
Abstract
Background
Many fruits, including watermelon, are proficient in carotenoid accumulation during ripening. While most genes encoding steps in the carotenoid biosynthetic pathway have been cloned, few transcriptional regulators of these genes have been defined to date. Here we describe the identification of a set of putative carotenoid-related transcription factors resulting from fresh watermelon carotenoid and transcriptome analysis during fruit development and ripening. Our goal is to both clarify the expression profiles of carotenoid pathway genes and to identify candidate regulators and molecular targets for crop improvement.
Results
Total carotenoids progressively increased during fruit ripening up to ~55 μg g-1 fw in red-ripe fruits. Trans-lycopene was the carotenoid that contributed most to this increase. Many of the genes related to carotenoid metabolism displayed changing expression levels during fruit ripening generating a metabolic flux toward carotenoid synthesis. Constitutive low expression of lycopene cyclase genes resulted in lycopene accumulation. RNA-seq expression profiling of watermelon fruit development yielded a set of transcription factors whose expression was correlated with ripening and carotenoid accumulation. Nineteen putative transcription factor genes from watermelon and homologous to tomato carotenoid-associated genes were identified. Among these, six were differentially expressed in the flesh of both species during fruit development and ripening.
Conclusions
Taken together the data suggest that, while the regulation of a common set of metabolic genes likely influences carotenoid synthesis and accumulation in watermelon and tomato fruits during development and ripening, specific and limiting regulators may differ between climacteric and non-climacteric fruits, possibly related to their differential susceptibility to and use of ethylene during ripening.
Similar content being viewed by others


Background
The isoprenoid (also known as terpenoid) pathway is one of the most important and well-studied biosynthetic pathways in plants. It involves cross-talk between the cytosolic mevalonate (MVA) and plastidial 2-C-methyl-d-erythritol 4-phosphate (MEP) pathways, to give rise to isopentenyl-diphosphate (IPP), the C5 building block required for the synthesis of a diverse group of natural products that perform numerous biochemical functions in plants. The main branch of the isoprenoid pathway leads to the synthesis and accumulation of carotenoids, C40 terpenoid compounds formed by the condensation of eight isoprene units, within plastids [1]. Carotenoids are important natural pigments found in all plants and algae, in many bacteria and fungi, as well as in some animals. In photosynthetic organisms, carotenoids are always present within chloroplasts associated with the light-harvesting complexes of photosystems, where they gather light energy and transfer it to the chlorophylls, in addition to contributing to protection of the photosynthetic apparatus from photooxidation [2]. Chromoplast synthesized carotenoids accumulate during fruit ripening resulting in dramatic changes in tissue pigmentation. Carotenoids also play an essential role in attracting insects and herbivores that act as floral pollinators and seed dispersion vehicles, including the consumption of plant organs such as ripe fruits as food by humans. When carotenoid-rich foods are ingested, carotenoids are assimilated, metabolized, transported in the plasma by lipoproteins, and stored in various tissues [3] where they display biological activities including acting as antioxidants and free-radical scavengers, reducing the risk of certain types of cancers, and promoting immune responses. In addition, some carotenoids (e.g. β-carotene, β-cryptoxanthin) are precursors of vitamin A, an essential nutrient for humans [4–9]. Carotenoids can undergo multiple structural modifications, namely, cyclization, hydroxylation, and epoxidation, yielding the great variety of carotenoids found in nature comprising more than 600 compounds [10].
Increasing interest is devoted to carotenoid content and composition of food crops because of their important roles in human health [11]. Cultivated watermelon [Citrullus lanatus (Thunb.) Matsum. & Nakai var. lanatus] is important in the diets of people throughout the world. More than 6% of the world's total area devoted to vegetable production is used for watermelon production [12]. Throughout the Mediterranean basin, watermelon is considered an extremely important agricultural crop, with over 106 tons produced in 2010 at a production value of approximately $3 billion US [12]. The ripening of watermelon fruit is a highly regulated process because color, flavor, aroma, and texture change in a coordinated manner. One of the most noticeable characteristics of watermelon ripening is the dramatic increase in carotenoids. The change in pigmentation is caused by synthesis and accumulation of lycopene within the chromoplasts in watermelon fruit resulting in their characteristic red coloration.
To maximize the health-promoting benefits of carotenoids through increased consumption, characterization of carotenoid synthesis and accumulation in important food crops such as watermelon is essential. Understanding the molecular and genetic components controlling the carotenoid biosynthetic pathway is fundamental for targeted breeding aimed at improving carotenoid-producing watermelon cultivars [13, 14].
In the past decade, most carotenoid biosynthesis genes in plants have been identified [15–17]. Identification of the cauliflower Or (Orange) gene further indicates the importance of providing a sink for newly synthesized carotenoids in determining net accumulation [18, 19]. Progress in elucidating the mechanisms that control carotenoid biosynthesis and accumulation in plants have been recently achieved using climacteric [e.g. tomato (Solanum lycopersicum Mill.)] and non-climateric [pepper (Capsicum annuum L.)] fruits as models [20–23], though limited information is available for other species including watermelon.
Different strategies to control carotenoid biosynthesis and accumulation in plant tissues have been reported [20–22]. During flower color development and fruit ripening, transcriptional regulation of carotenoid gene expression has been shown to be a major mechanism by which the biosynthesis and accumulation of specific carotenoids are regulated. Classic examples are found in tomato and pepper (both fruits and flowers), where the synthesis and accumulation of specific carotenoids parallel increased expression of precursor (upstream) carotenogenic genes and reduced expression of downstream (metabolism) genes of the accumulating carotenoids [16, 24, 25]. The mechanisms of these transcriptional control systems remain poorly understood. Post-transcriptional regulation at the enzymatic level also plays a role in controlling carotenoid biosynthesis and accumulation [26–28]. Metabolic turnover of carotenoids by carotenoid cleavage dioxygenases (CCDs) not only produces important signalling and accessory apocarotenoid molecules, but also helps to maintain the steady-state level of carotenoids in plant tissues. Expression of CCDs has been found to negatively regulate carotenoid accumulation [29, 30]. A body of evidence has shown that oxidative cleavage of carotenoids is induced under environmental stresses [31, 32]. Light and circadian rhythm have been shown to alter the expression of nearly all MEP genes and several carotenoid synthesis genes [33–35] as well as carotenoid catabolism genes [36]. Developmental cues also play important roles in conferring metabolic turnover of carotenoids [28]. In addition, altered plastid biogenesis leading to increased plastid compartment size was associated with elevated chlorophyll and carotenoid levels in hp (high-pigment) mutants [37–39].
A system analysis approach for transcriptome and metabolic data is presented here to identify putative transcription factors that may impact carotenoid accumulation during watermelon fruit ripening. The pattern of synthesis and accumulation of carotenoids and the expression of carotenoid- and isoprenoid-related genes (specifically, those coding for putative transcription factors) has been analyzed in fresh watermelon during fruit development and ripening in order to clarify the factors influencing accumulation of these bioactive molecules and to identify key regulators and molecular targets for crop improvement.
Results and discussion
An integrative study combining carotenoid profiles and whole genome transcriptome analysis was performed to gain insight into novel genes associated with and possibly regulating the synthesis and accumulation of carotenoids in watermelon plastids during fruit development and ripening. Analysis of carotenoids in the flesh of watermelon fruits at four successive stages of fruit development and ripening corresponding to the white, white-pink, pink and red-ripe color of the fruit flesh (see methods for details of these stages), are shown in Table 1. Phytoene, phytofluene 1, phytofluene 2, ζ-carotene 2, cis-lycopene, trans-lycopene, β-carotene, γ-carotene and lutein were identified. Other carotenoids such as ζ-carotene, cis-neurosporene, α- and δ-carotene, zeaxanthin and violaxanthin were not detected [LOD (Limit Of Detection) = 0.005 μg g-1 fresh weight (fw); LOQ (Limit Of Quantification = 0.015 μg g-1 fw) at any of the four stages analyzed.
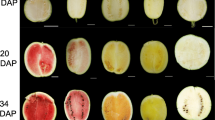
The amount of each identified carotenoid varied significantly during fruit ripening (p < 0.001), with the exception of phytofluene 2 and lutein that remained low in all ripening stages. As expected, the total amount of carotenoids (expressed as μg g-1 fw) progressively increased during fruit ripening from 0.100 μg g-1 fw to 55.253 μg g-1 fw at the white and red-ripe stages, respectively. Trans-lycopene contributed most to this increase, while the cis isomer(s), although increasing in proportion similar to trans, represented only 0.2% of the total carotenoid content from the white-pink (where it was detected first) to the red-ripe stage. In terms of color, at the white stage, there is no difference between the inner peel (outer mesocarp) and the placenta [flesh (inner mesocarp and endocarp) tissues] (Figure 1a), while, at the white-pink stage, the flesh tissue started to turn red (Figure 1b) due to lycopene accumulation. Trans-lycopene was, in fact, detected at a very low concentration (0.030 μg g-1 fw) at the white stage and predominated (3.950 μg g-1 fw) at the white-pink stage of ripening. The highest rate of change in accumulation of trans-lycopene was observed in the transitional phase between the white-pink and the pink stage where it reached 28.080 μg g-1 fw, ~7.1 times higher than that of the previous stage. Trans-lycopene increased less than two-fold (50.540 μg g-1 fw) by the next (red-ripe) stage. These results confirm our earlier studies, in which lycopene content of the same cultivar ranged between 0 and 47.1 μg g-1 fw from the white to the red-ripe stages of ripening [40]. In addition, the amount of lycopene measured at the red-ripe stage concurs and falls within the range (35–112 μg g-1 fw) reported for ripe red-fleshed commercial cultivars by Perkins-Veazie et al. [41, 42].

Fruits of watermelon cultivar Dumara at four different stages of ripening. Ripening stages were indicated as white (a), white-pink (b), pink (c) and red-ripe (d).
At the white stage, lutein, even if present in a very low amount (0.040 μg g-1 fw), was the most abundant carotenoid in the watermelon flesh, contributing to 40% of total carotenoids. Phytofluene 1 and 2 were the only acyclic carotenoids detectable at this stage although in amounts close to the LOQ. Starting from the white-pink ripening stage the acyclic carotenoids phytoene and phytofluene 1 increased gradually reaching their maximum (0.970 μg g-1 fw, 0.810 μg g-1 fw, respectively) in red-ripe fruit. ζ-carotene 2 showed its highest concentration (0.13 μg g-1 fw) in fully ripe watermelons but it was detectable only from the pink stage. Phytofluene 2, ζ-carotene 2, cis-lycopene and lutein remained at very low levels throughout watermelon fruit development and ripening. In photosynthetic tissues lutein plays a critical role in light-harvesting complex assembly and function, and in photoprotection of photosystems [43]. It is the most abundant carotenoid in the chloroplasts and often accounts for >50% of the total carotenoid pool. The small amounts of lutein found in the non-photosynthetic tissues of the flesh of the young, not yet fully developed, watermelon fruit, could have a role in the protection of the plastid membranes from oxidative stress.
