
Abstract
Background
As economically relevant traits, feeding behavior and food preference domestication determine production cost and profitability. Although there are intensive research efforts on feeding behavior and food intake, little is known about food preference. Mandarin fish accept only live prey fish and refuse dead prey fish or artificial diets. Very little is currently known about the genes regulating this unique food preference.
Results
Using transcriptome sequencing and digital gene expression profiling, we identified 1,986 and 4,526 differentially expressed genes in feeders and nonfeeders of dead prey fish, respectively. Up-regulation of Crbp, Rgr and Rdh8, and down-regulation of Gc expression, consistent with greater visual ability in feeders, could promote positive phototaxis. Altered expressions of period, casein kinase and Rev-erbα might reset circadian phase. Down-regulation of orexigenic and up-regulation of anorexigenic genes in feeders were associated with lower appetite. The mRNA levels of Creb, c-fos, C/EBP, zif268, Bdnf and Syt were dramatically decreased in feeders, which might result in significant deficiency in memory retention of its natural food preference (live prey fish). There were roughly 100 times more potential SNPs in feeders than in nonfeeders.
Conclusions
In summary, differential expression in the genes identified shed new light on why mandarin fish only feed on live prey fish, with pathways regulating retinal photosensitivity, circadian rhythm, appetite control, learning and memory involved. We also found dramatic difference in SNP abundance in feeders vs nonfeeders. These differences together might account for the different food preferences. Elucidating the genes regulating the unique food preference (live prey fish) in mandarin fish could lead to a better understanding of mechanisms controlling food preference in animals, including mammals.
Similar content being viewed by others


Background
As economically relevant traits, feeding behavior and food preference domestication determine production cost and profitability. Although there are intensive research efforts on feeding behavior and food intake [1, 2], little is known about food preference. Food preference is an innate behavioral trait subject to genetic influences. However, the genes and genetic factors that determine food preference are largely unknown. Although several genes, including genes encoding taste receptors such as Tas1r1 [3, 4], PKD2L1, PKD1L3 and CD36 [5], obesity-associated genes such as FTO [6] and APOA2 [7], and metabolism genes such as AMY1 [8] and RNASE1 [9], were shown to be involved in the determination of food preference, very little is currently known about the transcriptome determining the unique food preference, such as live prey.
Mandarin fish, as a demersal piscivore, is found only in the freshwaters of China and the River Amur along the Russian borderlands. It has very unique food preference. In the wild, once the fry start feeding, it feed solely on live fry of other fish species [10]. In rearing conditions, mandarin fish also accept only live prey fish, refusing dead prey fish or artificial diets [11]. Among major species of mandarin fish, Siniperca scherzeri, is much easier to accept dead prey fish compared to S. chuatsi, and the difference in acceptance of dead prey fish is further amplified in the hybrid F1 of S. chuatsi (♀) × S. scherzeri (♂). We hypothesize that changes in gene expression as well as SNPs account for this dramatic difference. Elucidating the genes regulating the unique food preference (live prey fish) in mandarin fish could lead to a better understanding of mechanisms controlling food preference in animals, including mammals.
In higher vertebrates such as mammals, the responses of primary centers are coordinated by correlation centres, and the cerebrum is the site of memory and considers the results of experiences on which intelligence and learning are based. In lower vertebrates such as fish, sense organs directly send signals to the primary centers to initiate behavior. Hence their behavior consists of reflex responses with little variation or modification by experience. Therefore compared with mammals, the study of feeding behavior and food preference in fish could enable us to observe direct effects of genes and genetic factors on food preference. To elucidate the relationship between gene expression and food preference, we performed de novo transcriptome sequencing and digital gene expression profiling (DGE) of feeders and nonfeeders of dead prey fish in mandarin fish. We showed that expression of genes in several pathways, including retinal photosensitivity, circadian rhythm, appetite control, learning and memory, were significantly different in feeders and nonfeeders. These potential determinants provide a glimpse of genetic architecture of the unique food preference.
Results
High-throughput sequencing and annotation of unigenes
To obtain an overview of gene expression profile in mandarin fish with different food preferences, cDNA libraries were constructed from dead prey fish feeders (SC_X) and nonfeeders (SC_W), and sequenced using the Illumina Hiseq2000 system. High quality reads of SC_X and SC_W were assembled, yielding 665,466, and 716,044 contigs, respectively (Table 1). After removing the partial overlapping sequences, a total of 118,218 distinct sequences were obtained (All-Unigene, mean size: 506 bp, N50: 611 bp) (Additional file 1). Among these unigenes, 69.5% (82,108) were between 100 and 500 bp in length, 30.5% (36,110) were longer than 500 bp, of which 9.8% (11,550) were longer than 1,000 bp. Six unigenes were aligned with either Sanger-derived sequences that we obtained by sequencing PCR products or reference sequences deposited in NCBI. Each sequence had more than 98% coverage validation (Additional files 2, 3, 4, 5, 6, 7). The sequencing data in this study have been deposited in the EBI ArrayExpress database (accession number: E-MTAB-1365).
