
Abstract
Background
The intestinal microbiota is a complex and diverse ecosystem that plays a significant role in maintaining the health and well-being of the mammalian host. During the last decade focus has increased on the importance of intestinal bacteria. Several molecular methods can be applied to describe the composition of the microbiota. This study used a new approach, the Gut Microbiotassay: an assembly of 24 primer sets targeting the main phyla and taxonomically related subgroups of the intestinal microbiota, to be used with the high-throughput qPCR chip ‘Access Array 48.48′, AA48.48, (Fluidigm®) followed by next generation sequencing. Primers were designed if necessary and all primer sets were screened against DNA extracted from pure cultures of 15 representative bacterial species. Subsequently the setup was tested on DNA extracted from small and large intestinal content from piglets with and without diarrhoea. The PCR amplicons from the 2304 reaction chambers were harvested from the AA48.48, purified, and sequenced using 454-technology.
Results
The Gut Microbiotassay was able to detect significant differences in the quantity and composition of the microbiota according to gut sections and diarrhoeic status. 454-sequencing confirmed the specificity of the primer sets. Diarrhoea was associated with a reduced number of members from the genus Streptococcus, and in particular S. alactolyticus.
Conclusion
The Gut Microbiotassay provides fast and affordable high-throughput quantification of the bacterial composition in many samples and enables further descriptive taxonomic information if combined with 454-sequencing.
Similar content being viewed by others


Background
Immediately after birth the mammalian gastrointestinal tract is colonized by a complex and diverse microbial ecosystem. The bacterial invasion and the following gut microbial composition has an enormous impact on its host’s health and well-being [1]. To gain a better understanding of this complex ecosystem, culture-independent methods are essential, as a considerable fraction of the intestinal microbiota has not yet been cultured [2]. One of the ongoing controversies is how to study the bacterial composition in complex ecosystems. To date, some of the widely used approaches to characterize the intestinal microbiota are: metagenomics, phylogenetic microarrays, DNA fingerprinting techniques, and qPCR [2–5]. These methods provide different degrees of taxonomic as well as quantitative information on the microbiota [5]. Nonetheless, variation in technical procedures and differences in data treatment and interpretation makes it challenging to compare results between studies. Also, the expenses and time consumption in relation to labour intensity and data analysis vary greatly. Especially metagenomic approaches are receiving increased attention in the study of microbial communities as a result of their shorter sequencing speed, extended read length, and lower costs [5, 6]. However, the enormous amount of data generated becomes cumbersome to analyse, and requires lots of dedicated time as well as expertise to manage [6]. The Access Array 48.48, AA48.48, (Fluidigm Corporation, South San Francisco, CA, USA) creates an affordable link between high-throughput qPCR and next generation sequencing (NGS) and provides manageable data with valuable quantitative and taxonomic information.
Since the 1990’s, qPCR has been applied widely due to its quantitative precision, high specificity and sensitivity, broad dynamic range, good reproducibility, and relatively low costs [7]. In general, qPCR is quick to perform with a low to medium throughput, since most qPCR platforms have a capability format of 96 or 384 [7]. The high-throughput qPCR chip AA48.48 combines 48 detector inlets and 48 sample inlets by interconnecting channels into 2304 individual reaction chambers (singleplex) http://www.fluidigm.com/access-array-system.html. In contrast to most qPCR platforms which require reaction volumes between 5 to 100 μl [7], the AA48.48 operates with a reaction volume of approximately 35 nl [8]. By tagging primers at their 5′-end (using the ‘The Access Array 4-primer amplicon tagging strategy’ [8]), and including a 454 Barcode Library (454BL) with the samples, amplicon generation and library preparation is achieved in the same reaction. Afterwards, amplicons can be harvested directly from the AA48.48 sample inlets where the respective samples were initially loaded. The unique barcodes and specific primers enable segregation of the pooled samples in NGS analysis later on. The AA48.48 process time from start to finish is approximately five hours. After each qPCR run Fluidigm Real-Time PCR Analysis software (Fluidigm Corporation) generates a heatmap constructed as a grid of 48 × 48 small squares presenting all the 2304 primer sample combinations. The heatmap depicts the Cq-value for each reaction according to a colour scale, providing a good overview of each sample and the possibility to compare the bacterial profiles across all the samples.
This study describes the design, optimization, and validation of the Gut Microbiotassay: a primer setup consisting of 24 primer sets targeting the main bacterial phyla of the intestinal microbiota at various taxonomic levels, to be used with the AA48.48 in combination with NGS. Furthermore, it demonstrates the applicability of the Gut Microbiotassay on luminal content collected from the small and large intestine of piglets of different diarrhoeic status. Finally, it validates the specificity of the Gut Microbiotassay by sequencing these amplicons. To our knowledge, the AA48.48 has not previously been used to investigate microbial communities.
Methods
Development of the Gut Microbiotassay
Primer design
Inspired by Rajilic-Stojanovic et al. [9], the primer setup was designed to target the ribosomal RNA genes (16S or 23S) of the major bacterial groups present in the mammalian intestinal microbiota, including the phyla: Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria [10]. To gain insight into the bacterial composition, different taxonomic levels were represented: domain, phylum, class, family, genus, and species-specific primers (Table 1).
Primer specificity was checked in silico with the ‘probe match’ facility of the Ribosomal Database Project, release 10 (http://rdp.cme.msu.edu/) [11] and the ‘check’ application of ProbeCheck (http://131.130.66.200/cgi-bin/probecheck/content.pl?id=home) [12] using the programs’ default settings. Judged by the search results from these online databases, primers were renamed according to their main target. Some of the published primers were slightly modified to improve their specificity. New primers were designed using the ‘Probe Design Tool’ of the ARB Software package (http://www.arb-home.de/) [13], or Primrose [14] using default options, and further validated based on BLAST search (NCBI). Primers were designed to have approximately same length of nucleotides, GC-content, a minimum number of degenerate bases, and to produce amplicons between 200–500 bp long, compatible with the read length of the 454 GS FLX Titanium Sequencer. Similar properties were important, given that all the primers had to function under the same conditions when run on an AA48.48.