The content of β-carotene was at low levels until the red-ripe stage when β-carotene content was four times higher than the previous stage, representing 2.6% of total carotenoids. At the white-pink ripening stage the amount of γ-carotene was at least 10 times higher than that of β-carotene, but in subsequent stages their contents became similar. Tadmor et al. [44] reported that red watermelons generally have any of the three following carotenoid profiles: 1) high levels of lycopene and small amounts (less than 5% of total carotenoids) of β-carotene; 2) accumulation of mostly lycopene and significant (>10%) β-carotene and 3) lycopene exclusively, with no detectable β-carotene. The first carotenoid pattern seems to best characterize the Dumara cultivar at the red-ripe stage suggesting that mature fruit of this cultivar retain at least some lycopene β-cyclase activity.
The expression profiles of 19,324 genes, among the 23,440 predicted in the watermelon genome assembly [45, 46], were generated from trascriptome characterization of watermelon ripening fruits of the Dumara cultivar by Illumina RNA sequencing. Searching the resulting transcriptomics data for genes involved in the pathways related to carotenoid metabolism (MVA, MEP and carotenoid biosynthetic and catabolism pathways) (Figure 2), we identified 40 sequences coding for putative proteins with an e-value lower than 1e-10 and a minimal Reads Per Kilobase of exon model per Million mapped reads (RPKM) value of 8 in at least one of the four ripening stages. This threshold was arbitrarily chosen to filter the sequences with very low counts which may represent estimates of transcript abundance that are too unreliable for inclusion in differential gene expression analysis (see Additional file 1: Table S1 for the expression levels of additional isoprenoid pathway related putative genes for which reads below 8 RPKM, were found). Among the identified sequences, 23 genes were differentially expressed in the flesh tissue during fruit development and ripening (FDR ≤ 0.05) (Table 2).

Summary diagram of pathways related to carotenoid metabolism (MVA, MEP, carotenoid biosynthetic and catabolism pathways). See below for legend. 1. ACAT, acetyl-CoA acetyltransferase; 2. HMGS, 3-hydroxy-3-methylglutaryl-CoA synthase; 3. HMGR, 3-hydroxy-3-methylglutaryl-CoA reductase; 4. MVK, mevalonate kinase; 5. PMK, phosphomevalonate kinase; 6. PMD, diphosphomevalonate decarboxylase; 7. IDI, isopentenyl-diphosphate δ-isomerase; 8. GPS, geranyl-diphosphate synthase; 9. FPS, farnesyl diphosphate synthase; 10. DXS, 1-deoxy-d-xylulose-5-phosphate synthase; 11. DXR, 1-deoxy-d-xylulose-5-phosphate reductoisomerase; 12. MCT, 2-C-methyl-d-erythritol-4-phosphate cytidyltransferase; 13. CMK, 4-diphosphocytidyl-2-C-methyl-d-erythritol kinase; 14. MDS, 2-C-methyl-d-erythritol 2,4-cyclodiphosphate synthase; 15. HDS, 4-hydroxy-3-methylbut-2-en-1-yl diphosphate synthase; 16. HDR, 4-hydroxy-3-methylbut-2-enyl diphosphate reductase; 17. GGPS, geranylgeranyl diphosphate synthase; 18. KSB, ent-kaur-16-ene synthase; 19. GGPR, geranylgeranyl diphosphate reductase; 20. PREA, prenyl transferase; 21. PSY, phytoene synthase; 22. PDS, phytoene desaturase; 23. Z-ISO, ζ-carotene isomerase; 24. ZDS, ζ-carotene desaturase; 25. CRTISO, carotene isomerase; 26. LCYE, lycopene ϵ-cyclase; 27. LCYB, lycopene β-cyclase; 28. CHYB, β-carotene hydroxylase; 29. CHYE, ϵ-carotene hydroxylase; 30. ZEP, zeaxanthin epoxidase; 31. VDE, violaxanthin de-epoxidase; 32. NXS, neoxanthin synthase; 33. XISO, xanthophyll isomerase; 34. NCED, 9-cis-epoxycarotenoid dioxygenase; 35. SDR, short chain dehydrogenase; 36. AAO-MoCo, abscisic aldehyde oxidase-molybdenum co-factor. In the diagram, the enzymes with down-regulated gene expression level are indicated in red, the enzymes with up-regulated gene expression level in blue, while the enzymes with gene expression level that increases in the intermediate stages and decreases in the last stage in green. The asterisks identify enzymes encoded by several gene isoforms; the gene expression level of the predominant isoform is shown in the diagram.
Of the 11 sequences thought to be involved in the upstream MVA pathway leading to isopentenyl-diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) biosynthesis, only one (Cla010997) coding for a putative acetyl-CoA acetyltransferase 2 (ACAT2) was differentially expressed during watermelon ripening. Changes in its expression levels in RPKM, are reported in Figure 3.

Expression level of mevalonic acid (MVA) pathway genes during watermelon ripening. Data were obtained by Illumina RNA Sequencing using two biological replicas and are expressed as Reads Per Kilobase of exon model per Million mapped reads (RPKM). The figure includes only the genes differentially expressed during watermelon fruit ripening with a FDR (False Discovery Rate) ≤ 0.05.
ACAT2 catalyses the condensation of two acetyl-CoA subunits to form acetoacetyl-CoA thus directing this central metabolite to the MVA pathway. Interestingly ACAT2 mRNA expression was up-regulated approximately three-fold during watermelon ripening suggesting that this enzyme may divert the metabolic flux of acetyl-CoA from the biosynthesis of fatty acids and amino acids toward the synthesis of isoprenoids. 3-Hydroxy-3-methylglutaryl-CoA reductase (HMGR) is a key regulatory enzyme in the pathway and catalyses the formation of MVA from 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA). It is a highly regulated enzyme, being subjected to transcriptional, translational, and post-translational control [47]. Although a sequence (Cla015723) coding for a putative HMGR1 is differentially expressed in ripening watermelon fruits (FDR = 0.0137) (Additional file 1: Table S1), it was not taken into account in this study having RPKM far below our defined minimal value of 8. However, the very low expression of hmgr 1 (0–0.3 RPKM) in ripening watermelon fruits supports the assertion that the conversion of HMG-CoA to MVA is the rate limiting step in the biosynthesis of sterols and other isoprenoids [48].
Two different genes (Cla009550 and Cla007929) encoding putative isopentenyl-diphosphate δ-isomerases (IDI1 and IDI2, respectively) were identified. These enzymes convert the relatively un-reactive IPP to the more-reactive electrophile DMAPP. Both genes were stably expressed during watermelon ripening but IDI1 expression was considerably higher than that of IDI2 (708 vs 60 RPKM, as the average value of the four ripening stages, respectively). In Arabidopsis thaliana (L.) Heynh. IDI1 and IDI2 are expressed in all organs, with IDI1 less abundant than IDI2. Examination of green fluorescent protein fusions established that IDI1 is mainly located in the plastids, whereas IDI2 is in the mitochondria. Both proteins are also present in the cytosol as a result of their translation from naturally occurring shorter transcripts lacking transit peptides [49].
Three sequences were found to be associated with putative enzymes involved in the IPP down-stream MVA pathway (Table 2). Of these, two (Cla016321 and Cla003330) were differentially expressed during watermelon ripening (Figure 3). The expression of Cla016321, coding for a putative geranyl-diphosphate synthase (GPS), was strongly inhibited as soon as the ripening process started. This enzyme is responsible for the conversion of DMAPP in the presence of IPP into geranyl-diphosphate (GPP) which is further converted into farnesyl diphosphate (FPP) in the presence of another molecule of IPP by farnesyl diphosphate synthase (FPS1, the putative protein encoded by Cla003330). It is well established that cytosolic IPP, which is then isomerized to DMAPP by the activity of IDIs, contributes to the formation of functional plastidic isoprenoids [50, 51]. Down-regulation of Cla016321 could determine a shift of the MVA produced IPP towards carotenoid biosynthesis. Squalene synthase (encoded in watermelon by Cla016602; e-value = 9e-46; FDR = 0.13483; RPKM = 25 ± 7, as the average of the four ripening stages) catalyses the head-to-head condensation of two FPP molecules to form squalene, the first committed step in sterol biosynthesis, and as such was not considered further here.
In plants, IPP and DMAPP entering in the biosynthesis of carotenoids are mainly synthesized by the MEP pathway in the plastids [1]. In this work 11 sequences were found that are likely to be involved in the up-stream pathway leading to IPP and DMAPP biosynthesis, and 8 were differentially expressed during watermelon fruit development and ripening (Table 2; Figure 4). 1-Deoxy-d-xylulose-5-phosphate synthase (DXS) has been shown to catalyze one of the rate-limiting steps of the MEP pathway [52]. It generates 1-deoxy-d-xylulose-5-phosphate (DXP) by the transketolase-type condensation of pyruvate and d-glyceraldehyde 3-phosphate and is also involved in the biosynthesis of thiamine and pyridoxal (vitamin B1 and B6, respectively) in bacteria and higher plants [53–55]. Therefore DXS appears to play a key role linking isoprenoid and vitamin biosynthesis. Four genes (Cla009346, Cla009347, Cla009348 and Cla022299) were found to encode for putative DXS isoforms. Most enzymes of the MEP pathway are encoded by single-copy genes in flowering plants, whereas DXS is typically encoded by a small gene family [35, 49, 56, 57]. The genes of the DXS family display differential expression during plant development and in specific organs, suggesting a non-redundant function and possibly a role in production of particular isoprenoids [58, 59]. With the exception of Cla022299, whose expression did not vary during ripening (FDR = 0.1204; RPKM = 10 ± 3, as the average of the four ripening stages), the other three sequences were differentially expressed (FDR ≤ 0.05) and induced during watermelon fruit ripening (Table 2; Figure 4). This is in agreement with what was found in tomato and pepper fruits [60, 61]. In tomato, the highest level of DXS transcripts was detected at the breaker stage, and then decreased during later ripening [62]. DXS mRNA was found to be most abundant in young Arabidopsis chs5 mutant, maize (Zea mays L.) and peppermint (Mentha × piperita L.) leaves suggesting that its activity is of critical importance at the early stages of leaf and chloroplast development and confirming its organ and tissue specificity [55, 59, 63]. The fact that multiple watermelon DXS genes are induced during ripening suggests a predominant role of members of this family in driving fruit carotenoid accumulation.