49,155 unigenes (41.6% of All-Unigene) were aligned function by BLASTx, and 69,063 (58.4%) assembled sequences could not be matched to any known protein. 48,796 annotated sequences had significant matches with 27,354 unique accession numbers. The BLASTx top-hit species distribution of gene annotations showed highest homology to zebrafish (Danio rerio, 52%), followed by Atlantic salmon (Salmo salar, 9%) and spotted green pufferfish (Tetraodon nigroviridis, 4%) (Table 2). In addition, the mandarin fish sequences also had homologies to four other fish species including Japanese pufferfish (Takifugu rubripes), sablefish (Anoplopoma fimbria), rainbow smelt (Osmerus mordax) and medaka (Oryzias latipes). These results indicated a high level of phylogenetic conservation between mandarin fish and other fish species, especially zebrafish. 182 unigenes of our transcriptome libraries matched the published mandarin fish protein sequences (645) currently available in NCBI database, suggesting the identification of a large number of new genes by transcriptome sequencing reported here.
Of 49,155 annotated sequences in mandarin fish transcriptome, 14,228 (28.9%) were assigned with one or more gene ontology (GO) terms. In total, 106,024 GO assignments were finally obtained, with 46.1% for biological processes, 34.8% for cellular components, and 19.1% for molecular functions. Level 2 GO functional categories were summarized in Figure 1. The number and assortment of allocated GO categories reflected the diversity and complexity of genes expressed in mandarin fish. We mapped the 49,155 annotated sequences to the reference canonical pathways in Kyoto Encyclopedia of Genes and Genomes (KEGG) to identify the biological pathways. In total, we assigned 30,964 sequences to 205 known metabolic or signaling pathways. The representative pathways with the differentially expressed genes were mitogen-activated protein kinases (MAPK) signaling pathway (1222 members), calcium signaling pathway (913 members), insulin signaling pathway (711 members), long-term potentiation (LTP) (542 members), long-term depression (369 members), taste transduction (173 members) and mammalian circadian rhythm (92 members) (Additional file 8).

Functional annotation of mandarin fish transcripts based on GO categorization. The left y-axis indicates the percentage of a specific category of genes in that main category. The right y-axis indicates the number of genes in a category.
SNP and SSR discovery
The transcript/EST-based markers are important resource for determining functional genetic variation [12]. We detected 4,768 potential SNPs in feeders and 41 potential SNPs in nonfeeders. The overall frequency of predicted SNPs in the mandarin fish transcriptome was one per 12,430 bp. A total of 4,809 SNPs were identified, including 1,592 transitions and 3,217 transversions; 2,510 of these SNPs had been annotated. Of these SNPs, 2,062 were identified from unigenes covered by ten or more reads, suggesting that 42.9% of SNPs found in this transcriptome were covered with sufficient sequencing depth and more likely to represent ‘true’ SNPs. Of 19 SNP loci predicted to reside in the 10 amplified sequences, 15 sites were validated (Additional file 9). There were roughly 100 times more potential SNPs in feeders than in nonfeeders. Whether the difference in SNP prevalence accounts for the different acceptance of dead prey fish in hybrid F1 warrants further investigation. For identification of Simple Sequence Repeats (SSRs), all 118,218 unigenes generated in this study were applied to determine potential microsatellites using Batchprimer3.0 (http://probes.pw.usda.gov/batchprimer3/index.html). We identified a total of 22,418 potential SSRs in 17,933 (15.2%) unigenes with frequency of one SSR per 2.7 kb of the unigenes. Of 17,933 SSR-containing sequences, 7,585 (42.3%) had been annotated, and could be considered as preferred candidates for marker development.
Identification of differentially expressed genes
We found 1,986 and 4,526 unigenes to be differentially expressed between feeders and nonfeeders from transcriptome and DGE analysis (False Discovery Rate (FDR) ≤ 0.001, fold-change ≥ 2, Figures 2 and 3), respectively. Analysis of these genes revealed the signaling pathways involved, including retinal photosensitivity (retinal G protein-coupled receptor (Rgr), retinol dehydrogenase 8 (Rdh8), cellular retinol-binding protein (Crbp) and guanylate cyclase (Gc)), circadian rhythm (period 1 (Per1), period 2 (Per2), Rev-erbα, casein kinase (Ck) and nocturnin), appetite control (neuropeptide Y (Npy), growth hormone (Gh), pro-opiomelanocortin (Pomc), peptide YY (Pyy), insulin and leptin), learning and memory (cyclic AMP-response element-binding protein (Creb), c-fos, fos-related antigen 2 (Fra-2), CCAAT enhancer binding protein (C/EBP), zif268, brain-derived neurotrophic factor (Bdnf) and synaptotagmin (Syt)) (Additional file 10, Figure 4). The importance of these genes was further supported by the identification of significant potential SNP, SSR and antisense transcripts in these genes. To compare the two tissues (liver and brain) that are of interest for food preference, we identified 11,433 unigenes that were differentially expressed between liver and brain in feeders, and 12,085 unigenes in nonfeeders (Figure 3) by DGE. In addition, DGE analysis of mandarin fish with the two different food preferences generated 9,597,700 clean tags from brain and 9,964,672 clean tags from liver, respectively (Additional file 11).