All primers, except for the species-specific ones, were tagged at their 5′-end to enable incorporation of a 454BL necessary for 454-sequencing: forward tag: 5′-ACACTGACGACATGGTTCTACA-3′, and reverse tag: 5′-TACGGTAGCAGAGACTTGGTCT-3′ in accordance to the ‘Access Array™ System User Guide’ [8]. Primers were purchased from Eurofins MWG Synthesis GmbH (Ebersberg, Germany) and stored at -20°C.
Empirical testing of primers
All primers were tested against 15 strains of pure-cultured reference bacteria, representing targets for one or more of the different primer sets (Table 1). Thus the reference bacterial DNA functioned as both positive and negative controls for the individual primer sets.
DNA extraction
Chromosomal DNA from cultured reference bacteria (Additional file 1: Table S1) was isolated using the Easy-DNA™ Kit (Invitrogen, Carlsbad, CA, USA), for further details on the DNA extraction protocol see Additional file 2. DNA was precipitated with ethanol, and resuspended in 60 μl TE buffer. The reference bacteria were cultured as recommended by DSMZ (http://www.dsmz.de/).
An interplate calibrator (IPC) was included in all AA48.48, consisting of bacterial DNA extracted from ~ 100 mg colonic content from a healthy conventional pig, 14-week-old, Danish landrace. Intestinal content was collected immediately after euthanization and frozen at – 80°C. A 10% PBS suspension was made from the intestinal content, and from here on, the protocol was identical to the one used for the reference bacteria.
DNA concentration and purity were assessed by the 260/280 nm-ratio using the Nanodrop® ND-1000 (NanoDrop Technologies Inc., Wilmington, Germany) spectrophotometer (Additional file 2). DNA was stored at -20°C until needed.
Verifying the Gut Microbiotassay on the Access Array 48.48
Tenfold serial dilutions ranging from 0.5 pg/μl to 50 ng/μl were made from DNA extracted from 15 reference bacteria. The AA48.48 was processed following the ‘Access Array System™ User Guide’ [8]. In short: primer sets were mixed in equal concentrations and diluted to 4 μM with 20 × Access Array Loading Reagent (Fluidigm, South San Francisco, CA, USA) and nuclease-free water (Ambion Inc., Austin, USA). Master Mix was prepared as described in the instructions, and sample solutions were produced from 4 μl Master Mix and 1 μl DNA. The AA48.48 was primed in a ‘pre-PCR’ IFC controller AX (Fluidigm), before it was processed in the Biomark (Fluidigm) using the Fluidigm ‘AA 48 × 48 Standard v1’-protocol listed in Table 2. Subsequently the amplicons were harvested in a ‘post-PCR’ IFC controller AX (Fluidigm).
Harvested amplicons were measured on the Agilent DNA 1000 chip (Agilent Technologies, Waldbronn, Germany) with the Agilent 2100 Bioanalyzer (Agilent Technologies) to verify the specificity of the Gut Microbiotassay. This was assessed from the size and number of amplicons generated by the primer sets listed in Table 1, with the reference bacteria as targets.
Three primer concentrations (1, 2 and 4 μM), and three primer:454BL-ratios (1:0.2, 1:0.4 and 1:0.8 μM) were tested, before settling on the final protocol.
Testing the Gut Microbiotassay on complex samples
Samples and sampling
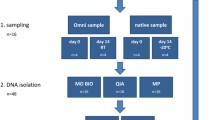
To explore the efficiency range of the Gut Microbiotassay, and test its sensitivities under varying circumstances, the study included 12 three-day-old piglets from conventional pig-farms (Danish landrace); seven with and five without clinical diarrhoea (Additional file 3: Table S2). None of the animals had received any treatment. Piglets were sacrificed and luminal content from the small and large intestine were obtained immediately after and frozen at -80°C. All handling of the animals was in accordance with regulations from The Danish Centre for Animal Welfare.
DNA extraction
100 mg intestinal content was suspended in 900 μl PBS and bead-beated in 2 ml microcentrifuge tubes containing a 5 mm steel bead (Qiagen, Hilden, Germany) at 15.0 hertz for 2.3 min (Tissuelyser II, Qiagen). Tubes were centrifuged at 10,000 × g, 90 s, 20°C, and 350 μl of supernatant were transferred to new tubes. These were placed in the QIAsymphony SP (Qiagen) for DNA extraction using the QIAsymphony Virus/Bacteria Mini Kit (Qiagen) with the protocol ’Pathogen complex 200′ (Qiagen), elution volume: 60 μl. DNA was measured as previously mentioned and stored at -20°C until further processing (Additional file 3: Table S2).
Analysing complex samples on the AA48.48 with the Gut Microbiotassay
To test the amplification efficiency on DNA extracted from intestinal content ‘sample-calibration-curves’ were constructed from tenfold serial dilutions (0.25 pg/μl - 25 ng/μl). These were made from pooled sample material of 1 μl extracted DNA from each of the samples listed in Additional file 3: Table S2. ‘Control-calibration-curves’ were constructed from the IPC, and ‘reference-calibration-curves’ were generated by pooling all reference bacteria in equal concentrations.
DNA extracted from the pig samples included in the study was diluted to 50 ng/μl with nuclease-free water (Ambion). To test how the addition of a 454BL affected the Cq values, an AA48.48 with a 454BL and one without a 454BL were run under the exact same conditions. With a 454BL, each sample mixture consisted of 3 μl Master Mix, 1 μl sample (DNA, 50 ng/μl), and 1 μl Access Array Barcode Library for the 454 GS FLX Titanium Sequencer (Fluidigm), 2 μM. Afterwards the amplicons were harvested and stored at -20°C until needed.