Expression level of 2- C -methyl- d -erythritol-4-phosphate (MEP) pathway genes during watermelon ripening. Data were obtained by Illumina RNA Sequencing using two biological replicas and are expressed as Reads Per Kilobase of exon model per Million mapped reads (RPKM). The figure includes only the genes differentially expressed during watermelon fruit ripening with a FDR (False Discovery Rate) ≤ 0.05.
DXP is converted to MEP by the enzyme DXP reductoisomerase (DXR) encoded in watermelon by the gene sequence Cla019193 whose mRNA expression profile remained stable during fruit ripening (average RPKM = 39 ± 7). MEP is subsequently converted into IPP and DMAPP by the consecutive action of five independent enzymes: 2-C-methyl-d-erythritol 4-phosphate cytidyltransferase (MCT), 4-diphosphocytidyl-2-C-methyl-d-erythritol kinase (CMK), 2-C-methyl-d-erythritol 2,4-cyclodiphosphate synthase (MDS), 4-hydroxy-3-methylbut-2-en-1-yl diphosphate synthase (HDS), and 4-hydroxy-3-methylbut-2-enyl diphosphate reductase (HDR). Isomerization of IPP and DMAPP is catalyzed by the two isomerases IDI1 and IDI2 (encoded by Cla009550 and Cla007929, respectively). The sequences Cla004566, Cla014654, Cla005033 and both Cla010297 and Cla015963 coding, respectively, for the putative enzymes MCT, MDS, HDS and HDR all showed a significant increase in expression levels during ripening at least up to the pink stage (Figure 4). Cla011088 encoding for a putative CMK, was stably expressed throughout watermelon fruit ripening (average RPKM = 14 ± 4; FDR = 0.0718).
Geranylgeranyl diphosphate (GGPP), the precursor in the synthesis of all plastid isoprenoids, is generated by geranylgeranyl diphosphate synthase (GGPS) that catalyses the condensation of three IPP and one DMAPP units. The expression of Cla020121, encoding for a putative GGPS, increased (approx. six-fold) in the transition between the white and the white-pink stage of ripening, in agreement with the higher rate of synthesis of lycopene between these transitional phases, then progressively decreased at the pink and the ripe-red stages. In other fruits such as mango (Mangifera indica L.), GGPS levels were stable throughout the fruit life [64].
Since GGPP is a common precursor for the synthesis of phyllochinones, tocopherols, plastoquinones, chlorophylls, gibberellins and carotenoids the expression profiles of genes which may affect carotenoid biosynthesis through competition with phytoene synthase (PSY) for GGPP were also analyzed (Table 3; Figure 5). In watermelon, two sequences (Cla019109 and Cla003139) were found to encode putative geranylgeranyl diphosphate reductases (GGPR) that convert GGPP to phytyl diphosphate in the tocopherol and chlorophyll biosynthetic pathways. Both were differentially regulated during watermelon ripening (Figure 5). While, the expression of Cla005482 and Cla005390, coding for ent-kaur-16-ene synthase (KSB) and prenyl transferase (PREA), diverting the GGPP flux towards the synthesis of gibberellins and plastoquinones, respectively, were stably maintained low during watermelon fruit ripening (average RPKM = 8 ± 2 and 22 ± 5, respectively) presumably to guarantee the synthesis of these fundamental metabolites.

Expression level of genes related to alternative geranylgeranyl diphosphate (GGPP) catabolism during watermelon ripening. Data were obtained by Illumina RNA Sequencing using two biological replicas and are expressed as Reads Per Kilobase of exon model per Million mapped reads (RPKM). The figure includes only the genes differentially expressed during watermelon fruit ripening with a FDR (False Discovery Rate) ≤ 0.05.
Of the 11 sequences associated with putative enzymes of the carotenoid biosynthetic pathway, 8 were differentially expressed during watermelon ripening (Table 2; Figure 6). As in tomato, two members of the PSY gene family were identified and with different expression patterns. Cla009122 expression level was very low at the white stage but increased sharply as the fruit matured, peaking at the pink and then decreasing at the red-ripe stage. A similar expression profile of a putative Psy-1 (WMU38667) was described by Guo et al. [45] in watermelons of the inbred line 97103. In contrast, Cla005425 maintained low expression throughout fruit development and ripening, decreasing further in the later stages. The expression profiles are similar to those of tomato psy-1 and psy-2, respectively. Psy-1 is induced in ripening tomato fruit in association with elevated lycopene accumulation. Psy-2, on the other hand, is low in fruit and highly expressed in leaf [65]. Therefore it appears that the PSY encoded by Cla005425 has a function similar to tomato PSY-2 whereas the product of Cla009122 is similar to PSY-1.

Expression level of carotenoid metabolism pathway genes during watermelon ripening. Data were obtained by Illumina RNA Sequencing using two biological replicas and are expressed as Reads Per Kilobase of exon model per Million mapped reads (RPKM). The figure includes only the genes differentially expressed during watermelon fruit ripening with a FDR (False Discovery Rate) ≤ 0.05.
The expression level of phytoene desaturase (PDS; Cla010898), carotene cis-trans isomerase (CrtISO; Cla017593) and ζ-carotene desaturase (Zds; Cla003751) genes increased during fruit development and ripening up to the pink stage and then remained constant (Figure 6). It is well known that PDS catalyzes the desaturation steps, sequentially producing phytofluene and ζ-carotene [66] from phytoene. ZDS with CRTISO are both involved in the steps which sequentially convert 9,9'-di-cis-ζ-carotene to pro-lycopene (7,9,7',9'-tetra-cis-lycopene) and to all-trans-lycopene. Isaacson et al. [67] found that the function of CRTISO paralleled that of the 9,9'-di-cis-ζ-carotene desaturase (ZDS) to convert 7,9,9'-tri-cis-neurosporene to 9'-cis-neurosporene and 7,9,7',9'-tetra-cis-lycopene to all-trans-lycopene. In tomato, Isaacson et al. [68] reported that deletions in the promoter region and coding region of CrtISO resulted in two different color mutants of tangerine accumulating pro-lycopene and ζ-carotene instead of all-trans-lycopene. This strongly suggests that watermelon CrtISO mutations might also cause the salmon yellow or orange mutation that accumulates pro-lycopene and ζ-carotene as major fruit carotenoids [69].
Lycopene β-cyclase (Lcyb; Cla005011) and lycopene ϵ-cyclase (Lcye; Cla016840) expression levels were low at the white stage and did not change during watermelon ripening (average RPKM = 23 ± 5 and 6 ± 5, respectively). LCYB is one of the crucial enzymes for carotenoid biosynthesis. LCYB along with LCYE bring about the cyclization of lycopene. Activities of both of these enzymes make α-carotene via δ-carotene, while activity of LCYB alone leads to formation of β-carotene via γ-carotene [70]. In tomato, the down-regulation of this gene may generate a blockade downstream, leading to the accumulation of lycopene in red-ripe fruits [71]. The low expression level of LYCB mRNA that we found in the Dumara cultivar, may permanently maintain low metabolic flux toward cyclic carotenes and xanthophylls during ripening. In contrast to tomato, during watermelon ripening no chloroplast-to-chromoplast transition occurs, rather chromoplasts originate from the differentiation of proplastids. Therefore constant synthesis of β-carotene and lutein, which are present in significant quantities in the purified chloroplasts of unripe tomatoes [72], is not required in watermelon fruits. However, a dramatic reduction in the expression of Lcyb and β-carotene hydroxylase gene (Chyb), although with differences in the amount of transcript level variation, was recently reported in red-fleshed “ZAOHUA” and pink-fleshed “96B41” watermelon varieties 20–30 days after pollination and related to lycopene accumulation during ripening [73], suggesting that the regulation of Lcyb is influenced by watermelon genotype. A gradual decrease of Lcyb, which resulted undetectable at the over-ripe stage, was also reported by Guo et al. [45] during watermelon inbred line 97103 ripening.
Two sequences (Cla011420 and Cla006149) coding for putative CHYB isoforms were identified with a similar but quantitatively different expression pattern (Figure 6). The high expression levels of downstream genes Chyb and zeaxanthin epoxidase (Zep; Cla020214), whose expression increased early during watermelon fruit ripening and remained stable over time, may help maintain the amounts of γ- and β-carotene at low levels as intermediate metabolites for other compounds. Similarly the lack of either zeaxanthin and violaxanthin, products of CHYB and ZEP activities, in the watermelon carotenoid profiles at any stage of ripening may be due to their rapid catabolism by dioxygenases [eg. into abscisic acid (ABA)] [73].
Two members of 9-cis-epoxy-carotenoid dioxygenase family, Nced1s (Cla005404 and Cla009779), involved in the cleavage of 9-cis-violaxanthin or 9-cis-neoxanthin to form C25 epoxy-apocarotenal and xanthoxin (C15), a precursor of ABA [74, 75], increased during watermelon ripening with a different expression profile. The transcript Cla005404 reached the peak of expression at the pink stage whereas the transcript Cla009779 peaked at the white-pink stage and then slightly declined at the pink and red-ripe stages (Table 2; Figure 7). In addition, one member of CCD family [CCD1 (Cla015245)], preferentially involved in volatile compound production from different carotenoid substrates [76, 77], was found to increase, reaching the highest values in the pink and red-ripe fruit ripening stages (Figure 7). Now these data suggest that a complex balance between up-regulation and down-regulation of genes belonging to different central pathways of plant cell metabolism generate a flux of metabolic precursors towards lycopene synthesis during watermelon fruit development and ripening. Concurrently, a constitutive low expression level of lycopene cyclase genes creates a blockade downstream, leading to the accumulation of lycopene and demonstrates differential and coordinate regulation of such genes.

Expression level of carotenoid catabolism genes during watermelon ripening. Data were obtained by Illumina RNA Sequencing using two biological replicas and are expressed as Reads Per Kilobase of exon model per Million mapped reads (RPKM). The figure includes only the genes differentially expressed during watermelon fruit ripening with a FDR (False Discovery Rate) ≤ 0.05.
In tomato, many regulatory factors operating in the fine control of the isoprenoid pathway have been elucidated [14, 22, 78]. Many genes (LeRIN-MADS, TAGL1, SlERF6, AP2, MC, NAC-NOR, DET1, DDB1, CUL4, CNR, GLK2, HB-1, U/GLK2 and TDR4) have been shown to encode transcriptional regulators affecting the expression of other genes responsible for ripening phenotype, including isoprenoid biosynthesis and accumulation [79–87]. We screened the transcript set of watermelon for similarity against the known mRNA sequences of the above-mentioned tomato transcription factors by BLAST using an e-value threshold of ≤1e − 10. Nineteen sequences encoding putative transcription factors with a RPKM value ≥8 in at least one of the four watermelon ripening stages assayed were identified. Among these, only 6 were differentially expressed in the flesh tissue during fruit development and ripening (FDR ≤ 0.05) (Table 4).