Scatter plot showing the correlation between the expression levels of feeders and nonfeeders by transcriptome sequencing. SC_X and SC_W indicate feeders and nonfeeders, respectively. The x-axis contains Log10 of Reads Per Kb per Million reads (RPKM) of feeders and the y-axis indicates Log10 of RPKM of nonfeeders. Limitations were based on FDR ≤ 0.001, and the absolute value of Log2 (SC_X / SC_W) ≥ 1.

Differential expression analysis of unigenes by DGE. WL and WB indicate liver and brain in nonfeeders, respectively, while XL and XB indicate liver and brain in feeders. The superscripts of each column represent the number of differentially expressed genes between groups.

Differentially expressed genes between feeders and nonfeeders from transcriptome and DGE analysis. The most important pathways related to live prey food preference included retinal photosensitivity (A), circadian rhythm (B), appetite control (C), learning and memory (D). The colors of ellipses were shaded according to significance level (bright red: the mRNA expression levels of feeders were significantly higher than those in nonfeeders (FDR ≤ 0.001, the absolute value of log2[Ratio] ≥ 1); pink: the mRNA expression levels of feeders were slightly higher than those in nonfeeders (FDR ≤ 0.5, the absolute value of log2[Ratio] ≥ 0.5); bright green: the mRNA expression levels of feeders were significantly lower than those in nonfeeders (FDR ≤ 0.001, the absolute value of log2[Ratio] ≥ 1); pale green: the mRNA expression levels of feeders were slightly lower than those in nonfeeders (FDR ≤ 0.5, the absolute value of log2[Ratio] ≥ 0.5).
Real-time RT-PCR was frequently used to confirm data obtained from high-throughput sequencing [13, 14]. Here, we also used Real-time RT-PCR to confirm the differential expression of 18 genes in S. chuatsi that went through the same training procedure. The data obtained were consistent with those obtained from the transcriptome sequencing and DGE analysis (Figure 5).

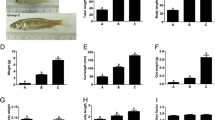
Validation of differentially expressed genes with Real-time RT-PCR. A. The relative mRNA abundance of retinal photosensitivity genes (Rgr, Rdh8, Crbp and Gc) in feeders and nonfeeders was determined by Real-time RT-PCR. Compared with nonfeeders, the mRNA levels of Rgr and Crbp were significantly increased, whereas Gc was decreased significantly. B. The relative mRNA abundance of clock genes (Per1, Per2, Clock and Bmal1) in feeders and nonfeeders was determined by Real-time RT-PCR. Compared with nonfeeders, the mRNA levels of Per1 and Per2 were decreased significantly and slightly, respectively, whereas Clock and Bmal1 were slightly increased. C. The relative mRNA abundance of appetite control genes (Pomc, Pyy, leptin, Npy and ghrelin) in feeders and nonfeeders was determined by Real-time RT-PCR. Compared with nonfeeders, the mRNA levels of Pomc, Pyy and leptin were significantly increased, whereas Npy and ghrelin were decreased significantly and slightly, respectively. D. The relative mRNA abundance of learning and memory genes (Creb, Ncam, c-fos and Bdnf) in feeders and nonfeeders was determined by Real-time RT-PCR. Compared with nonfeeders, the mRNA levels of Creb and Ncam were slightly decreased, c-fos and Bdnf were decreased significantly. Data are presented as mean ± standard error (n = 6). * indicates significant differences between groups based on one-way analysis of variance (ANOVA) followed by the post hoc test (P < 0.05).
Discussion
Little is known about the genes and biological mechanisms controlling food preference in animals. In this study, by profiling the transcriptomes of dead prey fish feeders and nonfeeders in mandarin fish, we identified differentially expressed genes potentially influencing the unique food preference of live prey, including those affecting retinal photosensitivity, circadian rhythm, appetite control, learning and memory. Real-time RT-PCR confirmed the differential expression in selected genes. We also found dramatic difference in SNP abundance in feeders vs nonfeeders. These differences together might account for the different food preferences.
Differentially expressed genes involved in retinal photosensitivity pathway
Animals make food choices based on a number of physiological, nutritional, environmental, and sociocultural factors [5]. Sensitivity of sensory system is critical to food preference. It is thus advantageous for mandarin fish to catch prey fish at night through the perception of motion and shape with the help of its well-developed scotopic vision [11]. Although Salmo spp. also shows the selection of food motion and shape to some extent, the offered food pellet can be captured immediately before it falls down to the bottom of the tank because they have high visual acuity, thus can feed swiftly by darting [15–17]. This contrasts with mandarin fish, which has low visual acuity and feed only by stalking. Mandarin fish might not be able to accomplish its relatively long process of prey recognition before the offered food pellet has fallen down to the bottom of the tank and can no longer be perceived by its sensory organs. The more strict selection of prey motion and shape also makes it more difficult to feed mandarin fish with artificial diets and dead prey fish. Hence much better developed visual ability could improve the mandarin fish to accept dead prey fish or artificial diets [11].