Amplicon preparation for NGS
To normalize the harvested sample amplicons for NGS, they were measured as described earlier and pooled in equal concentrations. This resulted in a total volume of 144.27 μl of 16 ng/μl which was purified to remove any PCR by-products. First the volume was reduced to 15 μl by extracting the DNA with phenol chloroform using a standard procedure [15]. Next, the extracted DNA was run in a 0.7% Seakem® LE Agorose gel (Lonza Rockland, Rockland, ME, USA) for 86 min, 90 V, and incubated for 30 min in 0.0004% ethidium bromide. Bands were visualized with UV-radiation using the Bio-Rad Universal hood II (Segrate, Milan, Italy) and bands in the size range 200–900 bp were excised, equalling expected amplicon sizes. DNA was extracted from the excised gel using the Qiaquick Gel Extraction Kit (Qiagen) in accordance with the kit manual.
The final pool of purified DNA from the 12 piglets to be sequenced was 723.25 ng (260/280-nm ratio: 1.96). This was run on a quarter PicoTiterPlate™ on a 454 GS FLX Titanium Sequencer (Roche) by LGC Genomics (GmbH, Berlin, Germany).
Data analysis
From raw Cq values to relative quantitative data
Cq values generated from the AA48.48 without a 454BL added were exported from the ‘Fluidigm Real-Time PCR Analysis software’, version 3.0.2 (Fluidigm), to Excel. Cq values were corrected to the IPC included in all runs, and those exceeding primer specific cut-off values, determined from the verification step of the Gut Microbiotassay (Additional file 4: Table S3), were excluded. Relative quantification was calculated from the mean of the technical replicates using the Livak-method [16]. This method was chosen based on the theory that the total amount of bacteria, targeted by the primer set domain Bacteria B, constituted 100% of the microbiota in the individual gut section at all times. Hence, the Cq values of all primer sets for each sample were normalised against the Cq value of their respective domain Bacteria B primer set: . To compare total bacteria detected with the domain Bacteria B primer set, these Cq values were related to the final number of thermal cycles run, 35: . Normalization was further done to total mean of all primer sets for each sample with similar results (data not shown).
454-sequencing data
Sequence data, available at NCBI Sequence Read Archive under Accession SRA061551, was analysed using BION, a yet unpublished open source program. For more information on the BION software, its functions, and the main statistics for the raw results see Additional file 5. In short: the sequence dataset was converted to FASTQ, split according to sample barcodes and primer sequences, and trimmed in the same process. Next, sequences were cleaned using a cut-off for minimum quality of 96%, and a minimum sequence length of 200 bp. The remaining sequences were clustered using UCLUST, based on a minimum seed similarity of 99.5%. Query sequences were compared to Greengenes Gene Database [17] using the k-mer matching program Simrank2, an improved version of Simrank [18]. Simrank2 returns the n % best similarities, no matter how low they are. Also, it only produces 8-mers from regions above a given quality, and skips sequences with too few 8-mers. This last feature helps improve the data quality. Simrank2 was set to return the best 1% similarities with a similarity cut-off of 50%. Taxonomy was generated by transforming percentages for the Greengenes OTUs to scores, read densities, the sum of which was 1. This was done in a weighted manner, so that OTUs of the highest similarity scored a high number and vice versa, the sum of which was the original reads. Only phylotypes with a primer-specific read density of ≥ 1% were included in the statistical analysis.
Statistics
Initially, all data were transformed with the natural logarithm. Primer sets with more than half of the data missing were removed from the dataset. If primer sets in taxonomic lineage showed pair-wise correlations above 0.99, only the primer set of the highest taxonomic level in the lineage was retained for analysis, and any conclusion drawn from this also accounted for the excluded sub-level primer sets. To include the information from remaining data-deficient primer sets, for which the fraction of missing data was low and never above 0.35, the EM-algorithm [19] was used to substitute missing values with imputed ones, by applying a multivariate Gaussian model. Each primer set was allowed to depend freely on the others and also for dependency on gut section, diarrhoeic status, and interaction between these. Model fit of the multivariate model for the primer sets after imputation was assessed by transforming the model residuals with the inverse of the square root of the estimated covariance matrix between the primer sets, and applying standard model control to these standardized residuals. These analyses were consistent with standard model behaviour.
Effect of gut section and diarrhoeic status was tested with the Likelihood Ratio Method using the Wilks test [20]. Sequence data were compared for effect of diarrhoeic status using the non-parametric Wilcoxon test [21].
P-values < 0.05 were considered statistically significant.
Results
Designing the Gut Microbiotassay
The Gut Microbiotassay was constructed from 49 primers constituting 24 primers sets (Table 1) in order to target the main bacterial Phyla (Actinobacteria, Bacteroides, Firmicutes, Fusobacteria, Proteobacteria and Verrucomicrobia) and some of the highly important bacterial groups and species reported in the mammalian intestinal tract [9]. 12 primers were designed de novo, and 37 primers were from published literature, of which ten were modified. Five of the 24 primer sets were unmodified pairs from published literature, whereas the rest were combinations composed in this study. As it was not possible to find or design specific primer sets targeting the 16S rRNA gene for class β- and γ-proteobacteria, and genus Lactobacillus, these were designed to target the 23S rRNA gene instead.
Sensitivity and specificity of the Gut Microbiotassay
Tenfold serial dilutions of DNA extracted from 15 reference bacteria were used to evaluate the specificity and sensitivity of the Gut Microbiotassay (Figure 1).

Heatmap generated by the Fluidigm Real-Time PCR Analysis program from raw Cq data. At the top horizontal: Tenfold serial dilutions of DNA (ranging from 50 ng/μl down to 0.5 pg/μl) extracted from 15 different reference bacteria. Raised numbers in parentheses represent the respective numbers given to each reference bacterium in Table 1. These were used to test the specificity and sensitivity of the primer sets included in the Gut Microbiotassay, listed vertically on the left. Primers were run in duplicates. On the right: a colour scale depicting the respective Cq values (the software uses commas instead of points as decimal separator for Cq values).