Two genes (Cla010815 and Cla000691) were related to the Solanum lycopersicum MADS-box transcription factor RIPENING INHIBITOR (LeRIN-MADS), a member of the SEPALLATA (SEP) subfamily. Both sequences were constitutively expressed at high levels during watermelon ripening, with average RPKM values of 339 ± 57 (FDR = 0.1738) and 299 ± 24 (FDR = 0.6762), respectively. MADS-BOX genes are normally associated with floral development, but RIN-MADS is an essential regulator of tomato fruit ripening. RIN-MADS controls tomato softening and ethylene production by the direct transcriptional regulation of cell-wall-modifying genes and ACS (1-aminocyclopropane-1-carboxylate synthase) genes, respectively. In addition, recently, it has been demonstrated to interact with promoters of many genes involved in the major pathways associated with ripening, including carotenoid biosynthesis and accumulation, to both initiate and maintain their expression throughout the ripening process [88–90]. RIN-MADS protein and mRNA are first detected slightly before the breaker stage and maintained throughout ripening [89]. In Ailsa Craig tomato fruits, expression of LeRIN-MADS increased 16-fold during ripening [91]. Similarly, expression of a strawberry (Fragaria x ananassa Duch.) LeRIN-MADS homologous gene, identified by screening of a strawberry fruit cDNA library with a tomato LeRIN-MADS cDNA, was enhanced during ripening, suggesting that transcriptional control of ripening is conserved among climacteric and non-climacteric species [79, 92, 93]. The identification of watermelon LeRIN-MADS homolog genes, expressed at high levels throughout the process of fruit ripening, further supports this hypothesis.
Similarly, neither of the two sequences (Cla009725 and Cla019630) homologous to MADS-box transcription factor TOMATO AGAMOUS-LIKE 1 (TAGL1), was differentially expressed during watermelon fruit ripening (FDR = 0.7333 and 0.6273, respectively) (Table 4). The expression of both had average values of 88 ± 10 RPKM and 65 ± 7 RPKM, respectively. In tomato TAGL1 is induced in the early stages of carpel development and later at the onset of ripening, suggesting it is involved in both processes. Its expression increases during ripening, peaking at the orange stage of fruit development, possibly in relation to ethylene biosynthesis [84, 94]. Part of the TAGL1 activity in tomato fruit ripening is, in fact, exerted through regulation of the ACS2 gene coding for 1-aminocyclopropane-1-carboxylate synthase, the rate-limiting enzyme in ethylene biosynthesis [95]. Ethylene biosynthesis is not essential for watermelon fruit ripening, although varying patterns of ethylene production have been reported in non-climacteric fruits, including watermelons [96] and may explain in part the difference in TAGL1 expression profiles between tomato and watermelon. The early TAGL1 activity in tomato was shown to be related to expansion of the carpel [84, 94]. The large size of watermelon and the constitutive expression of the homologous genes may reflect activity related to the exceptional size of the mature watermelon fruit. In tomato, TAGL1 requires RIN-MADS activity for the induction of lycopene accumulation in ripe fruit. In watermelon, both RIN and TAGL1 are expressed at a substantial level during ripening supporting the idea they have a role in carotenoid synthesis and accumulation.
TDR4 is another member of the MADS box transcription family, belonging to the SQUAMOSA (SQUA) subfamily, whose expression pattern suggests a possible role during tomato fruit ripening [80] in an ethylene-independent manner [97]. Although three sequences were identified in watermelon with a high similarity to TDR4 (Cla010813, Cla022037 and Cla006943 - data not shown), all were expressed at a very low level (below 8 RPKM) and, for this reason, were not considered further. TDR4, hence, seems not involved in isoprenoid accumulation during watermelon fruit ripening, but it may influence different biosynthetic pathways in other non-climacteric fruits. A TDR4 ortholog was, in fact, recently shown to influence anthocyanin biosynthesis during bilberry ripening [98].
COLORLESS NON RIPENING (CNR) encodes a transcription factor of the SQUAMOSA promoter-binding protein family (SPB/SPL). It likely controls expression of SQUA MADS box genes (such as TDR4) by interacting with their promoters. Tomato mutants in this gene show pleiotropic non-ripening phenotypes, including a mealy and pale pericarp [99–101]. Five related sequences were identified in watermelon (Table 4). Two (Cla013549 and Cla009630) exhibited a low and stable expression pattern with average RPKM values of 14.7 ± 1.3 (FDR = 0.9119) and 9.5 ± 1.4 (FDR = 0.7472), respectively. The other three sequences (Cla003384, Cla001754 and Cla019702) were differentially expressed during watermelon fruit ripening showing a sharp reduction already in early ripening (Figure 8). In Liberto and Ailsa Craig wild-type tomato fruits CNR was transiently expressed at the breaker stage of ripening [82]. CNR is necessary to induce ripening-associated increases in respiration and ethylene synthesis in tomato and other climacteric fruits, in non-climacteric fruits its role remain unclear. The down regulation of the putative CNR genes during watermelon ripening suggests it may act as a regulator of isoprenoid accumulation, but with mechanisms different by those operating in climacteric fruits.

Expression level of putative ripening transcriptional regulator genes during watermelon ripening. Data were obtained by Illumina RNA Sequencing and are expressed as Reads Per Kilobase of exon model per Million mapped reads (RPKM). The figure includes only the genes differentially expressed during watermelon fruit ripening with a FDR (False Discovery Rate) ≤ 0.05.
Another ripening regulator that pleiotropically controls many aspects of tomato ripening is NON-RIPENING (NAC-NOR). Cla023408 showed a high similitude with NAC-NOR. In watermelon the expression level of Cla023408 did not significantly change during fruit ripening (average RPKM = 27 ± 11) suggesting that NAC-NOR protein is not limiting in watermelon fruit ripening as it is in tomato [91].
In tomato APETALA2a (SlAP2a) transcription factor, a member of the APETALA2/ETHYLENE RESPONSE FACTOR (AP2/ERF) superfamily, influences fruit ripening via regulation of ethylene biosynthesis and signaling [86, 102]. In tomato, RIN-MADS, NAC-NOR and CNR positively regulate SIAP2a expression which is, in turn, a negative regulator of ripening and ethylene production. SIAP2a is expressed at a relatively low level in flowers and early fruit stages but it is strongly up-regulated between the mature green and breaker stages and is highly expressed to the red-ripe stage [86, 102]. Three homologs (Cla018268, Cla020243 and Cla000701) of SIAP2a were identified in the watermelon transcriptome. Cla018268 was expressed at a relatively low level and down-regulated during ripening (Figure 8). On the contrary, Cla020243 and Cla000701 expression was almost constant during ripening although with differences in their relative expression levels. Cla020243 was in fact expressed at a relatively low level (average RPKM = 22 ± 4; FDR = 0.3045) whereas Cla000701 was highly expressed throughout ripening (average RPKM = 294 ± 78; FDR = 0.1955) suggesting Cla000701 is the most likely functional ortholog of SlAP2a in watermelon, though its role in non-climacteric ripening may be in a different context than through regulation of ethylene response.
An additional member of the AP2/ERF superfamily, SlERF6, has been recently identified to play an important role in tomato fruit ripening and carotenoid accumulation acting as a negative regulator of two of the predominant nutritional compounds of tomato, lycopene and β-carotene [23]. Ethylene response factors (ERFs) are plant transcriptional regulators mediating ethylene-dependent gene expression via binding to the GCC motif found in the promoter region of ethylene-regulated genes. Two watermelon genes (Cla021765 and Cla003789) have high similarity with SlERF6. While Cla021765 was constitutively expressed during ripening with no significant changes over time (average RPKM = 42 ± 12), Cla003789 expression was up regulated during the late stages of watermelon ripening, peaking at the pink stage (Figure 8). It has been suggested that SlERF6 may integrate ethylene-dependent and independent regulatory activities to allow for the fine-tuning of signal outputs.
Putative homologs of tomato components of the light signal transduction pathway, such as DE-ETIOLATED1 (DET1; originally identified as HIGH PIGMENT [hp2]), UV-DAMAGED DNA BINDING PROTEIN1 (DDB1; originally hp1) and CULLIN-4 (CUL4) were identified in watermelon (sequences Cla021340, Cla019536 and Cla019583, respectively – Table 4). All three genes were expressed at a low level during watermelon fruit ripening with no significant variation (FDR > 0.05) suggesting they are probably not rate limiting in isoprenoid biosynthesis and associated fruit pigmentation.
GLK2 (Golden 2-like) transcription factor (of the GARP family Myb transcription factor) determines chlorophyll accumulation and distribution in developing tomato fruits by controlling chloroplast formation. In tomato it is expressed in fruits where it accumulates at the mature-green stage [87]. In watermelon fruits the sequence Cla003729 was identified as putative homolog of GLK2 (e-value = 3e-11) but no transcript reads were generated by the Illumina RNA sequencing technology suggesting it is not expressed during ripening. This is in accordance with the evidences that watermelon flesh chromoplasts do not de-differentiate from chloroplasts as occurs in tomatoes but, mainly, from undifferentiated proplastids [103].
Another transcription factor, the homeobox protein LeHB-1, was reported to regulate fruit ripening through transcriptional control of 1-aminocyclopropane carboxylic acid oxidase 1 (ACO1) expression [83]. LeHB-1 is highly expressed in tomato flower buds, senescing flowers, and developing immature and mature green fruits, but its mRNA declined during ripening and is maintained at a stable but relatively low level in red-ripe fruits [83]. Cla017080 the putative watermelon homolog of LeHB-1 was expressed in developing watermelon fruits at the white stage, down regulated at the beginning of carotenoid accumulation (white-pink stage) and up-regulated at the pink and red-ripe stages of ripening (Figure 8). Ethylene is not essential for non-climacteric fruits, however, recent studies indicate that ethylene and/or a modulated sensitivity to ethylene might participate in physiological changes during non-climacteric fruit development [104–107]. Indeed, many non-climacteric fruits, including watermelon, are highly sensitive to exogenous ethylene [108, 109]. In addition Cla017080 may regulate isoprenoid accumulation in an ethylene independent way by controlling other regulative factors. Putative LeHB-1 sites have been, in fact, found in the promoters of a number of ripening related genes, including LeRIN-MADS and NAC-NOR.
Conclusions
Taken together, these results suggest maintenance of many regulators in the watermelon genome in common with tomato, yet suggest a complex and, for many aspects, different regulatory system for isoprenoid biosynthesis between these fruits. While a common set of metabolic and regulatory genes influences carotenoid accumulation during development and ripening, specific regulatory systems may also differ possibly related to the different ripening physiologies of climacteric and non-climacteric fruits. As such, these data represent the starting point for characterizing conserved and distinct regulatory functions of isoprenoid biosynthesis in climacteric and non-climacteric species. They also provide information and targets for plant researchers and breeders to test as potential tools for watermelon crop improvement. Since this study was done only at the transcriptional level, subsequent experimentation is required to see if other changes arise at other levels of cellular regulation.