Adaptation to dark in most animals is associated with increased 11-cis-retinal generation and rhodopsin reconstitution. Factors that interfere with the rhodopsin cycle or its downstream signaling pathways will affect vision, especially scotopic vision . We observed differential expression of Crbp, Rgr, Rdh8 and Gc in brains of feeders vs nonfeeders. Crbp is involved in the initial processing of retinol from food [18]. Light-dependent formation of 11-cis-retinal by the retinal pigment epithelium and regeneration of rhodopsin under photic conditions involve the RGR opsin located in the retinal pigment epithelium [19]. RDH8 is a critical regulator of chromophore regeneration [20]. Gc, as a family of enzymes that catalyze the conversion of GTP to cGMP, are central in phototransduction cascade [21]. Our results suggested that the up-regulation of Crbp, Rgr and Rdh8, and down-regulation of Gc in feeders might lead to increased 11-cis-retinal and rhodopsin levels, deceased cGMP generation, leading to greater visual ability and light sensitivity. Thus the feeders could capture the dead prey fish before it falls to the bottom of the tank. Moreover, the mRNA expression levels of connexin 35 (Cx35), Cyp4v2 and chicken ovalbumin upstream promoter transcription factor 2 (Coup-tf2) genes in the brain of feeders were all higher than those of nonfeeders, potentially contributing to the food preference [22–25] (Figure 4A).
Differentially expressed genes involved in circadian rhythm pathway
Previous studies demonstrated that the molecular mechanisms of circadian rhythm generation in zebrafish appear to be generally consistent with the mammalian model [26]. We identified homologs of the mammalian clock genes in mandarin fish. We found differential expression in several clock genes, including Per1, Per2, cryptochrome (Cry), Clock, Bmal1, CkIα, CkIγ, CkIδ, CkIIβ, Rev-erbα, Skp1 and Rbx2 in feeders vs nonfeeders (Figure 4B). These genes are known to be critical regulators of circadian rhythm [27–33]. Taken together, changes in expression levels of these clock genes in feeders might reset circadian phase and contribute to the unique food preference.
Differentially expressed genes involved in appetite control pathway
Previous studies provide a framework for understanding the regulation of food intake in mammals and fish. Peripheral signals such as leptin from adipocytes, insulin from endocrine pancreas, cholecystokinin and peptide YY from gastrointestinal tract are incorporated in the hypothalamus to generate orexigenic (such as NPY and ghrelin) or anorexigenic (such as α-melanocyte stimulating hormone (α-MSH) derived from POMC) signals [34]. We observed lower expression of orexigenic genes (Npy, proenkephalin, Gh, uncoupling protein 2 (Ucp2), Creb, eukaryotic initiation factor (eIF) 4E binding protein (4Ebp1), tuberous sclerosis 1 (Tsc1), ghrelin and leptin receptor gene-related protein (Ob-rgrp)) and higher expression of anorexigenic genes (Pomc, Pyy, preprosomatostatin (Srif), insulin, leptin, cholecystokinin (Cck) and tachykinin 1) in feeders compared with nonfeeders (Figure 4C). These genes are well established regulators of energy homeostasis and play important roles in determination of food preferences [35–46]. The changes in gene expression suggested that feeders had decreased appetite.
Differentially expressed genes involved in learning and memory pathway
Food habits can develop with learning experience [47, 48]. Our previous study indicated that sensory modality and associative learning appear to be critical factors in food discrimination of mandarin fish [49]. However, study on genes involved in learning and memory of nocturnal piscivorous fish has received little attention. A number of molecules participate in learning and memory processes. The TORC1-mediated CREB regulation is a critical molecular step underlying synaptic plasticity and long-term memory [50, 51]. As a suppressor of CREB, protein phosphatase 1 (PP1) determines the efficacy of learning and memory by limiting acquisition and favoring memory decline [52]. Neural cell adhesion molecule (NCAM) plays an important role in axonal growth, learning, and memory through activating the phosphorylation of MAPKs and CREB [53]. In our study, compared with nonfeeders, the mRNA levels of Creb and Ncam were dramatically decreased and Pp1 were slightly increased in feeders, protentially resulting in significant deficiency in the maintenance of long-term memory of its natural food preference, and therefore feeders were able to accept novel food (dead prey fish) (Figure 4D).