The specificity for each primer set was assessed from the Cq values obtained for their respective target bacteria in addition to any cross reaction. For 13 of the 24 primer sets, specific positive reactions were registered on various taxonomic levels in agreement with the reference bacteria listed for each primer set in Table 1. The remaining primer sets showed different degrees of cross reaction (Figure 1). On average, the lowest unspecific and the highest specific Cq of same concentration differed by 9 Cq values, equalling less than 1% cross reaction. Apparently, the primer set phylum Firmicutes did not have a complete coverage, as it only amplified few bacteria from the Clostridia clusters. The highest specific Cq value was determined for each primer set from the representative target bacterial species and used as cut-off value in the data analysis (Additional file 4: Table S3).
The dynamic range for the specific primer sets spanned from 50 ng to 50 pg DNA/μl for two primer sets, to 5 pg DNA/μl for 15 primer sets, and for seven primer sets down to 0.5 pg DNA/μl (lowest concentration tested), Table 3. Linear regression of log-concentration versus Cq values for the different primer sets demonstrated r2 from 0.943 to 0.999 (SD 0.0074). Gel pictures and electropherograms confirmed specific amplification by demonstrating the expected number of amplicons corresponding to the number of taxonomic levels represented by the specific primer sets. Additionally, amplicon sizes (bp) were comparable to those listed in Table 1.
Fluidigm’s recommendation on DNA sample concentration is 25–50 ng/μl [8]. When testing the amplification efficiency on 23 calibration curves (ranges: 0.25 pg/μl – 50 ng/μl), there was no PCR inhibition for either of the primer sets with DNA of 25 or 50 ng/μl.
Addition of a 454 Barcode Library
Including a 454BL with the reactions did not affect the Cq values drastically. The biggest deviation was less than 1 Cq compared to the respective Cq values without a 454BL added, and 90 percent of the corresponding values from the two data sets were correlated by r2 > 0.90. Uncorrelated data were mainly seen if no or very few target bacteria were present in the reaction. In such cases the 454BL occasionally caused some interaction with itself or/and the present primer set, creating a signal that obscured the low or missing signal from the target bacteria. Because of this, samples were run twice: with and without a 454BL added. Cq values without a 454BL were used for data analysis, whereas amplicons with the 454BL incorporated were available for 454-sequencing.
The Gut Microbiotassay on complex samples
Piglets of different diarrhoeic status were sacrificed and luminal content was collected from the small and the large intestine. This resulted in a total of 23 samples divided into the following groups: small intestine without diarrhoea (S-) n = 5, small intestine with diarrhoea (S+) n = 7, large intestine without diarrhoea (L-) n = 4, and large intestine with diarrhoea (L+) n = 7.
AA48.48 general findings
The following primer sets were missing more than half of the data, and were consequently removed from the statistical data analysis: family Clostridium cluster IV, class ϵ-proteobacteria and δ-proteobacteria, phyla Verrucomicrobia, and domain Archeae. Primer sets showing pair-wise correlation above 0.99, were: class β- and γ- proteobacteria with family Enterobacteriacea, and species E. coli; and phylum Bacteroidetes with genus Bacteroides. Of the general bacteria primers only domain Bacteria B was tested as this was the primer set used for normalization. This left 15 primer sets for data analysis. The multivariate test for effect of gut section and diarrhoeic status, with effects allowed to differ with the values of gut section, revealed statistically significant effects (S/L p = 0.01, S+/S - p = 0.002, and L+/L - p = 0.006, respectively). However, effects were not limited to the primer sets that showed significant differences in the Gut Microbiotassay. If these were excluded, and the data analysis repeated, this still resulted in a significant effect from diarrhoeic status in the large intestine (L+/ L - p = 0.01), while it was borderline insignificant in the small intestine (S+/ S - p = 0.056).
Table 4 lists the estimated mean percentages and 95% confidence interval for each primer set included in the data analysis relative to total bacteria. Diarrhoeic gut sections contained fewer bacteria from phylum Firmicutes (genus Streptococcus and unclassified (p < 0.05)), but a higher fraction of genus Enterococcus (small intestine p = 0.04). There was a highly significant depletion of members from genus Streptococcus in the diseased compared to the healthy intestines. The same tendency was seen for Clostridium cluster I (small intestine p = 0.02). The diarrhoeic small intestine harboured fewer members from class β- and γ-proteobacteria than the healthy one, while this was reversed for the large intestine (p < 0.05). Generally healthy piglets had a gut microbiota dominated by Gram-positive bacteria, which was partly displaced by Gram-negative bacteria in diarrhoeic piglets.
454-Sequencing results
Sequencing barcoded sample amplicons resulted in 275,133 unprocessed consensuses, which dropped to 164,055 after quality trimming. Amplicons generated by the primer sets domain Bacteria A and B encompassed 16S rRNA gene sequences from both Gram-positive and Gram-negative bacteria genera plus from unclassified bacteria. Good congruency was found between the primer sets of the Gut Microbiotassay and the 454-sequencing results generated from their respective amplicons (Figure 2).

Specificity of the Gut Microbiotassay. The figure shows the sequencing results and corresponding read density scores (≥ 1%) for the individual primer sets of the Gut Microbiotassay targeting the 16S rRNA gene. Green colours indicate sequences corresponding to the primer sets’ target, whereas red colours represents unspecific amplification. The darker the colour, the higher the read density score.
