snel B, Huynen M, Bork P: Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem Sci. 1998, 23: 324-328. 10.1016/S0968-0004(98)01274-2.
Williams SG, Greenwood JA, Jones CW: Molecular analysis of the lac operon encoding the binding-protein-dependent lactose transport system and β-galactosidase in Agrobacterium radiobacter. Mol Microbiol. 1992, 6: 1755-1768. 10.1111/j.1365-2958.1992.tb01348.x.
Hara Y, Kadotani N, Izui H, Katshkina JI, Kuvaeva TM, Andreeva IG, Golubeva LI, Malko DB, Makeev VJ, Mashko SV, Kozlov YI: The complete genome sequence of Pantoea ananatis AJ13355, an organism with great biotechnological potential. Appl Microbiol Biotechnol. 2012, 93: 331-341. 10.1007/s00253-011-3713-5.
Kido K, Adachi R, Hasegawa M, Yano K, Hikichi Y, Takeuchi S, Atsuchi T, Takikawa Y: Internal fruite rot of netted melon caused by Pantoea ananatis (=Erwinia ananas) in Japan. J Gen Plant Pathol. 2008, 74: 302-312. 10.1007/s10327-008-0107-3.
Pukatzki S, Ma AT, Revel AT, Sturtevant D, Mekalanos JJ: Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc Natl Acad Sci U S A. 2007, 104: 15508-15513. 10.1073/pnas.0706532104.
Suarez G, Sierra JC, Erova TE, Sha J, Horneman AJ, Chopra AK: A type VI secretion system effector protein, VgrG1, from Aeromonas hydrophila that induces host cell toxicity by ADP ribosylation of actin. J Bacteriol. 2010, 192: 155-169. 10.1128/JB.01260-09.
Davies G, Henrissat B: Structure and mechanisms of glycosyl hydrolases. Structure. 1995, 3: 853-859. 10.1016/S0969-2126(01)00220-9.
Vocadlo DJ, Withers S: Detailed comparative analysis of the catalytic mechanisms of beta-N-acetylglucosaminidases from families 3 and 20 of glycoside hydrolases. Biogeosciences. 2005, 44: 12809-12818.
Shneider MM, Buth SA, Ho BT, Basler M, Mekalanos JJ, Leiman PG: PAAR-repeat proteins sharpen and diversify the type VI secretion system spike. Nature. 2013, 500: 350-353. 10.1038/nature12453.
Zhang D, de Souza RF, Anantharaman V, Iyer LM, Aravind L: Polymorphic toxin systems: comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol Direct. 2012, 7: 18-10.1186/1745-6150-7-18.
Youderian P, Hartzell PL: Triple mutants uncover three new genes required for social motility in Myxococcus xanthus. Genetics. 2007, 177: 557-566. 10.1534/genetics.107.076182.
Kung VL, Khare S, Stehlik C, Bacon EM, Hughes AJ, Hauser AR: An rhs gene of Pseudomonas aeruginosa encodes a virulence protein that activates the inflammasome. Proc Natl Acad Sci U S A. 2012, 109: 275-280.
Stavrinides J, No A, Ochman H: A single genetic locus in the phytopathogen Pantoea stewartii enables gut colonization and pathogenicity in an insect host. Environ Microbiol. 2010, 12: 147-155. 10.1111/j.1462-2920.2009.02056.x.
Hurst MR, Glare TR, Jackson TA, Ronson CW: Plasmid-located pathogenicity determinants of Serratia entomophila, the causal agent of amber disease of grass grub, show similarity to the insecticidal toxins of Photorhabdus luminescens. J Bacteriol. 2000, 182: 127-138.
Hurst MR, Jones SA, Binglin T, Harper LA, Jackson TA, Glare TR: The main virulence determinant of Yersinia entomophaga MH96 is a broad-host-range toxin complex active against insects. J Bacteriol. 2011, 193: 1966-1980. 10.1128/JB.01044-10.
Waterfield N, Hares M, Yang G, Dowling A, Ffrench-Constant R: Potentiation and cellular phenotypes of the insecticidal toxin complexes of Photorhabdus bacteria. Cell Microbiol. 2005, 7: 373-382. 10.1111/j.1462-5822.2004.00467.x.
McNulty C, Thompson J, Barrett B, Lord L, Andersen C, Roberts IS: The cell surface expression of group 2 capsular polysaccharides in Escherichia coli: the role of KpsD, RhsA and a multi-protein complex at the pole of the cell. Mol Microbiol. 2006, 59: 907-922. 10.1111/j.1365-2958.2005.05010.x.
Roh E, Heu S, Moon E: Genus-specific distribution and pathovar-specific variation of the glycinerin R gene homologs in Xanthomonas genomes. J Microbiol. 2008, 44: 681-686.