Along the same line, as members of immediate early gene and the Fos family of transcription factors, c-fos and Fra-2 mRNA expressions are up-regulated in response to a variety of neuronal activation protocols, including LTP [54–56]. In the current study, c-fos, Fra-2 and zif268 genes were expressed at significantly lower levels in feeders than in nonfeeders, suggesting disruption of LTP and memory formation in feeders [57]. We hypothesize that the acquisition of novel food preference might be closely associated with effacement of the original memory of natural food preference (live prey), and the enhanced learning capacity for new food preference (dead prey). We also showed that in mandarin fish, a number of genes necessary for memory formation and retention (C/EBP, Bdnf, Syt I, Syt IV and nitric oxide synthase (Nos)) were differentially expressed in feeders vs nonfeeders. They might also be involved in the determination of the unique food preference in mandarin fish (Figure 4D) [58–61].
Conclusions
In summary, our results showed that there were individual differernces in hybrid F1 of mandarin fish in their feeding response to dead prey fish. The acquisition of novel food preference (dead prey fish) might be due to enhanced visual ability, resetting of circadian phase, decreased appetite, deficiency in memory retention and more abundant variant alleles. Interaction of retinal photosensitivity, circadian rhythm, appetite control, learning and memory outputs might drive the feeding behavior.
Methods
Fish and sample preparation
The hybrid F1 of S. chuatsi (♀) × S. scherzeri (♂) (body length about 5 cm) were obtained from Guangdong Freshwater Fish Farm (Panyu, Guangdong Province, China). The training was performed for 3 days following the methods previously described by Liang et al. [49] using net-cages as the experimental culture. Fry of India mrigal Cirrhina mrigola were used as the live prey fish in this study, and the dead prey fish were frozen India mrigal fry. During the training period, the fish were visually sorted into feeders and nonfeeders on the basis of plumpness or emaciation, respectively. The training period did not cause the nonfeeders to starve to death because of the relatively short time and large size of the fish. To eliminate the influence of hunger on the mRNA levels of mandarin fish, both groups were fed with live prey fish for two days after training. And then 16 fish were randomly selected from each group. Total RNA was isolated from brain and liver tissues using SV total RNA isolation system (Promega, Madison, WI, USA) according to manufacturer's protocol. Equal amount of total RNA from brain and liver tissues of 16 fish in each group were pooled and used to construct the libraries for transcriptome and DGE analysis. The animal protocol was approved by Huazhong Agricutural University.
Transcriptome library preparation and Illumina sequencing
The samples for transcriptome analysis were prepared using Illumina's kit following manufacturer's instructions (San Diego, CA, USA). Poly(A) mRNA was purified from total RNA (a mixture of equal amount of RNA from brain and liver) using oligo-dT-attached magnetic beads. Paired-end cDNA libraries were sequenced using Illumina HiSeq2000 system. Image deconvolution and base calling were performed with the Illumina CASAVA v1.7. The reliability of the reads was 89.1% with average length of the reads at 90 bp. The library construction and sequencing were performed by the commercial service provider Beijing Genomics Institute at Shenzhen (Shenzhen, China). Transcriptome assembly was carried out with short reads assembling program SOAPdenovo [62] with k-mer length from 21 to 41 bp. Then the reads are mapped back to assembled contigs. By using the paired-end information, it is able to detect contigs from the same transcript as well as the distances between these contigs. We connected the contigs using N to represent unknown sequences between each pair of contigs, and then scaffolds were made. Paired-end reads were used again for gap filling of scaffolds to obtain sequences with least Ns and could not be extended on either end. Such sequences were defined as unigenes. Separate cDNA libraries were constructed from dead prey fish feeders (SC_X) and nonfeeders (SC_W), unigenes from each library were taken into further process of sequence splicing and redundancy removing with sequence clustering software to acquire non-redundant unigenes (All-Unigene) as long as possible. The assembly parameters were more than 89.1% identity over a minimum of 50 bases with a maximum of 2 bases of unmatched overhangs at sequence end.
To annotate the mandarin fish transcriptome, we performed the BLASTx alignment (e-value < 0.00001) between All-Unigene and protein databases such as NCBI, Swiss-Prot, KEGG and COG. Functional annotation by gene ontology terms (GO; http://www.geneontology.org) was accomplished with Blast2GO software. After obtaining GO annotation for each unigene, we applied WEGO software [63] to conduct GO functional classification for all unigenes. To assess the quality of sequencing data, 3 unigenes generated by transcriptome sequencing were randomly selected for cloning and sequence validation by PCR. All three PCR products were sequenced using an ABI PrismTM 377 (PerkinElmer, Waltham, MA, USA). To ascertain the quality of SOAPdenovo assembly of short reads into long contiguous RNA transcript sequences, we aligned 3 of the transcriptome-derived unigenes to the corresponding Sanger-sequenced, full-length, cloned mRNA sequences from mandarin fish in NCBI.
SSR and SNP analysis
To detect SNPs in the cDNA pool, the consensus assembly sequence generated from the two trancriptome libraries was employed as a reference sequence to which individual reads were aligned using SOAPsnp [64]. SNP identification was limited to the unigenes containing at least five reads. The minimum allele quality (accumulated sequence quality for every allele) was not lower than 20. We considered only SNPs, excluding all indels and variants involving more than one nucleotide. To validate the detected SNPs, we designed primers to amplify 10 unigenes containing potential SNPs. The PCR products were sequenced with both forward and reverse primers using ABI PrismTM 377 (PerkinElmer). Batchprimer3.0 was adopted to identify and localize microsatellite motifs, which were defined as di-nucleotide SSR with a minimum of six repetitions, four repetitions for trinucleotide, and three repetitions for tetra- to hexa-nucleotide motifs.