Comparing significant findings from AA48.48 with 454-sequencing data
Based on the Gut Microbiotassay data for the individual bacterial groups, four groups showed significant differences: domain Bacteria B, genus Streptococcus, phylum Actinobacteria, and class β- and γ-proteobacteria, Table 4. In the small intestine, diarrhoea was associated with a significantly reduced bacterial load (p = 0.003). Sequence data generated by the domain Bacteria B primer set indicated that diarrhoea was associated with a decreased number of members from genus Streptococcus, including S. alactolyticus in both the small intestine (p = 0.0061, and p = 0.0099, respectively) and the large intestine (p = 0.0061 and p = 0.013). When comparing the results for genus Streptococcus, there were significantly fewer Streptococci in the diseased intestines (S+/ S - p = 0.0003, L+/ L - p = 0.00002). The sequences generated by genus Streptococcus confirmed the previous results (S+/ S - p = 0.0061, L+/ L - p = 0.012). These were classified to: genus Streptococcus, species S. hyointestinalis, S. suis, and S. alactolyticus. However, at species level, only S. alactolyticus, showed coherently significant results with genus Streptococcus (S+/ S - p = 0.047, L+/ L - p = 0.012). Phylum Actinobacteria was more abundant in the diarrhoeic compared to the healthy small intestine (p = 0.034), however, this was not reflected in the sequencing data for the small intestine. The diarrhoeic large intestine possessed significantly more members from the class β- and γ-proteobacteria, including family Enterobacteriaceae and species E. coli, than the healthy one (p = 0.029). Nonetheless, for this gut section no significant differences showed up in the sequencing data from the family Enterobacteriaceae primer set.
Specificity of the Gut Microbiotassay
After sequencing the amplicons generated by the Gut Microbiotassay the specificity of the primer sets was revaluated (Figure 2), except for the primer sets targeting the 23S rRNA gene (genus Lactobacillus and class β- and γ-proteobacteria) as the freeware used to analyze the sequence data is currently based on 16S rRNA gene databases.
16 of the 24 primer sets only generated sequences from their intended target group, four showed cross reaction with a single group, and two primer sets cross reacted with more than two other taxonomic groups. The primer set phylum Firmicutes did not reveal any clostridia, as predicted earlier. In conclusion, the sequencing data confirmed the specificity of the primer sets found in the verification process of the Gut Microbiotassay when tested against DNA extracted from pure-cultured reference bacteria.
Discussion
Most qPCR studies describing the gut microbiota typically do this by using a general bacteria primer and a few group-specific ones [4, 22]. In contrast, this study developed the Gut Microbiotassay, an assay composed of 24 primer systems, capable of screening the microbiota for the most common bacteria in the mammalian intestine [9, 10] at various taxonomic levels. The Gut Microbiotassay was tested against representative reference bacteria, and next on complex intestinal samples from piglets of different diarrhoeic status. The sample amplicons were harvested and sequenced, and functioned as a proof of concept, evaluated the specificity of the Gut Microbiotassay by further elucidating the components of the gut microbiota.
This approach offers an alternative to current molecular methods employed to characterize the gut microbiota such as phylogenetic microarrays [23], and NGS [24]. In contrast to phylogenetic microarrays, the AA48.48 is highly flexible because primer sets can readily be replaced to meet the needs of a current research study. In addition, the AA48.48 outmatches the phylogenetic microarray on sample capacity, as well as sensitivity [25]. Also, no pre-amplification is needed when running the Gut Microbiotassay with the AA48.48, which reduces the workload, and also the risk of introducing technical variation. The effect of such technical variation can be reduced by normalization. In the present study each sample were normalized against the Cq value of their respective domain Bacteria B primer set. Impact of normalization against their respective domain Bacteria B was tested by performing a second normalization procedure; at this point data was also normalized to total mean of all primer sets for each sample individually, with similar end results (data not shown). As the choice of normalization in the present study (Domain Bacteria B) or total mean did not have a great impact on final results we assume that the technical variation was low or that we managed to normalize efficiently using both methods. Further, data from domain Bacteria A and domain Bacteria B were highly correlated also pointing to domain Bacteria B as a reasonable reference primer efficiency of qPCR is of great importance and should ideally be within the range of 85% to 110%. The majority of the primers used in the Gut Microbiotassay (21 out of the 23 primers included in Table 3) had and acceptable efficiency between 80-110%. However, the primer Doman bacteria A was found to differ to much with regards to efficiency and dynamic range, thus data from this primer pair was only used to support data generated from the primer Doman bacteria B. Likewise primer set Phylum Firmicutes produced to high efficiency when tested on two reference bacteria Roseburia sp. and F. Prausnitzii. However Roseburia sp. and F. Prausnitzii are covered by several other well performing primers including Clostridium cluster XIV and Clostridium cluster IV respectively. Therefore the Gut Microbiotassay cannot be used as an absolute quantitative assay across the different primer sets. In order to make it a truly quantitative assay it will be necessary either to have a defined start sample regarding species composition or only to use specific primers on the array where high efficiency has been proven.
The heatmap generated by the software ‘Fluidigm Real-Time PCR Analysis’ (Fluidigm Corporation) depicts the raw Cq values for each reaction. This makes it feasible to quickly evaluate and visually compare the bacterial profiles across a large number of samples. As sample amplicons are harvested individually following qPCR, it is possible to pinpoint which samples to sequence for further taxonomic information. Selective sequencing reduces costs compared to non-selective sequencing. Also, the dataset generated from sequencing the Gut Microbiotassay PCR amplicons produces a much more manageable dataset compared to metagenomic approaches. The Gut Microbiotassay provides a quantitative picture of the distribution of the known gut microbiota represented by the primers. Moreover, if combined with 454-sequencing, it enables detection of bacteria with unidentified sequences [25]. A limitation of the assay when running on complex bacterial samples is that the primers will have different efficiencies and dynamic ranges due to imperfect matches with some of the target sequences. The Gut Microbiotassay has therefore the most value for analysing high-throughput quantification of the bacterial composition in many samples or samples with defined biomarkers.
The validation of the Gut Microbiotassay by sequencing the amplicons from intestinal content of two different gut sections from piglets of different diarrhoeic status demonstrated the potential to further elucidate the components of the gut microbiota. This study used the results from the Gut Microbiotassay to quantify the taxonomical groups, and NGS to access the bacterial constituents.