Sisto A, Cipriani MG, Morea M, Lonigro SL, Valerio F, Lavermicocca P: An Rhs like element is involved in bacteriocin production by Pseudomonas savastanoi pv. savastanoi. Antonie Van Leeuwenhoek. 2010, 98: 505-517. 10.1007/s10482-010-9468-7.
Poole SJ, Diner EJ, Aoki SK, Braaten BA, de Roodenbeke TC, Low DA, Hayes CS: Identification of functional toxin/immunity genes linked to contact-dependent growth inhibition (CDI) and rearrangement hotspot (Rhs) systems. PLoS Genet. 2011, 7: e1002217-10.1371/journal.pgen.1002217.
Koskiniemia S, Lamoureuxa JG, Nikolakakisb KC, de Roodenbekea C, Kaplana MD, Lowa DA, Hayesa CS: Rhs proteins from diverse bacteria mediate intercellular competition. Proc Natl Acad Sci U S A. 2013, 110: 7032-7037. 10.1073/pnas.1300627110.
Hill CW: Large genomic sequence repetitions in bacteria: lessons from rRNA operons and Rhs elements. Res Microbiol. 1999, 150: 665-674. 10.1016/S0923-2508(99)00125-4.
Jackson AP, Thomas GH, Parkhill J, Thomson N: Evolutionary diversification of an ancient gene family (rhs) through C-terminal displacement. BMC Genomics. 2009, 10: 584-10.1186/1471-2164-10-584.
Landsber MJ, Jones SA, Rothnagel R, Busby JN, Marshall SD, Simpson RM, Lott JS, Hankamer B, Hurst MR: 3D structure of the Yersinia entomophaga toxin complex and implications for insecticidal activity. Proc Natl Acad Sci U S A. 2011, 108: 20544-20529. 10.1073/pnas.1111155108.
Petersen TN, Brunak S, von Heijne G, Nielsen H: SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011, 8: 785-786. 10.1038/nmeth.1701.
Kroght A, Larsson B, von Heijne G, Sonnhamer EL: Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001, 305: 567-580. 10.1006/jmbi.2000.4315.
Gardy JL, Laird MR, Chen F, Rey S, Walsh CJ, Ester M, Brinkman FSL: PSORTb v. 2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics. 2005, 21: 617-623. 10.1093/bioinformatics/bti057.
Fokine A, Miroshnikov KA, Shneider MM, Mesvanzhinov VV, Rossmann MG: Structure of the bacteriophage phi KZ lytic transglycosylase gp144. J Biol Chem. 2008, 283: 7242-7250. 10.1074/jbc.M709398200.
Patzer SI, Albrecht R, Braun V, Zeth K: Structural and mechanistic studies of pesticin, a bacterial homolog of phage lysozymes. J Biol Chem. 2012, 287: 23381-23396. 10.1074/jbc.M112.362913.
Vollmer W, Pilsl H, Hantke K, Holtje J-V, Braun V: Pesticin displays muramidase activity. J Bacteriol. 1997, 179: 1580-1583.
Scheurwater E, Reid CW, Clarke AJ: Lytic transglycosylases: bacterial space-making autolysin. Int J Biochem Cell Biol. 2008, 40: 586-591. 10.1016/j.biocel.2007.03.018.
Vollmer W, Joris B, Charlier P, Foster S: Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol Rev. 2008, 32: 259-286. 10.1111/j.1574-6976.2007.00099.x.
Russell AB, Hood RD, Bui NK, LeRoux M, Vollmer W, Mougous JD: Type VI secretion delivers bacteriolytic effectors to target cells. Nature. 2011, 475: 34-347.
Davies JK, Reeves P: Genetics of resistance to colicins in Escherichia coli K-12: cross-resistance among colicins of group A. J Bacteriol. 1975, 123: 102-117.
Pilsl H, Smajs D, Braun V: Characterization of colicin S4 and its receptor, OmpW, a minor protein of the Escherichia coli outer membrane. J Bacteriol. 1999, 181: 3578-3581.
Hoell IA, Dalhus B, Heggset EB, Aspmo SI, Eijsink VGH: Crystal structure and enzymatic properties of a bacterial family 19 chitinase reveal differences from plant enzymes. FEBS J. 2006, 273: 4889-4900. 10.1111/j.1742-4658.2006.05487.x.
Wang J, Gebre AK, Anderson RA, Parks JS: Cloning and in vitro expression of rat lecithin: cholesterol acyltransferase. Biochim Biophys Acta. 1997, 1346: 207-211. 10.1016/S0005-2760(97)00055-6.
Barret M, Egan F, Fargier E, Morrissey JP, O’Gara F: Genomic analysis of the type VI secretion systems in Pseudomonas spp: novel clusters and putative effectors uncovered. Microbiology. 2011, 157: 1726-1740. 10.1099/mic.0.048645-0.