Digital gene expression profiling
The four tag libraries of mandarin fish (the liver tissues of feeders and nonfeeders, the brain tissues of feeders and nonfeeders, respectively) were constructed in parallel using the Digital Gene Expression Tag Profile Kit (Illumina) according to the manufacturer's instructions. Six μg of total RNA per sample was used for mRNA capture using oligo-dT magnetic bead adsorption and oligo-dT was used to synthesize the first and second-strand cDNA. The 5' ends of tags were generated by endonuclease NlaIII, which recognizes and cuts off the CATG sites on cDNA. The cDNA fragments with 3' ends were then purified with magnetic beads and Illumina adapter 1 was ligated to the sticky 5' ends. The junction of Illumina adapter 1 and CATG site is the recognition site of MmeI, which cuts 17 bp downstream of the CATG site, producing tags with adapter 1. After removing 3' fragments with magnetic beads precipitation, the 21-bp unique tags with adaptor 1 were purified and ligated to adaptor 2 to form a cDNA tag library. Sequencing by synthesis (SBS) was performed using the Illumina HiSeq2000 system. All high quality tags were mapped to the reference sequence generated by transcriptome sequencing, and only 1 bp mismatch was permitted. For gene expression analysis, the number of unambiguous clean tags for each gene was calculated and then normalized to TPM (number of transcripts per million tags) [65].
Identification of differentially expressed genes
Gene expression levels were measured through short reads mapping in Reads Per Kb per Million reads (RPKM) [66]. GO functional analysis and KEGG pathway analysis were then carried out in differentially expressed genes. SYBR Green Real-time RT-PCR was performed to validate the transcriptome and DGE data (Additional file 12). In addition to the hybrid F1 of S. chuatsi (♀) × S. scherzeri (♂), total RNA was prepared from S. chuatsi that went through the same training procedure and used for the validation of differentially expressed genes. Beta-actin was amplified in parallel as an internal control. There were six biological and three technical replicates respectively.
Statistical analysis
We used FDR ≤ 0.001 and the absolute value of log2[Ratio] ≥ 1 as the threshold to judge the significance of gene expression difference. Statistical analysis was performed with SPSS13.0 software. Data normality and homogeneity of variances were analyzed. Results were presented as the means ± S.E. (n = 6) for each group. One-way analysis of variance (ANOVA) followed by the post hoc test were carried out to determine whether the differences between groups were significant (P < 0.05).
References
Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW: Central nervous system control of food intake and body weight. Nature. 2006, 443: 289-295. 10.1038/nature05026.
Cone RD: Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005, 8: 571-578. 10.1038/nn1455.
Zhao H, Yang JR, Xu H, Zhang J: Pseudogenization of the umami taste receptor gene Tas1r1 in the giant panda coincided with its dietary switch to bamboo. Mol Biol Evol. 2010, 27: 2669-2673. 10.1093/molbev/msq153.
Li R, Fan W, Tian G, Zhu H, He L, Cai J, Huang Q, Cai Q, Li B, Bai Y, Zhang Z, Zhang Y, Wang W, Li J, Wei F, Li H, Jian M, Li J, Zhang Z, Nielsen R, Li D, Gu W, Yang Z, Xuan Z, Ryder OA, Leung FC, Zhou Y, Cao J, Sun X, Fu Y: The sequence and de novo assembly of the giant panda genome. Nature. 2010, 463: 311-317. 10.1038/nature08696.
Garcia-Bailo B, Toguri C, Eny KM, El-Sohemy A: Genetic variation in taste and its influence on food selection. OMICS. 2009, 13: 69-80. 10.1089/omi.2008.0031.
McCaffery JM, Papandonatos GD, Peter I, Huggins GS, Raynor HA, Delahanty LM, Cheskin LJ, Balasubramanyam A, Wagenknecht LE, Wing RR, Genetic Subgroup of Look AHEAD: Obesity susceptibility loci and dietary intake in the Look AHEAD Trial. Am J Clin Nutr. 2012, 95: 1477-1486. 10.3945/ajcn.111.026955.
Corella D, Arnett DK, Tsai MY, Kabagambe EK, Peacock JM, Hixson JE, Straka RJ, Province M, Lai CQ, Parnell LD, Borecki I, Ordovas JM: The -256T > C polymorphism in the apolipoprotein A-II gene promoter is associated with body mass index and food intake in the genetics of lipid lowering drugs and diet network study. Clin Chem. 2007, 53: 1144-1152. 10.1373/clinchem.2006.084863.
Shadan S: You are what you ate. Nature. 2007, 449: 155-10.1038/449155a.