Common intestinal bacteria in the neonatal piglet include members of: Clostridia, Streptococcaceae, Lactobacillaceae, Enterobacteriaceae, Fusobacteria and sometimes Bacteroidetes [26, 27]. These bacterial groups were also found in the gut microbiota of three-day-old piglets using the Gut Microbiotassay. The Gut Microbiotassay indicated that a healthy gut microbiota was dominated by Gram-positive bacteria, which were partly replaced by Gram-negative bacteria in the large intestine of diarrhoeic piglets. Robinson et al. [28] came to a similar conclusion in a study investigating the intestinal microbiota of pig colons experimentally induced with swine dysentery. Consistent significant findings from the Gut Microbiotassay and the 454-sequencing results implied that diarrhoea was associated with a depletion of members from the genus Streptococcus, and previous research has shown that Streptococci is an important member of a healthy gut microbiota [26, 28, 29]. A detailed review of the aetiology behind the piglet diarrhoea is beyond the scope of this paper and prevented by the limited number of piglets analysed, as the primary focus of this paper has been on the verification and application of the Gut Microbiotassay.
Conclusions
The Gut Microbiotassay offers affordable quantitative screening of the microbiota with the AA48.48. It has been thoroughly tested and strict criteria for data analysis have been outlined. It provides a high sample capacity, a wide dynamic range, and it facilitates selective 454-sequencing afterwards. Hence, it is timesaving and economical due to the easy library preparation, the low consumption of master mix, and the optional selective sequencing. These features make the Gut Microbiotassay a worthy high-throughput competitor to the current alternative methods used for investigating diverse ecosystems.
Abbreviations
- AA48.48:
-
Access array 48.48
- 454BL:
-
454 Barcode library
- IPC:
-
Interplate calibrator
- L:
-
Large intestine
- L+:
-
Large intestine with diarrhoea
- L-:
-
Large intestine without diarrhoea
- NGS:
-
Next generation sequencing
- S:
-
Small intestine
- S+:
-
Small intestine with diarrhoea
- S-:
-
Small intestine without diarrhoea.
References
Sekirov I, Russell SL, Antunes LCM, Finlay BB: Gut microbiota in health and disease. Physiol Rev. 2010, 90: 859-904. 10.1152/physrev.00045.2009.
Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD, Dore J: Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol. 1999, 65: 4799-4807.
Dowd SF, Sun Y, Wolcott RD, Domingo A, Carroll JA: Bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) for microbiome studies: bacterial diversity in the ileum of newly weaned Salmonella-infected pigs. Foodborne Pathog Dis. 2008, 5: 459-472. 10.1089/fpd.2008.0107.
van den Bogert B, de Vos WM, Zoetendal EG, Kleerebezem M: Microarray analysis and barcoded pyrosequencing provide consistent microbial profiles depending on the source of human intestinal samples. Appl Environ Microbiol. 2011, 77: 2071-2080. 10.1128/AEM.02477-10.
Inglis G, Thomas MC, Thomas DK, Kalmokoff ML, Brooks SP, Selinger L: Molecular methods to measure intestinal bacteria: a review. J AOAC Int. 2012, 95: 5-23. 10.5740/jaoacint.SGE_Inglis.
Lamendella R, VerBerkmoes N, Jansson JK: ‘Omics’ of the mammalian gut - new insights into function. Curr Opin Biotechnol. 2012, 23: 491-500. 10.1016/j.copbio.2012.01.016.
Logan JMJ, Edwards KJ: An overview of real-time PCR platforms. Real-Time PCR Current Technology and Application. Edited by: Logan JMJ, Edwards KJ, Saunders NA. 2009, Norfolk, UK: Caister Academic Press, 7-22.
Fluidigm: Access array system™ user guide v3. Part#: 68000158, Rev B. 2010, http://www.fluidigm.com/user-document-request.html,
Rajilic-Stojanovic M, Smidt H, de Vos WM: Diversity of the human gastrointestinal tract microbiota revisited. Environ Microbiol. 2007, 9: 2125-2136. 10.1111/j.1462-2920.2007.01369.x.
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI: Evolution of mammals and their gut microbes. Science. 2008, 320: 1647-1651. 10.1126/science.1155725.
Cole JR, Chai B, Farris RJ, Wang Q, Kulam SA, McGarrell DM, Garrity GM, Tiedje JM: The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res. 2005, 33: D294-D296.
Loy A, Arnold R, Tischler P, Rattei T, Wagner M, Horn M: ProbeCheck - a central resource for evaluating oligonucleotide probe coverage and specificity. Environ Microbiol. 2008, 10: 2894-2896. 10.1111/j.1462-2920.2008.01706.x.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar , Buchner A, Lai T, Steppi S, Jobb G, Forster W, Brettske I, Gerber S, Ginhart AW, Gross O, Grumann S, Hermann S, Jost R, Konig A, Liss T, Lussmann R, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T, et al: ARB: a software environment for sequence data. Nucleic Acids Res. 2004, 32: 1363-1371. 10.1093/nar/gkh293.
Ashelford KE, Weightman AJ, Fry JC: Nucleic Acids Res. 2002, 30 (15): 3481-9. 10.1093/nar/gkf450. Aug 1, PMID: 12140334 [PubMed - indexed for MEDLINE] Free PMC Article
Sambrook J, Russell DW: Purification of nucleic acids by extraction with phenol:chloroform. Cold Spring Harb Protoc. 2006
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods. 2001, 25: 402-408. 10.1006/meth.2001.1262.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL: Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006, 72: 5069-5072. 10.1128/AEM.03006-05.
DeSantis T, Keller K, Karaoz U, Alekseyenko A, Singh N, Brodie E, Pei Z, Andersen G, Larsen N: Simrank: rapid and sensitive general-purpose k-mer search tool. BMC Ecol. 2011, 11: 11-10.1186/1472-6785-11-11.