Kawano M, Aravind L, Storz G: An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Mol Microbiol. 2007, 64: 738-754. 10.1111/j.1365-2958.2007.05688.x.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S: MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013, 30: 2725-2729. 10.1093/molbev/mst197.
Abascal F, Zardoya R, Posada D: ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005, 21: 2104-2105. 10.1093/bioinformatics/bti263.
Le SQ, Gascuel O: An improved general amino acid replacement matrix. Mol Biol Evol. 2008, 25: 1307-1320. 10.1093/molbev/msn067.
Mattinen L, Nissinen R, TeroRiipi T, Kalkkinen N, Pirhonen M: Host-extract induced changes in the secretome of the plant pathogenic bacterium Pectobacterium atrosepticum. Proteomics. 2007, 7: 3527-3537. 10.1002/pmic.200600759.
Mattinen L, Somervuo P, Nykyri P, Nissinen R, Kouvonen P, Corthals G, Auvinen P, Aittamaa M, Valkonen JPT, Pirhonen M: Microarray profiling of host-extract induced genes and characterization of the type VI secretion cluster in the potato pathogen Pectobacterium atrosepticum. Microbiol. 2008, 154: 2387-2396. 10.1099/mic.0.2008/017582-0.
Liu H, Coulhurst SJ, Pritchard L, Hedley PE, Ravensdale M, Humphris S, Burr T, Takle G, Brurberg MB, Birch PR, Salmond GPC, Toth IK: Quorum sensing coordinates brute force and stealth modes of infection in the plant pathogen Pectobacterium atrosepticum. PLoS Pathog. 2008, 4: e1000093-10.1371/journal.ppat.1000093.
Records AR, Gross DC: Sensor kinases RetS and LadS regulate Pseudomonas syringae type VI secretion and virulence factors. J Bacteriol. 2010, 192: 3584-3596. 10.1128/JB.00114-10.
Dimmic MW, Rest JS: Mindell DP: rtREV: an amino acid substitution matrix for inference of retrovirus and reverse transcriptase phylogeny. J Mol Evol. 2002, 55: 65-73. 10.1007/s00239-001-2304-y.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
Zdobnov EM, Apweiler R: InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001, 17: 847-848. 10.1093/bioinformatics/17.9.847.
Marchler-Bauer A, Anderson JB, Derbyshire MK, DeWeese-Scott C, Gonzales NR, Gwadz M, Hao L, He S, Hurwitz DI, Jackson JD, Ke Z, Krylov D, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Thanki N, Yamashita RA, Yin JJ, Zhang D, Bryant SH: CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res. 2007, 35: D237-D240. 10.1093/nar/gkl951.
Söding J, Biegert A, Lupas AN: The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33: W244-W248. 10.1093/nar/gki408.
Tatusov RL, Galperin MY, Natale DA, Koonin EV: The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28: 33-36. 10.1093/nar/28.1.33.
Acknowledgements
This study was partially supported by the University of Pretoria, the National Research Foundation (NRF), the Forestry and Agricultural Biotechnology Institute (FABI), the Tree Protection Co-operative Programme (TPCP), the NRF/Department of Science and Technology Centre of Excellence in Tree Health Biotechnology (CTHB), and the THRIP support program of the Department of Trade and Industry, South Africa. We acknowledge Zander Human for assistance with phylogenetic analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DYS, LNM, SNV, IKT and TAC conceived the study. DYS performed experiments and analysis. DYS, LNM, SNV, IKT and TAC wrote the original manuscript. All authors read and approved the final version.