Methods
Plant cultivation
Watermelon (Citrullus lanatus (Thunb.) Matsum. & Nakai var. lanatus) cultivar Dumara was used in the present study. Dumara is a twenty year old cultivar but it is still one of the most important commercial selections worldwide and produces elongated fruits characterized by green skin with dark green stripes and sweet, firm flesh containing seeds. Sowing was carried out on 19 February 2010 in plug-seedling trays. One-month-old watermelon seedlings were transplanted at a spacing of approximately 200 cm and 250 cm between rows into a sandy soil of an open-field in the province of Lecce in southern Italy (latitude 40°23’16”80 N, longitude 17°57’41”40E; decimal degrees 40.3881; 17.9615). After transplanting, drip irrigation was applied with 4 L h-1, for 1–3 h, at 1–2-day intervals, as determined by potential evapotranspiration at the research station, climate data and crop coefficients as defined by FAO [110]. Drippers were placed at 0.4 m intervals along the irrigation line. Chemical fertilizer solution (145 kg N ha-1, 140 kg P2O5 ha-1, 210 kg K2O ha-1) was added to water irrigation by pump injection twice a week. The production methods also included hand-weeding and plant-pathogen control with synthetic chemical pesticides. Imidacloprid (200 g L-1) was used to reduce aphids, acetamiprid (200 g L-1) was applied to reduce thrips and abamectine (18 g L-1) was used to reduce mites.
Fruit sampling
Watermelon fruits were harvested from the rows at different ripening stages. Three independent samples of at least 3 injury-free watermelon fruits were hand harvested randomly at four ripening stages indicated as white [~10 days after pollination (DAP)]: small fruit size (approx. 18 cm long × 10 cm wide) and white flesh; white-pink (~18 DAP): not yet mature medium sized fruit (approx. 31×18 cm) with white-pink flesh; pink (~28 DAP): large fruit size (approx. 35×23 cm) with pink flesh and green tendril; red-ripe (~34 DAP): fully expanded (approx. 40×28 cm) mature fruit with red flesh, brown tendril and yellow ground spot (Figure 1). Watermelon fruits were quickly delivered to the laboratory and cut longitudinally from the stem-end to the blossom-end through the ground spot.
The soluble solid content (°Brix) was measured immediately by cutting a wedge of flesh from the heart area (between locules and the fruit centre) and squeezing the juice into a digital refractometer (Atago PR-100, NSG Precision Cells, Inc, Farming dale, NY, US) calibrated with a 10% sucrose solution. Since soluble solid content increases during watermelon ripening, the measured values were used to identify the four ripening stages as follows: white stage (2-3°Brix), white-pink stage (4-5°Brix), pink stage (7-8°Brix) and red-ripe stage (10-12°Brix).
For all further analyses, flesh samples were taken from the heart area of each watermelon. These tissues were immediately frozen in liquid nitrogen and stored at −80°C until use.
Carotenoid extraction and HPLC analysis
Frozen flesh samples from each fruit stage were rapidly homogenized with a laboratory blender (Waring Laboratory and Science, Torrington, CT, US). Carotenoid extraction and determination were conducted as described by Alba et al. [111]. Frozen homogenates (300 mg) were subjected to extraction of carotenoids with 300 mL of tetrahydrofuran and 50 μL of Mg carbonate (0.3 g/mL). The samples were homogenized in a FastPrep machine (FastPrep FP120, Qbiogene, Inc., Carlsbad, CA, US) and resulting homogenates were filtered with a Spin-X filter (Corning International K.K., Tokyo, Japan). The samples were re-extracted with 300 μL of 5% w/v butylated hydroxytoluene in methanol.
Carotenoids were partitioned into 375 μL of petroleum ether using 150 mL of 25% NaCl. The extract was evaporated to near dryness using a Vacufuge 5301 Centrifugal Vacuum Concentrator (Krackeler Scientific Inc., Albany, NY, US), suspended in 500 μL di methyl t-butyl ether and 475 μL di methanol and passed through a syringe filter (GE Osmonics, Minnetonka, MN, US) prior to injection onto a C30 carotenoid column (Waters, Milford, MA, US).
HPLC employed a Summit HPLC system and a PDA-100 photodiode array detector (Dionex, Sunnyvale, CA, US). The elution gradient consisted of 5 min at 100% methanol, a 20-min ramp to 95% t-butyl ether, 5 min at 95% t-butyl ether, and a 5-min ramp returning the system to 100% methanol. The column was equilibrated with 100% methanol for 10 min before each run. Spectra were collected at 348, 434, 450 and 471 nm and pigments were identified via co-migration with purified standards and/or by their pigment-specific absorbance spectra. Results are presented as mean value ± standard deviation of at least three independent replicated experiments (n = 3). Statistical analysis was based on a one-way ANOVA test. The post-hoc method by Holm-Sidak was applied to establish significant differences between means with a confidence level of 95%. All statistical comparisons were performed using the SigmaStat Version 3.11 software (Systat Software Inc., Chicago, IL, US).
RNA-Seq experiment
Total RNA isolation
Total RNA was isolated from frozen flesh homogenates from every fruit stage using the RNeasy Plant Mini kit (Qiagen, Hilden, Germany). RNA quality and quantity were determined using a NanoDrop spectrophotometer and denaturing agarose gel electrophoresis [112]. Only RNAs with an OD260:OD280 ratio >1.80 and no discernible degradation were used for preparing samples for sequencing of mRNA.
Preparation of cDNA libraries and sequencing (RNA-seq)
Sample preparation and multiplex sequencing was essentially as described in Zhong et al. [113]. In summary, samples for sequencing of mRNA were prepared using mRNA-Seq Sample Prep Kit (Illumina, San Diego, CA, US) following manufacturer’s instructions. PolyA+RNA was extracted from 10 μg of each total RNA sample using poly-T oligo-attached magnetic beads. The mRNA was eluted in 10 mM Tris–HCl and fragmentated in small pieces using divalent cations under elevated temperature. For the first strand of cDNA synthesis, cleaved mRNA fragments were mixed with random primers, incubated at 70°C for 5 minutes, and then transferred to an ice bath. 5× First strand buffer, 100 mM DTT, 25 mM dNTP mix and RNase OUT were added to the previous mix obtaining a total volume of 19 μl; this reaction mix was incubated for 2 minutes at 25°C. Then, SuperScript II (Invitrogen, Carlsbad, CA, US) was added to the sample that was incubated at 25°C for 10 minutes, 42°C for 50 minutes, 70°C for 15 minutes. The resulting first strand cDNA was used to make second strand cDNA in a reaction mix containing GEX Second strand buffer, 25 mM dNTPs, DNA polymerase I, RNase H in a total volume of 100 μl; this reaction mix was incubated for 2.5 hours at 16°C. The resulting double stranded cDNA was then purified using the QIAquick PCR purification kit (Qiagen), following the manufacturer’s instructions. The cDNA was blunt ended with End Repair Enzyme (NEB) in the presence of 2.5 mM dNTPs (NEB) and 10 mM ATP. Adenine nucleotide was ed to the 3' ends of the blunt ended cDNA with Klenow DNA Polymerase (3' to 5' exominus) in the presence of 1 mM dATP (NEB) by incubating at 37°C for 30 minutes. The end labeled double stranded cDNA was purified with a MinElute PCR purification kit (Qiagen). The double stranded cDNA with A-nucleotides on 3' ends was ligated with adapters (Illumina) using T4 DNA ligase at room temperature for 15 minutes. The samples were then purified with MinElute PCR purification kit (Qiagen). The products of the ligation reaction were purified on 2% agarose gel selecting 200 bp (±25 bp) templates. Subsequently, the cDNA was amplified with two adapter primers (Illumina) with initial denaturing step at 98°C for 30 seconds, followed by 15 cycles at 98°C for 10 seconds, 65°C for 30 seconds, 72°C for 30 seconds with a final extension cycle at 72°C for 5 minutes. The PCR product was purified with Qiaquick PCR purification kit. DNA size, purity and concentration were checked by an Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA, US). Libraries were barcoded and multiplexed in collections of four samples per lane of sequencing. Sequencing was performed on an Illumina GAII at the Cornell Weill Medical School campus in New York City. A total of 5.7-10.7 million reads were obtained for each library. Raw RNA-seq reads have been deposited into the NCBI sequence read archive (SRA) under accession SRA102510.
Gene expression analysis of RNA-Seq data
RNA-Seq reads were first aligned to ribosomal RNA (rRNA) sequence database [114] using Bowtie allowing up to two mismatches [115], to remove any possible rRNA contaminations. The resulting filtered reads were aligned to the watermelon reference genome [116] using TopHat [117] allowing one segment mismatch. Following alignments, raw counts for each watermelon gene were normalized to Reads Per Kilobase of exon model per Million mapped reads (RPKM). Two biological replicas from distinct watermelon fruits (n = 2) were performed.
To identify differentially expressed genes during watermelon fruit development, the RNA-seq expression data were first transformed using the getVarianceStabilizedData function in the DESeq package [118]. The variance-stabilizing transformed RNA-Seq expression data were then fed to the LIMMA package, and F tests were performed [119]. Raw p-values of multiple tests were corrected using FDR [120]. Genes with FDRs less than 0.05 were identified as differentially expressed genes.