Little RJA, Rubin DB: Statistical analysis with missing data. 1987, New York: Wiley
Anderson TW: An introduction to multivariate statistical analysis. 1984, New York: John Wiley, 2
Lehmann EL: Nonparametrics. Statistical Methods Based on Ranks, Revised edition. 2006, New York: Springer
Castillo M, Martin-Orue SM, Manzanilla EG, Badiola I, Martin M, Gasa J: Quantification of total bacteria, enterobacteria and lactobacilli populations in pig digesta by real-time PCR. Vet Microbiol. 2006, 114: 165-170. 10.1016/j.vetmic.2005.11.055.
Paliy O, Agans R: Application of phylogenetic microarrays to interrogation of human microbiota. Fems Microbiol Ecol. 2012, 79: 2-11. 10.1111/j.1574-6941.2011.01222.x.
Murray DC, Bunce M, Cannell BL, Oliver R, Houston J, White NE, Barrero RA, Bellgard MI, Haile J: DNA-based faecal dietary analysis: a comparison of qPCR and high throughput sequencing approaches. Plos One. 2011, 6 (10): e25776-10.1371/journal.pone.0025776. doi: 10.1371/journal.pone.0025776
Everett KR, Rees-George J, Pushparajah IPS, Janssen BJ, Luo Z: Advantages and disadvantages of microarrays to study microbial population dynamics - a minireview. New Zealand Plant Protection. 2010, 63: 1-6.
Petri D, Hill JE, Van Kessel AG: Microbial succession in the gastrointestinal tract (GIT) of the preweaned pig. Livest Sci. 2010, 133: 107-109. 10.1016/j.livsci.2010.06.037.
Ducluzeau R: Implantation and development of the gut flora in the newborn animal. Ann Rech Vet. 1983, 14: 354-359.
Robinson IM, Whipp SC, Bucklin JA, Allison MJ: Characterization of predominant bacteria from the colons of normal and dysenteric pigs. Appl Environ Microbiol. 1984, 48: 964-969.
Leser TD, Amenuvor JZ, Jensen TK, Lindecrona RH, Boye M, Moller K: Culture-independent analysis of gut bacteria: the pig gastrointestinal tract microbiota revisited. Appl Environ Microbiol. 2002, 68: 673-690. 10.1128/AEM.68.2.673-690.2002.
Liu ZZ, DeSantis TZ, Andersen GL, Knight R: Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res. 2008, 36: e120-10.1093/nar/gkn491.
Schwieger F, Tebbe CC: A new approach to utilize PCR-single-strand-conformation polymorphism for 16s rRNA gene-based microbial community analysis. Appl Environ Microbiol. 1998, 64: 4870-4876.
Haakensen M, Dobson CM, Deneer H, Ziola B: Real-time PCR detection of bacteria belonging to the Firmicutes Phylum. Int J Food Microbiol. 2008, 125: 236-241. 10.1016/j.ijfoodmicro.2008.04.002.
Mühling M, Woolven-Allen J, Murrell JC, Joint I: Improved group-specific PCR primers for denaturing gradient gel electrophoresis analysis of the genetic diversity of complex microbial communities. ISME J. 2008, 2: 379-392. 10.1038/ismej.2007.97.
Felske A, Akkermans ADL, de Vos WM: In situ detection of an uncultured predominant bacillus in Dutch grassland soils. Appl Environ Microbiol. 1998, 64: 4588-4590.
Behr T, Koob C, Schedl M, Mehlen A, Meier H, Knopp D, Frahm E, Obst U, Schleifer KH, Niessner R, Ludwig W: A nested array of rRNA targeted probes for the detection and identification of enterococci by reverse hybridization. Syst Appl Microbiol. 2000, 23: 563-572. 10.1016/S0723-2020(00)80031-4.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG: Clustal W and clustal X version 2.0. Bioinformatics. 2007, 23: 2947-2948. 10.1093/bioinformatics/btm404.
Rinttila T, Kassinen A, Malinen E, Krogius L, Palva A: Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J Appl Microbiol. 2004, 97: 1166-1177. 10.1111/j.1365-2672.2004.02409.x.
Hung CH, Cheng CH, Cheng LH, Liang CM, Lin CY: Application of Clostridium-specific PCR primers on the analysis of dark fermentation hydrogen-producing bacterial community. Int J Hydrog Energy. 2008, 33: 1586-1592. 10.1016/j.ijhydene.2007.09.037.
Skånseng B, Kaldhusdal M, Rudi K: Comparison of chicken gut colonisation by the pathogens Campylobacter jejuni and Clostridium perfringens by real-time quantitative PCR. Mol Cell Probes. 2006, 20: 269-279. 10.1016/j.mcp.2006.02.001.
Ramirez-Farias C, Slezak K, Fuller Z, Duncan A, Holtrop G, Louis P: Effect of inulin on the human gut microbiota: stimulation of Bifidobacterium adolescentis and Faecalibacterium prausnitzii. Br J Nutr. 2009, 101: 541-550.
Matsuki T, Watanabe K, Fujimoto J, Takada T, Tanaka R: Use of 16S rRNA gene-targeted group-specific primers for real-time PCR analysis of predominant bacteria in human feces. Appl Environ Microbiol. 2004, 70: 7220-7228. 10.1128/AEM.70.12.7220-7228.2004.
Matsuki T, Watanabe K, Fujimoto J, Miyamoto Y, Takada T, Matsumoto K, Oyaizu H, Tanaka R: Development of 16S rRNA-gene-targeted group-specific primers for the detection and identification of predominant bacteria in human feces. Appl Environ Microbiol. 2002, 68: 5445-5451. 10.1128/AEM.68.11.5445-5451.2002.
Manz W, Amann R, Ludwig W, Vancanneyt M, Schleifer KH: Application of a suite of 16S rRNA-specific oligonucleotide probes designed to investigate bacteria of the phylum cytophaga-flavobacter-bacteroides in the natural environment. Microbiology. 1996, 142: 1097-1106. 10.1099/13500872-142-5-1097.