Electronic supplementary material
12864_2013_5789_MOESM1_ESM.xls
Additional file 1: Table S1: BLASTP analysis of type VI secretion system (T6SS) homologs found in Pantoea ananatis strain LMG 20103. This table shows the percentage amino acid identify between the T6SS gene products of P. ananatis T6SS-1 to 3. The gene products of LMG 20103 T6SS-1 were used as bait for BlastP analysis. (XLS 32 KB)
12864_2013_5789_MOESM2_ESM.xls
Additional file 2: Table S2: List of primers used for PCR amplification of type VI secretion system genes from different strains of Pantoea ananatis. This file contains PCR primer sequences and the T6SS genes of P. ananatis which were targeted for Polymerase Chain Reaction (PCR) amplification. (XLS 34 KB)
12864_2013_5789_MOESM3_ESM.xls
Additional file 3: Table S3: Strains of Pantoea ananatis tested for the presence of T6SS gene clusters. This file contains the list of all strains of Pantoea ananatis screened for the presence or absence type VI secretion system gene homologs. The file also contains information on the host of isolation and country from which the different strains of P. ananatis were isolated from. (XLS 36 KB)
12864_2013_5789_MOESM4_ESM.xls
Additional file 4: Table S4: Distribution of type VI secretion system gene clusters in 46 strains of Pantoea ananatis. This file contains results of the PCR screen, which clearly shows the distribution of type VI secretion system genes in all strains of Pantoea ananatis analysed in this study. (XLS 52 KB)
12864_2013_5789_MOESM5_ESM.doc
Additional file 5: Figure S1: Synteny between Pantoea ananatis type VI secretion system 1, 2 and 3 (PA T6SS-1, 2, 3). This file contains pairwise alignment of the homologous type VI secretion system gene clusters found in sequenced strains of P. ananatis. Alignments were generated using Mauve v.2.3.1 and show variable and conserved regions between the homologous T6SS gene clusters from different strains of P. ananatis. (DOC 584 KB)
12864_2013_5789_MOESM6_ESM.xls
Additional file 6: Table S5-Table S12: Gene content of type VI secretion system 1 (T6SS-1) found in all sequenced strains of Pantoea ananatis. This file contains all genes found within the contiguous region representing type VI secretion system 1 (PA T6SS-1) in all eight sequenced strains of Pantoea ananatis analysed in this study. For each strain of P. ananatis, the genes found in T6SS-1 are shown, including the product sizes, cluster of orthologous groups of proteins (COG) classification, conserved domain, subcellular localization, and the gene locus tags. (XLS 135 KB)
12864_2013_5789_MOESM7_ESM.xls
Additional file 7: Table S13-Table S15: Gene content of type VI secretion system 2 (T6SS-2) in sequenced strains of Pantoea ananatis. This file contains all genes found within the contiguous region representing type VI secretion system 2 (PA T6SS-2) in different strains of Pantoea ananatis. For each strain of P. ananatis, the genes found in T6SS-2 are shown, including the product sizes, COG classification, conserved domain, subcellular localization, and the gene locus tags. (XLS 64 KB)
12864_2013_5789_MOESM8_ESM.docx
Additional file 8: Figure S2: Alignment of the RhsD and RhsI proteins associated with Pantoea ananatis type VI secretion system 1. A) Shows alignment of the variable C-terminal domain of RhsD proteins from eight different strains of P. ananatis. This variable C-terminal domain is demarcated from the conserved N-terminal domain by a PxxxxxxDPxGL peptide motif indicated in the figure by blue stars. B) Shows alignment of the full length RhI proteins encoded by rhsI genes located downstream of the rhsD gene. Conserved residues are indicated by the different shadings. P. ananatis strains B1-9, LMG 2665T and LMG 5342 have identical C-terminal extensions and encode identical RhsI homologs. (DOCX 363 KB)
12864_2013_5789_MOESM9_ESM.doc
Additional file 9: Figure S3: Alignment of all representative Hcp proteins encoded by strains of Pantoea ananatis. Representative Hcp amino acid sequences were aligned in BioEdit using ClustalW2 with default settings. The orphan Hcp proteins of P. ananatis strain LMG 20103 encoded by PANA_2446 and PANA_2447 are highly similar to the T6SS-associated Hcp protein encoded by PANA_2364. P. ananatis strain PA-13 encodes an additional orphan Hcp protein (PAGR_g3636) which is unique to this strain and is highly divergent from all other Hcp proteins. (DOC 59 KB)
12864_2013_5789_MOESM10_ESM.xls
Additional file 10: Table S16: List of bacterial strains used for TssBC phylogenetic analysis. This file contains the list of TssB and TssC homologs from different bacterial species which were used for phylogenetic analysis. The accession numbers of all bacterial strains used in this study including the tssB and tssD gene locus tags are provided. (XLS 48 KB)
12864_2013_5789_MOESM11_ESM.xls
Additional file 11: Table S17: List of bacterial strains used for Hcp phylogenetic analysis. This file contains all TssD homologs used for phylogenetic analysis. The accession numbers of all bacterial strains used in this study including the tssD gene locus tags are provided. (XLS 33 KB)
12864_2013_5789_MOESM12_ESM.doc
Additional file 12: Figure S4: Hcp phylogenetic tree. The figure shows the evolutionary history of Hcp and “orphan” Hcp proteins of Pantoea ananatis. Phylogenetic analysis were conducted in MEGA6 [90]. The evolutionary history was inferred using the Maximum Likelihood method based on the General Reverse Transcriptase (rtRev) + I + G + F amino acid substitution models, as determined by ProtTest [91, 97]. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. (DOC 46 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Shyntum, D.Y., Venter, S.N., Moleleki, L.N. et al. Comparative genomics of type VI secretion systems in strains of Pantoea ananatisfrom different environments. BMC Genomics 15, 163 (2014). https://doi.org/10.1186/1471-2164-15-163
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-163