Abbreviations
- ABA:
-
Abscisic acid
- ACAT:
-
Acetyl-CoA acetyltransferase
- ACO:
-
1-aminocyclopropane carboxylic acid oxidase
- ACS:
-
1-aminocyclopropane-1-carboxylate synthase
- ANOVA:
-
Analysis of variance
- CCDs:
-
Carotenoid cleavage dioxygenases
- CHYB:
-
β-carotene hydroxylase
- CMK:
-
4-diphosphocytidyl-2-C-methyl-d-erythritol kinase
- DAP:
-
Day after pollination
- CRTISO:
-
Carotene cis-trans isomerase
- DMAPP:
-
Dimethylallyl diphosphate
- DXP:
-
1-deoxy-d-xylulose-5-phosphate
- DXR:
-
1-deoxy-d-xylulose-5-phosphate reductoisomerase
- DXS:
-
1-deoxy-d-xylulose-5-phosphate synthase
- ERFs:
-
Ethylene response factors
- FPS:
-
Farnesyl diphosphate synthase
- FDR:
-
False discovery rate
- FPP:
-
Farnesyl diphosphate
- GGPP:
-
Geranylgeranyl diphosphate
- GGPR:
-
Geranylgeranyl diphosphate reductase
- GGPS:
-
geranylgeranyl diphosphate synthase
- GPP:
-
Geranyl-diphosphate
- GPS:
-
Geranyl-diphosphate synthase
- HDR:
-
4-hydroxy-3-methylbut-2-enyl diphosphate reductase
- HDS:
-
4-hydroxy-3-methylbut-2-en-1-yl diphosphate synthase
- HMG-CoA:
-
3-hydroxy-3-methyl-glutaryl-CoA
- HMGR:
-
3-hydroxy-3-methylglutaryl-CoA reductase
- hp:
-
High-pigment
- IDI:
-
Isopentenyl-diphosphate δ-isomerase
- IPP:
-
Isopentenyl-diphosphate
- KSB:
-
Ent-kaur-16-ene synthase
- LCYB:
-
Lycopene β-cyclase
- LCYE:
-
Lycopene ϵ-cyclase
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantification
- MCT:
-
2-C-methyl-d-erythritol 4-phosphate cytidyltransferase
- MDS:
-
2-C-methyl-d-erythritol 2,4-cyclodiphosphate synthase
- MEP:
-
2-C-methyl-d-erythritol 4-phosphate
- MVA:
-
Mevalonate
- PDS:
-
Phytoene desaturase
- PREA:
-
Prenyl transferase
- PSY:
-
Phytoene synthase
- RPKM:
-
Reads per kilobase of exon model per million mapped reads
- ZDS:
-
ζ-carotene desaturase
- ZEP:
-
Zeaxanthin epoxidase.
References
Vranová E, Coman D, Gruissem W: Structure and dynamics of the isoprenoid pathway network. Mol Plant. 2012, 5: 318-333. 10.1093/mp/sss015.
Frank HA, Cogdell RJ: Photochemistry of carotenoids. Carotenoids in Photosynthesis. Edited by: Young A, Britton G. 1993, London: Chapman & Hall, 253-326.
Parker RS: Carotenoids. 4. Absorption, metabolism, and transport of carotenoids. FASEB J. 1996, 10: 542-551.
Bendich A, Olson JA: Biological actions of carotenoids. FASEB J. 1998, 3: 1927-1932.
Ziegler RG: A review of epidemiologic evidence that carotenoids reduce the risk of cancer. J Nutr. 1989, 119: 116-122.
Edge R, McGarvey DJ, Truscott TG: The carotenoids as anti-oxidants - a review. J Photochem Photobiol B. 1997, 41: 189-200. 10.1016/S1011-1344(97)00092-4.
Rao AV, Agarwal S: Role of antioxidant lycopene in cancer and heart disease. J Am Coll Nutr. 2000, 19: 563-569. 10.1080/07315724.2000.10718953.
Palozza P, Parrone N, Catalano A, Simone R: Tomato lycopene and inflammatory cascade: basic interactions and clinical implications. Curr Med Chem. 2010, 17: 2547-2563. 10.2174/092986710791556041.
Palozza P, Simone R, Catalano A, Monego G, Barini A, Mele MC, Parrone N, Trombino S, Picci N, Ranelletti FO: Lycopene prevention of oxysterol-induced proinflammatory cytokine cascade in human macrophages: inhibition of NF-κB nuclear binding and increase in PPARγ expression. J Nutr Biochem. 2011, 22: 259-268. 10.1016/j.jnutbio.2010.02.003.
Britton G: Overview of carotenoid biosynthesis. Carotenoids: biosynthesis and metabolism, Volume 3. Edited by: Britton G, Liaaen-Jensen S, Pfander H. 1998, Basel: Birkhauser, 13-147.
Rao AV, Rao LG: Carotenoids and human health. Pharmacol Res. 2007, 55: 207-216. 10.1016/j.phrs.2007.01.012.
FAOSTAT-Agriculture.http://faostat.fao.org,
Botella-Pavía P, Rodríguez-Concepción M: Carotenoid biotechnology in plants for nutritionally improved foods. Physiol Plantarum. 2006, 126: 369-381. 10.1111/j.1399-3054.2006.00632.x.
Fraser PD, Enfissi EMA, Bramley PM: Genetic engineering of carotenoid formation in tomato fruit and the potential application of systems and synthetic biology approaches. Arch Biochem Biophys. 2009, 483: 196-204. 10.1016/j.abb.2008.10.009.
Cunningham FX, Gantt E: Genes and enzymes of carotenoid biosynthesis in plants. Annu Rev Plant Physiol Plant Mol Biol. 1998, 49: 557-583. 10.1146/annurev.arplant.49.1.557.
Hirschberg J: Carotenoid biosynthesis in flowering plants. Curr Opin Plant Biol. 2001, 4: 210-218. 10.1016/S1369-5266(00)00163-1.
Howitt CA, Pogson BJ: Carotenoid accumulation and function in seeds and non-green tissues. Plant Cell Environ. 2006, 29: 435-445. 10.1111/j.1365-3040.2005.01492.x.
Lu S, Van Eck J, Zhou X: The cauliflower Or gene encodes a DnaJ cysteine-rich domain containing protein that mediates high levels of beta-carotene accumulation. Plant Cell. 2006, 18: 3594-3605. 10.1105/tpc.106.046417.
Li L, Yanga Y, Xu Q: The Or gene enhances carotenoid accumulation and stability during post-harvest storage of potato tubers. Mol Plant. 2012, 5: 339-352. 10.1093/mp/ssr099.
Alexander L, Grierson D: Ethylene biosynthesis and action in tomato: a model for climacteric fruit ripening. J Exp Bot. 2002, 53: 2039-2055. 10.1093/jxb/erf072.
Bramley PM: Regulation of carotenoid formation during tomato fruit ripening and development. J Exp Bot. 2002, 53: 2107-2113. 10.1093/jxb/erf059.
Cazzonelli C, Pogson B: Source to sink: regulation of carotenoid biosynthesis in plants. Trends Plant Sci. 2010, 15: 266-274. 10.1016/j.tplants.2010.02.003.
Lee JM, Joung J-G, McQuinn R, Chung M-Y, Fei Z, Tieman D, Klee H, Giovannoni JJ: Combined transcriptome, genetic diversity and metabolite profiling in tomato fruit reveals that the ethylene response factor SlERF6 plays an important role in ripening and carotenoid accumulation. Plant J. 2012, 70: 191-204. 10.1111/j.1365-313X.2011.04863.x.
Kahlau S, Bock R: Plastid transcriptomics and translatomics of tomato fruit development and chloroplast-to-chromoplast differentiation: chromoplast gene expression largely serves the production of a single protein. Plant Cell. 2008, 20: 856-874. 10.1105/tpc.107.055202.
Apel W, Bock R: Enhancement of carotenoid biosynthesis in transplastomic tomatoes by induced lycopene-to-provitamin A conversion. Plant Physiol. 2009, 151: 59-66. 10.1104/pp.109.140533.
Bartley GE, Scolnik PA, Giuliano G: Molecular biology of carotenoid biosynthesis in plants. Annu Rev Plant Physiol Plant Mol Biol. 1994, 45: 287-301. 10.1146/annurev.pp.45.060194.001443.
Bartley GE, Scolnik PA: Plant carotenoids: pigments for photoprotection, visual attraction, and human health. Plant Cell. 1995, 7: 1027-1038.
Lu S, Li L: Carotenoid metabolism: biosynthesis, regulation, and beyond. J Integr Plant Biol. 2008, 50: 778-785. 10.1111/j.1744-7909.2008.00708.x.
Kato M, Matsumoto H, Ikoma Y, Okuda H, Yano M: The role of carotenoid cleavage dioxygenases in the regulation of carotenoid profiles during maturation in citrus fruit. J Exp Bot. 2006, 57: 2153-2164. 10.1093/jxb/erj172.
Ohmiya A, Kishimoto S, Aida R, Yoshioka S, Sumitomo K: Carotenoid cleavage dioxygenase (CmCCD4a) contributes to white color formation in chrysanthemum petals. Plant Physiol. 2006, 142: 1193-1201. 10.1104/pp.106.087130.
Qin X, Zeevaart JAD: The 9-cis-epoxycarotenoid cleavage reaction is the key regulatory step of abscisic acid biosynthesis in water-stressed bean. Proc Natl Acad Sci USA. 1999, 96: 15354-15361. 10.1073/pnas.96.26.15354.
Iuchi S, Kobayashi M, Yamaguchi-Shinozaki K, Shinozaki K: A stress-inducible gene for 9-cis-epoxycarotenoid dioxygenase involved in abscisic acid biosynthesis under water stress in drought-tolerant cowpea. Plant Physiol. 2000, 123: 553-562. 10.1104/pp.123.2.553.
Liu Y, Roof S, Ye Z, Barry C, van Tuinen A, Vrebalov J, Bowler C, Giovannoni J: Manipulation of light signal transduction as a means of modifying fruit nutritional quality in tomato. Proc Natl Acad Sci USA. 2004, 101: 9897-9902. 10.1073/pnas.0400935101.
Kim BR, Kim SU, Chang YJ: Differential expression of three 1-deoxy-D-xylulose-5-phosphate synthase genes in rice. Biotechnol Lett. 2005, 27: 997-1001. 10.1007/s10529-005-7849-1.
Cordoba E, Salmi M, León P: Unravelling the regulatory mechanisms that modulate the MEP pathway in higher plants. J Exp Bot. 2009, 60: 2933-2943. 10.1093/jxb/erp190.
Simkin AJ, Schwartz SH, Auldridge M, Taylor MG, Klee HJ: The tomato carotenoid cleavage dioxygenase 1 genes contribute to the formation of the flavor volatiles β-ionone, pseudoionone, and geranylacetone. Plant J. 2004, 40: 882-892. 10.1111/j.1365-313X.2004.02263.x.
Cookson PJ, Kiano JW, Shipton CA, Fraser PD, Romer S, Schuch W, Bramley PM, Pyke KA: Increases in cell elongation, plastid compartment size and phytoene synthase activity underlie the phenotype of the high pigment-1 mutant of tomato. Planta. 2003, 217: 896-903. 10.1007/s00425-003-1065-9.
Kolotilin I, Koltai H, Tadmor Y, Bar-Or C, Reuveni M, Meir A, Nahon S, Shlomo H, Chen L, Levin I: Transcriptional profiling of high pigment-2dg tomato mutant links early fruit plastid biogenesis with its overproduction of phytonutrients. Plant Physiol. 2007, 145: 389-401. 10.1104/pp.107.102962.
Galpaz N, Wang Q, Menda N, Zamir D, Hirschberg J: Abscisic acid deficiency in the tomato mutant high-pigment 3 leading to increased plastid number and higher fruit lycopene content. Plant J. 2008, 53: 717-730. 10.1111/j.1365-313X.2007.03362.x.