Franks AH, Harmsen HJM, Raangs GC, Jansen GJ, Schut F, Welling GW: Variations of bacterial populations in human feces measured by fluorescent in situ hybridization with group-specific 16S rRNA-Targeted oligonucleotide probes. Appl Environ Microbiol. 1998, 64: 3336-3345.
Glockner FO, Zaichikov E, Belkova N, Denissova L, Pernthaler J, Pernthaler A, Amann R: Comparative 16S rRNA analysis of lake bacterioplankton reveals globally distributed phylogenetic clusters including an abundant group of actinobacteria. Appl Environ Microbiol. 2000, 66: 5053-5065. 10.1128/AEM.66.11.5053-5065.2000.
Erhart R: PhD thesis. In situ Analyse mikrobieller Biozönosen in Abwasserreinigungsanlagen. 1997, Technical University of Munich: Department of Microbiology
Delroisse JM, Boulvin AL, Parmentier I, Dauphin RD, Vandenbol M, Portetelle D: Quantification of Bifidobacterium spp. and Lactobacillus spp. in rat fecal samples by real-time PCR. Microbiol Res. 2008, 163: 663-670. 10.1016/j.micres.2006.09.004.
Friedrich U, Van Langenhove H, Altendorf K, Lipski A: Microbial community and physicochemical analysis of an industrial waste gas biofilter and design of 16S rRNA-targeting oligonucleotide probes. Environ Microbiol. 2003, 5: 183-201. 10.1046/j.1462-2920.2003.00397.x.
Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO: Development of the human infant intestinal microbiota. PLoS Biol. 2007, 5: 1556-1573.
Malinen E, Kassinen A, Rinttila T, Palva A: Comparison of real-time PCR with SYBR Green I or 5 ′-nuclease assays and dot-blot hybridization with rDNA-targeted oligonucleotide probes in quantification of selected faecal bacteria. Microbiology. 2003, 149: 269-277. 10.1099/mic.0.25975-0.
Bui XT, Wolff A, Madsen M, Bang DD: Reverse transcriptase real-time PCR for detection and quantification of viable Campylobacter jejuni directly from poultry faecal samples. Res Microbiol. 2012, 163: 64-72. 10.1016/j.resmic.2011.10.007.
Sanguin H, Remenant B, Dechesne A, Thioulouse J, Vogel TM, Nesme X, Moenne-Loccoz Y, Grundmann GL: Potential of a 16S rRNA-based taxonomic microarray for analyzing the rhizosphere effects of maize on Agrobacterium spp. and bacterial communities. Appl Environ Microbiol. 2006, 72: 4302-4312. 10.1128/AEM.02686-05.
Walter J, Margosch D, Hammes WP, Hertel C: Detection of Fusobacterium species in human feces using genus-specific PCR primers and denaturing gradient gel electrophoresis. Microb Ecol Health Dis. 2002, 14: 129-132. 10.1080/089106002320644294.
Ranjan K: Master’s thesis. Verrucomicrobia: A model phylum to study the effects of deforestation on microbial diversity in the Amazon forest. 2010, University of Texas at Arlington: Environmental & Earth Science
Choi BK, Nattermann H, Grund S, Haider W, Gobel UB: Spirochetes from digital dermatitis lesions in cattle are closely related to treponemes associated with human periodontitis. Int J Syst Bacteriol. 1997, 47: 175-181. 10.1099/00207713-47-1-175.
Giovannoni SJ, Delong EF, Olsen GJ, Pace NR: Phylogenetic Group-Specific Oligodeoxynucleotide Probes for Identification of Single Microbial-Cells. J Bacteriol. 1988, 170: 720-726.
Delong EF: Archaea in Coastal Marine Environments. Proc Natl Acad Sci USA. 1992, 89: 5685-5689. 10.1073/pnas.89.12.5685.
Acknowledgements
The authors wish to thank Øystein Angen (Danish Veterinary Institute) for culturing the reference bacteria and Birgitta Svensmark (Pig Research Centre) for collecting the intestinal content samples. This work was supported by the Act on Innovation no. 421 of 31/05/2000 granted by the Ministry of Food, Agriculture and Fisheries of Denmark, the Danish Veterinary Institute (Technical University of Denmark), and the Pig Research Centre (Ministry of Food, Agriculture and Fisheries of Denmark).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MLHB designed the Gut Microbiotassay, conducted all the experiments, and wrote the manuscript. KS supervised the study and contributed to the origin of this manuscript. AS performed the statistical calculations and provided all information on statistical issues. NL analysed the 454-sequence data using his BION software. LM has been the primus of this project and has guided and supervised the progress of both the project and the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2013_7161_MOESM1_ESM.doc
Additional file 1: Table S1: Concentration and purity of DNA extracted from the reference bacteria and the interplate calibrator. (DOC 84 KB)
12864_2013_7161_MOESM4_ESM.doc
Additional file 4: Table S3: Cross reactions detected between primer systems and reference bacteria tested. Highest specific Cq value determined from the respective target reference bacteria has been used as cut-off value for the different primer systems in the data analysis. (DOC 78 KB)
12864_2013_7161_MOESM5_ESM.doc
Additional file 5: BION analysis of 454-sequencing data. Detailed information on the BION software, its functions, and the main statistics for the raw results is included in the file Results_referee.zip. When the file is unpacked it creates the directory Results_referee. This contains 24 subfolders containing the main statistics for each primer pair. However, the species-folders are empty, since the species-specific primers were not tagged. The remaining subfolder ‘Software’ and files are explained in the README-file. The entire BION-meta package (200 Mb) is not included, and there is currently not a stable link to it. But if interested, it can be downloaded from the following link: https://www.dropbox.com/sh/fumscuqpanqaqvu/_4H--XBxHQ. (DOC 60 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hermann-Bank, M.L., Skovgaard, K., Stockmarr, A. et al. The Gut Microbiotassay: a high-throughput qPCR approach combinable with next generation sequencing to study gut microbial diversity. BMC Genomics 14, 788 (2013). https://doi.org/10.1186/1471-2164-14-788
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-14-788