Zhang J, Zhang YP, Rosenberg HF: Adaptive evolution of a duplicated pancreatic ribonuclease gene in a leaf-eating monkey. Nat Genet. 2002, 30: 411-415. 10.1038/ng852.
Chiang IK: On the biology of mandarin fish, Siniperca chuatsi of Liang-Tze Lake. Acta Hydrobiol Sinica. 1959, 3: 375-385.
Liang XF, Liu JK, Huang BY: The role of sense organs in the feeding behaviour of Chinese perch. J Fish Biol. 1998, 52: 1058-1067. 10.1111/j.1095-8649.1998.tb00603.x.
Garg R, Patel RK, Tyagi AK, Jain M: De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 2011, 18: 53-63. 10.1093/dnares/dsq028.
Chen S, Yang PC, Jiang F, Wei YY, Ma ZY, Kang L: De novo analysis of transcriptome dynamics in the migratory locust during the development of phase traits. PloS ONE. 2010, 5: 1-15.
Kalavacharla V, Liu ZJ, Meyers BC, Thimmapuram J, Melmaiee K: Identification and analysis of common bean (Phaseolus vulgaris L.) transcriptomes by massively parallel pyrosequencing. BMC Plant Biol. 2011, 11: 135-10.1186/1471-2229-11-135.
Irvine JR, Northcote TG: Selection by young rainbow trout (Salmo gairdneri) in simulated stream environments for live and dead prey of different sizes. Can J Fish Aquat Sci. 1983, 40: 1745-1749. 10.1139/f83-202.
Stradmeyer L, Metcalfe NB, Thorpe JE: Effect of food pellet shape and texture on the feeding response of juvenile Atlantic salmon. Aquaculture. 1988, 73: 217-228. 10.1016/0044-8486(88)90056-7.
Stradmeyer L: A behavioural method to test feeding responses of fish to pelleted diets. Aquaculture. 1989, 79: 303-310. 10.1016/0044-8486(89)90471-7.
Noy N: Retinoid-binding proteins: mediators of retinoid action. Biochem J. 2000, 348: 481-495. 10.1042/0264-6021:3480481.
Yang M, Fong HK: Synthesis of the all-trans-retinal chromophore of retinal G protein-coupled receptor opsin in cultured pigment epithelial cells. J Biol Chem. 2002, 277: 3318-3324. 10.1074/jbc.M108946200.
Maeda A, Golczak M, Maeda T, Palczewski K: Limited roles of Rdh8, Rdh12, and Abca4 in all-trans-retinal clearance in mouse retina. Invest Ophthalmol Vis Sci. 2009, 50: 5435-5443. 10.1167/iovs.09-3944.
Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S, Chepenik KP, Waldman SA: Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000, 52: 375-414.
Liu CR, Xu L, Zhong YM, Li RX, Yang XL: Expression of connexin 35/36 in retinal horizontal and bipolar cells of carp. Neuroscience. 2009, 164: 1161-1169. 10.1016/j.neuroscience.2009.09.035.
Lin J, Nishiguchi KM, Nakamura M, Dryja TP, Berson EL, Miyake Y: Recessive mutations in the CYP4V2 gene in East Asian and Middle Eastern patients with Bietti crystalline corneoretinal dystrophy. J Med Genet. 2005, 42: e38-10.1136/jmg.2004.029066.
Tang K, Xie X, Park JI, Jamrich M, Tsai S, Tsai MJ: COUP-TFs regulate eye development by controlling factors essential for optic vesicle morphogenesis. Development. 2010, 137: 725-734. 10.1242/dev.040568.
Viczian AS, Vignali R, Zuber ME, Barsacchi G, Harris WA: XOtx5b and XOtx2 regulate photoreceptor and bipolar fates in the Xenopus retina. Development. 2003, 130: 1281-1294. 10.1242/dev.00343.
Cahill GM: Clock mechanisms in zebrafish. Cell Tissue Res. 2002, 309: 27-34. 10.1007/s00441-002-0570-7.
Reppert SM, Weaver DR: Coordination of circadian timing in mammals. Nature. 2002, 418: 935-941. 10.1038/nature00965.
Zylka MJ, Shearman LP, Weaver DR, Reppert SM: Three period homologs in mammals: differential light responses in the suprachiasmatic circadian clock and oscillating transcripts outside of brain. Neuron. 1998, 20: 1103-1110. 10.1016/S0896-6273(00)80492-4.
Akashi M, Tsuchiya Y, Yoshino T, Nishida E: Control of intracellular dynamics of mammalian period proteins by casein kinase I epsilon (CKI epsilon) and CKI delta in cultured cells. Mol Cell Biol. 2002, 22: 1693-1703. 10.1128/MCB.22.6.1693-1703.2002.
Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, Schibler U: The orphan nuclear receptor REV-ERBα controls circadian transcription within the Positive limb of the mammalian circadian oscillator. Cell. 2002, 110: 251-260. 10.1016/S0092-8674(02)00825-5.