Tlili I, Hdider C, Lenucci MS, Ilahy R, Jebari H, Dalessandro G: Bioactive compounds and antioxidant activities during fruit ripening of watermelon cultivars. J Food Comp Anal. 2011, 24: 923-928. 10.1016/j.jfca.2011.03.016.
Perkins-Veazie P, Collins JK, Pair SD, Roberts W: Lycopene content differs among red-fleshed watermelon cultivars. J Sci Food Agric. 2001, 81: 983-987. 10.1002/jsfa.880.
Perkins-Veazie P, Collins JK, Davis AR, Roberts BW: Carotenoid content of 50 watermelon cultivars. J Agric Food Chem. 2006, 54: 2593-2597. 10.1021/jf052066p.
Pogson B, McDonald KA, Truong M, Britton G, DellaPenna D: Arabidopsis carotenoid mutants demonstrate that lutein is not essential for photosynthesis in higher plants. Plant Cell. 1996, 8: 1627-1639.
Tadmor Y, Katzir N, King S, Levi A, Davis A, Hirschberg J: Fruit coloration in watermelon: Lessons from the tomato. Progress in Cucurbit Genetics and Breeding Research. Edited by: Lebeda A, Paris HS. 2004, Olomouc (Chech Republic): Palacky University in Olomouc, 181-185.
Guo S, Liu J, Zheng Y: Characterization of transcriptome dynamics during watermelon fruit development: sequencing, assembly, annotation and gene expression profiles. Genomics. 2011, 12: 454-466.
Cucurbit Genomics Database.http://www.icugi.org,
Goldstein JL, Brown MS: Regulation of the mevalonate pathway. Nature. 1990, 343: 425-430. 10.1038/343425a0.
Friesen JA, Rodwell VW: The 3-hydroxy-3-methylglutaryl coenzyme-A (HMG-CoA) reductases. Genome Biol. 2004, 5: 248-10.1186/gb-2004-5-11-248.
Phillips MA, D'Auria JC, Gershenzona J, Pichersky E: The Arabidopsis thaliana type I isopentenyl diphosphate isomerases are targeted to multiple subcellular compartments and have overlapping functions in isoprenoid biosynthesis. Plant Cell. 2008, 20: 677-696. 10.1105/tpc.107.053926.
Schwarz M, Arigoni D: Ginkgolide biosynthesis. Comprehensive natural products chemistry. Volume 2 Isoprenoids including carotenoids and steroids. Edited by: Barton SD, Nakanishi K, Meth-Cohn O, Cane DE. 1999, Amsterdam: Elsevier, 367-401.
Nagata N, Suzuki M, Yoshida S, Muranaka T: Mevalonic acid partially restores chloroplast and etioplast development in Arabidopsis lacking the non-mevalonate pathway. Planta. 2002, 216: 345-350. 10.1007/s00425-002-0871-9.
Estévez JM, Cantero A, Reindl A, Reichler S, León P: 1-Deoxy-d-xylulose-5-phosphate synthase, a limiting enzyme for plastidic isoprenoid biosynthesis in plants. J Biol Chem. 2001, 276: 22901-22909. 10.1074/jbc.M100854200.
Julliard JH, Douce R: Biosynthesis of the thiazole moiety of thiamin (vitamin B1) in higher plant chloroplasts. Proc Natl Acad Sci USA. 1991, 88: 2042-2045. 10.1073/pnas.88.6.2042.
Zhao G, Winkler ME: An Escherichia coli K-12 tktA tktB mutant deficient in transketolase activity requires pyridoxine (vitamin B6) as well as the aromatic amino acids and vitamins for growth. J Bacteriol. 1994, 176: 6134-6138.
Araki N, Kusumi K, Masamoto K, Niwa Y, Iba K: Temperature-sensitive Arabidopsis mutant defective in 1-deoxy-d-xylulose 5-phosphate synthase within the plastid non-mevalonate pathway of isoprenoid biosynthesis. Physiol Plantarum. 2000, 108: 19-24.
Walter MH, Hans J, Strack D: Two distantly related genes encoding 1-deoxy-d-xylulose 5-phosphate synthases: differential regulation in shoots and apocarotenoid-accumulating mycorrhizal roots. Plant J. 2002, 31: 243-254. 10.1046/j.1365-313X.2002.01352.x.
Rodríguez-Concepción M, Forés O, Martínez-García JF, Gozález V, Phillips MA, Ferrer A, Boronat A: Distinct light-mediated pathways regulate the biosynthesis and exchange of isoprenoid precursors during Arabidopsis seedling development. Plant Cell. 2004, 16: 144-156. 10.1105/tpc.016204.
Phillips MA, Walter MH, Ralph SG: Functional identification and differential expression of 1-deoxy-d-xylulose 5-phosphate synthase in induced terpenoid resin formation of Norway spruce (Picea abies). Plant Mol Biol. 2007, 65: 243-257. 10.1007/s11103-007-9212-5.
Cordoba E, Porta H, Arroyo A, San Román C, Medina L, Rodríguez-Concepción M, León P: Functional characterization of the three genes encoding 1-deoxy-d-xylulose 5-phosphate synthase in maize. J Exp Bot. 2011, 62: 2023-2038. 10.1093/jxb/erq393.
Bouvier F, Harlingue A, Suire C, Backhaus R, Camara B: Dedicated roles of plastid transketolases during the early onset of isoprenoid biogenesis in pepper fruits. Plant Physiol. 1998, 117: 1423-1431. 10.1104/pp.117.4.1423.
Enfissi EMA, Fraser PD, Lois LM, Boronat A, Schuch W, Bramley PM: Metabolic engineering of the mevalonate and non-mevalonate isopentenyl diphosphate-forming pathways for the production of health-promoting isoprenoids. Plant Biotech J. 2005, 3: 17-27.
Lois LM, Rodríguez-Concepción M, Gallego F, Campos N, Boronat A: Carotenoid biosynthesis during tomato fruit development: regulatory role of 1-deoxy-d-xylulose 5-phosphate synthase. Plant J. 2000, 22: 503-513. 10.1046/j.1365-313x.2000.00764.x.
Lange BM, Wildung MR, McCaskill D, Croteau R: A family of transketolases that directs isoprenoid biosynthesis via a mevalonate-independent pathway. Proc Natl Acad Sci USA. 1998, 95: 2100-2104. 10.1073/pnas.95.5.2100.
Pandit SS, Kulkarni RS, Giri AP, Köllner TG, Degenhardt J, Gershenzon J, Gupta VS: Expression profiling of various genes during the fruit development and ripening of mango. Plant Physiol Biochem. 2010, 48: 426-433. 10.1016/j.plaphy.2010.02.012.
Fraser PD, Enfissi EMA, Halket JM, Truesdale MR, Yu D, Gerrish C, Bramley PM: Manipulation of phytoene levels in tomato fruit: effects on isoprenoids, plastids, and intermediary metabolism. Plant Cell. 2007, 19: 3194-3211. 10.1105/tpc.106.049817.
Van den Berg H, Faulks R, Fernando Granado H, Hirschberg J, Olmedilla B, Sandmann G, Southon S, Stahl W: The potential for the improvement of carotenoid levels in foods and the likely systemic effects. J Sci Food Agric. 2000, 80: 880-912. 10.1002/(SICI)1097-0010(20000515)80:7<880::AID-JSFA646>3.0.CO;2-1.
Isaacson T, Ohad GI, Beyer P, Hirschberg J: Analysis in vitro of the enzyme CRTISO establishes a poly-cis-carotenoid biosynthesis pathway in plants. Plant Physiol. 2004, 136: 4246-4255. 10.1104/pp.104.052092.
Isaacson T, Ronen G, Zamir D, Hirschberg J: Cloning of tangerine from tomato reveals a carotenoid isomerase essential for the production of beta-carotene and xanthophylls in plants. Plant Cell. 2002, 14: 333-342. 10.1105/tpc.010303.
Tadmor Y, King S, Levi A, Davis A, Meir A, Wasserman B, Hirschberg J, Lewinsohn E: Comparative fruit colouration in watermelon and tomato. Food Res Int. 2005, 38: 837-841. 10.1016/j.foodres.2004.07.011.
Giuliano G, Tavazza R, Diretto G, Beyer P, Taylor MA: Metabolic engineering of carotenoid biosynthesis in plants. Trends Biotechnol. 2008, 26: 139-145. 10.1016/j.tibtech.2007.12.003.
Pecker I, Gabbay R, Cunningham FX, Hirschberg J: Cloning and characterization of the cDNA for lycopene β-cyclase from tomato reveals decrease in its expression during fruit ripening. Plant Mol Biol. 1996, 30: 807-819. 10.1007/BF00019013.
Lenucci MS, Serrone L, De Caroli M, Fraser PD, Bramley PM, Piro G, Dalessandro G: Isoprenoid, lipid, and protein contents in intact plastids isolated from mesocarp cells of traditional and high-pigment tomato cultivars at different ripening stages. J Agric Food Chem. 2012, 60: 1764-1775. 10.1021/jf204189z.
Kang B, Zhao W, Hou Y, Tian P: Expression of carotenogenic genes during the development and ripening of watermelon fruit. Sci Hort. 2010, 124: 368-375. 10.1016/j.scienta.2010.01.027.
Schwartz SH, Tan BC, Gage DA, Ztevaart JAD, McCarty DR: Specific oxidative cleavage of carotenoids by VP14 of maize. Science. 1997, 276: 1872-1874. 10.1126/science.276.5320.1872.
Schwartz SH, Qin X, Zeevaart JAD: Elucidation of the indirect pathway of abscisic acid biosynthesis by mutants, genes, and enzymes. Plant Physiol. 2003, 131: 1591-1601. 10.1104/pp.102.017921.
Auldridge ME, McCarty DR, Klee HJ: Plant carotenoid cleavage oxygenases and their apocarotenoid products. Curr Opin Plant Biol. 2006, 9: 315-321. 10.1016/j.pbi.2006.03.005.
Vogel JT, Tan B, McCarty DR, Klee HJ: The carotenoid cleavage dioxygenase 1 enzyme has broad substrate specificity, cleaving multiple carotenoids at two different bond positions. J Biol Chem. 2008, 283: 11364-11373. 10.1074/jbc.M710106200.
Enfissi EMA, Barneche F, Ahmed I: Integrative transcript and metabolite analysis of nutritionally enhanced DE-ETIOLATED1 downregulated tomato fruit. Plant Cell. 2010, 22: 1190-1215. 10.1105/tpc.110.073866.
Vrebalov J, Ruezinsky D, Padmanabhan V, White R, Medrano D, Drake R, Schuch W, Giovannoni J: A MADS-box gene necessary for fruit ripening at the tomato ripening-inhibitor (Rin) locus. Science. 2002, 296: 343-346. 10.1126/science.1068181.
Busi MV, Bustamante C, D’Angelo C, Hidalgo-Cuevas M, Boggio SB, Valle EM, Zabaleta E: MADS-box genes expressed during tomato seed and fruit development. Plant Mol Biol. 2003, 52: 801-815. 10.1023/A:1025001402838.
Giovannoni JJ: Genetic regulation of fruit development and ripening. Plant Cell. 2004, 16: S170-S180. 10.1105/tpc.019158.
Manning K, Tor M, Poole M, Hong Y, Thompson AJ, King GJ, Giovannoni JJ, Seymour GB: A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nature Genet. 2006, 38: 948-952. 10.1038/ng1841.
Lin Z, Hong Y, Yin M, Li C, Zhang K, Grierson D: A tomato HD-Zip homeobox protein, LeHB-1, plays an important role in floral organogenesis and ripening. Plant J. 2008, 55: 301-310. 10.1111/j.1365-313X.2008.03505.x.
Itkin M, Seybold H, Breitel D, Rogachev I, Meir S, Aharoni A: TOMATO AGAMOUS-LIKE 1 is a component of the fruit ripening regulatory network. Plant J. 2009, 60: 1081-1095. 10.1111/j.1365-313X.2009.04064.x.
Pan IL, McQuinn R, Giovannoni JJ, Irish VF: Functional diversification of AGAMOUS lineage genes in regulating tomato flower and fruit development. J Exp Bot. 2010, 61: 1795-1806. 10.1093/jxb/erq046.
Chung MY, Vrebalov J, Alba R, Lee J, McQuinn R, Chung JD, Klein P, Giovannoni J: A tomato (Solanum lycopersicum) APETALA2/ERF gene, SlAP2a, is a negative regulator of fruit ripening. Plant J. 2010, 64: 936-947. 10.1111/j.1365-313X.2010.04384.x.
Powell AL, Nguyen CV, Hill T: Uniform ripening encodes a Golden 2-like transcription factor regulating tomato fruit chloroplast development. Science. 2012, 336: 1711-1715. 10.1126/science.1222218.
Fujisawa M, Nakano T, Ito Y: Identification of potential target genes for the tomato fruit-ripening regulator RIN by chromatin immunoprecipitation. BMC Plant Biol. 2011, 11: 26-40. 10.1186/1471-2229-11-26.
Martel C, Vrebalov J, Tafelmeyer P, Giovannoni JJ: The tomato MADS-box transcription factor RIPENING INHIBITOR interacts with promoters involved in numerous ripening processes in a COLORLESS NONRIPENING-dependent manner. Plant Physiol. 2011, 157: 1568-1579. 10.1104/pp.111.181107.
Zhong S, Fei Z, Chen YR: Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nature Biotechnol. 2013, 31: 154-159. 10.1038/nbt.2462.
Cantu D, Blanco-Ulate B, Yang L, Labavitch JM, Bennett AB, Powell ALT: Ripening-regulated susceptibility of tomato fruit to Botrytis cinerea requires NOR but not RIN or ethylene. Plant Physiol. 2009, 150: 1434-1449. 10.1104/pp.109.138701.
Klee HJ, Giovannoni JJ: Genetics and control of tomato fruit ripening and quality attributes. Annu Rev Genet. 2011, 45: 41-59. 10.1146/annurev-genet-110410-132507.
Seymour GB, Ryder CD, Cevik V, Hammond JP, Popovich A, King GJ, Vrebalov J, Giovannoni JJ, Manning K: A SEPALLATA gene is involved in the development and ripening of strawberry (Fragaria × ananassa Duch.) fruit, a non-climacteric tissue. J Exp Bot. 2011, 62: 1179-1188. 10.1093/jxb/erq360.
Vrebalov J, Pan IL, Arroyo AJ: Fleshy fruit expansion and ripening are regulated by the tomato SHATTERPROOF gene TAGL1. Plant Cell. 2009, 21: 3041-3062. 10.1105/tpc.109.066936.
Wang KL-C, Li H, Ecker JR: Ethylene biosynthesis and signaling networks. Plant Cell. 2002, 14: S131-S151.
Perkins-Veazie P, Collins JK, Cartwright B: Ethylene production in watermelon fruit varies with cultivar and fruit tissue. Hort Science. 1995, 30: 825-826.
Bemer M, Karlova R, Ballester AR, Tikunov YM, Bovy AG, Wolters-Arts M, de Barros Rossetto P, Angenent GC, de Maagd RA: The tomato FRUITFULL homologs TDR4/FUL1 and MBP7/FUL2 regulate ethylene-independent aspects of fruit ripening. Plant Cel. 2012, 24: 4437-4451. 10.1105/tpc.112.103283.
Jaakola L, Poole M, Jones MO: A SQUAMOSA MADS box gene involved in the regulation of anthocyanin accumulation in bilberry fruits. Plant Physiol. 2010, 153: 1619-1629. 10.1104/pp.110.158279.
Fraser PD, Bramley P, Seymour GB: Effect of the Cnr mutation on carotenoid formation during tomato fruit ripening. Phytochem. 2001, 58: 75-79. 10.1016/S0031-9422(01)00175-3.
Orfila C, Huisman MM, Willats WG, van Alebeek GJ, Schols HA, Seymour GB, Knox JP: Altered cell wall disassembly during ripening of Cnr tomato fruit: implications for cell adhesion and fruit softening. Planta. 2002, 215: 440-447. 10.1007/s00425-002-0753-1.
Eriksson EM, Bovy A, Manning K, Harrison L, Andrews J, De Silva J, Tucker GA, Seymour GB: Effect of the Colorless non-ripening mutation on cell wall biochemistry and gene expression during tomato fruit development and ripening. Plant Physiol. 2004, 136: 4184-4197. 10.1104/pp.104.045765.
Karlova R, Rosin FM, Busscher-Lange J, Parapunova V, Do PT, Fernie AR, Fraser PD, Baxter C, Angenent GC, de Maagd RA: Transcriptome and metabolite profiling show that APETALA2a is a major regulator of tomato fruit ripening. Plant Cell. 2011, 23: 923-941. 10.1105/tpc.110.081273.
Bangalore DV, McGlynn WG, Scott DD: Effects of fruit maturity on watermelon ultrastructure and intracellular lycopene distribution. J Food Sci. 2008, 73: S222-S228. 10.1111/j.1750-3841.2008.00778.x.
Chervin C, El-Kereamy A, Roustan JP, Latche A, Lamon J, Bouzayen M: Ethylene seems required for the berry development and ripening in grape, a non-climacteric fruit. Plant Sci. 2004, 167: 1301-1305. 10.1016/j.plantsci.2004.06.026.
Katz E, Lagunes PM, Riov J, Weiss D, Goldschmidt EE: Molecular and physiological evidence suggests the existence of a system II-like pathway of ethylene production in non-climacteric Citrus fruit. Planta. 2004, 219: 243-252. 10.1007/s00425-004-1228-3.
Tesniere C, Pradal M, El-Kereamy A, Torregrosa L, Chatelet P, Roustan JP, Chervin C: Involvement of ethylene signalling in a non-climacteric fruit: new elements regarding the regulation of ADH expression in grapevine. J Exp Bot. 2004, 55: 2235-2240. 10.1093/jxb/erh244.
Trainotti L, Pavanello A, Casadoro G: Different ethylene receptors show an increased expression during the ripening of strawberries: does such an increment imply a role for ethylene in the ripening of these non-climacteric fruits?. J Exp Bot. 2005, 56: 2037-2046. 10.1093/jxb/eri202.
Karakurt Y, Huber D: Cell wall-degrading enzymes and pectin solubility and depolymerization in immature and ripe watermelon (Citrullus lanatus) fruit in response to exogenous ethylene. Physiol Plantarum. 2002, 116: 398-405. 10.1034/j.1399-3054.2002.1160316.x.
Wechter WP, Levi A, Harris KR: Gene expression in developing watermelon fruit. BMC Genomics. 2008, 9: 275-288. 10.1186/1471-2164-9-275.
FAO Water.http://www.fao.org/nr/water/cropinfo_watermelon.html,
Alba R, Payton P, Fei Z, McQuinn R, Debbie P, Martin GB, Tanksley SD, Giovannoni JJ: Transcriptome and selected metabolite analyses reveal multiple points of ethylene control during tomato fruit development. Plant Cell. 2005, 17: 2954-2965. 10.1105/tpc.105.036053.
Levi A, Galau GA, Wetzstein HY: A rapid procedure for the isolation of RNA from high-phenolic-containing tissues of pecan. Hort Science. 1992, 27: 1316-1318.
Zhong S, Joung JG, Zheng Y, Chen YR, Liu B, Shao Y, Xiang JZ, Fei Z, Giovannoni JJ: High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harb Protoc. 2011, 2011: 940-949.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO: The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucl Acids Res. 2013, 41: D590-D596. 10.1093/nar/gks1219.
Langmead B, Trapnell C, Pop M, Salzberg SL: Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10: R25-10.1186/gb-2009-10-3-r25.
Guo S, Zhang J, Sun H: The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nature Genet. 2013, 45: 51-58.
Trapnell C, Pachter L, Salzberg SL: TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009, 25: 1105-1111. 10.1093/bioinformatics/btp120.
Anders S, Huber W: Differential expression analysis for sequence count data. Genome Biol. 2010, 11: R106-10.1186/gb-2010-11-10-r106.
Smyth GK: Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004, 3: 3-
Benjamini Y, Hochberg Y: Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B. 1995, 57: 289-300.
Acknowledgement
The authors wish to thank Dr. Gaetano Carrozzo for technical assistance in watermelon plant cultivation and harvesting.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SG carried out most of the biochemical and molecular studies and helped to draft the manuscript, GP gave a substantial contributions to the conception and design of the experimental work, JML contributed to the RNA-sequencing experiments, YZ and ZF substantially contributed to the gene expression analysis of RNA-sequencing data and performed most of the statistical analysis, GD and JJG made substantial contributions to the interpretation of data, revised critically the manuscript for important intellectual content, and gave the final approval of the version to be published, MSL conceived the study, participated in its design and coordination, carried out some of the biochemical studies and wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2013_5494_MOESM1_ESM.docx
Additional file 1: Table S1: Expression levels of additional putative genes related to the isoprenoid pathway for which no reads (or reads below 8 RPKM) were found.Genes were identified through Illumina Sequencing Technology in watermelon fruit using two biological replicas. In bold are indicated the genes differentially expressed during watermelon ripening with a FDR (False Discovery Rate) ≤ 0.05. (DOCX 16 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Grassi, S., Piro, G., Lee, J.M. et al. Comparative genomics reveals candidate carotenoid pathway regulators of ripening watermelon fruit. BMC Genomics 14, 781 (2013). https://doi.org/10.1186/1471-2164-14-781
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-14-781