Ueda HR, Chen W, Adachi A, Wakamatsu H, Hayashi S, Takasugi T, Nagano M, Nakahama K, Suzuki Y, Sugano S, Iino M, Shigeyoshi Y, Hashimoto S: A transcription factor response element for gene expression during circadian night. Nature. 2002, 418: 534-539. 10.1038/nature00906.
Somers DE, Schultz TF, Milnamow M, Kay SA: ZEITLUPE encodes a novel clock-associated PAS protein from Arabidopsis. Cell. 2000, 101: 319-329. 10.1016/S0092-8674(00)80841-7.
Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, Conaway RC, Conaway JW, Harper JW, Pavletich NP: Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002, 416: 703-709. 10.1038/416703a.
Tao YX, Yuan ZH, Xie J: G protein-coupled receptors as regulators of energy homeostasis. Prog Mol Biol Transl Sci. 2013, 114: 1-43.
Kalra SP, Dube MG, Pu S, Xu B, Horvath TL, Kalra PS: Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr Rev. 1999, 20: 68-100. 10.1210/er.20.1.68.
Schiltz CA, Bremer QZ, Landry CF, Kelley AE: Food-associated cues alter forebrain functional connectivity as assessed with immediate early gene and proenkephalin expression. BMC Biol. 2007, 5: 16-10.1186/1741-7007-5-16.
Lin X, Volkoff H, Narnaware Y, Bernier NJ, Peyon P, Peter RE: Brain regulation of feeding behavior and food intake in fish. Comp Biochem Physiol A. 2000, 126: 415-434.
Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K: Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999, 402: 656-660. 10.1038/45230.
Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, Tschöp MH, Shanabrough M, Cline G, Shulman GI, Coppola A, Gao XB, Horvath TL, Diano S: UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature. 2008, 454: 846-851. 10.1038/nature07181.
Tao YX: The melanocortin-4 receptor: Physiology, pharmacology and pathophysiology. Endocr Rev. 2010, 31: 506-543. 10.1210/er.2009-0037.
Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes AE, Low MJ, Ghatei MA, Cone RD, Bloom SR: Gut hormone PYY3-36 physiologically inhibits food intake. Nature. 2002, 418: 650-654. 10.1038/nature00887.
Fan W, Ellacott KL, Halatchev IG, Takahashi K, Yu P, Cone RD: Cholecystokinin-mediated suppression of feeding involves the brainstem melanocortin system. Nat Neurosci. 2004, 7: 335-336. 10.1038/nn1214.
Feifel D, Vaccarino FJ: Central somatostatin: a re-examination of its effects on feeding. Brain Res. 1990, 535: 189-194. 10.1016/0006-8993(90)91600-L.
Couturier C, Sarkis C, Séron K, Belouzard S, Chen P, Lenain A, Corset L, Dam J, Vauthier V, Dubart A, Mallet J, Froguel P, Rouillé Y, Jockers R: Silencing of OB-RGRP in mouse hypothalamic arcuate nucleus increases leptin receptor signaling and prevents diet-induced obesity. Proc Natl Acad Sci U S A. 2007, 104: 19476-19481. 10.1073/pnas.0706671104.
Brunn GJ, Hudson CC, Sekulić A, Williams JM, Hosoi H, Houghton PJ, Lawrence JC, Abraham RT: Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science. 1997, 277: 99-101. 10.1126/science.277.5322.99.
Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ: Hypothalamic mTOR signaling regulates food intake. Science. 2006, 312: 927-930. 10.1126/science.1124147.
Brown JA: The adaptive significance of behavioural ontogeny in some centrarchid fishes. Environ Biol Fish. 1985, 13: 25-34. 10.1007/BF00004853.
Warburton K: Learning of foraging skills by fish. Fish Fish. 2003, 4: 203-215. 10.1046/j.1467-2979.2003.00125.x.
Liang XF, Oku H, Ogata HY, Liu J, He X: Weaning Chinese perch Siniperca chuatsi (Basilewsky) onto artificial diets based upon its specific sensory modality in feeding. Aquac Res. 2001, 32: 76-82.
Kovács KA, Steullet P, Steinmann M, Do KQ, Magistretti PJ, Halfon O, Cardinaux JR: TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity. Proc Natl Acad Sci U S A. 2007, 104: 4700-4705. 10.1073/pnas.0607524104.
Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ: Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994, 79: 59-68. 10.1016/0092-8674(94)90400-6.
Genoux D, Haditsch U, Knobloch M, Michalon A, Storm D, Mansuy IM: Protein phosphatase 1 is a molecular constraint on learning and memory. Nature. 2002, 418: 970-975. 10.1038/nature00928.
Schmid RS, Graff RD, Schaller MD, Chen S, Schachner M, Hemperly JJ, Maness PF: NCAM stimulates the ras-MAPK pathway and CREB phosphorylation in neuronal cells. J Neurobiol. 1999, 38: 542-558. 10.1002/(SICI)1097-4695(199903)38:4<542::AID-NEU9>3.0.CO;2-1.
Alberini CM: Transcription factors in long-term memory and synaptic plasticity. Physiol Rev. 2009, 89: 121-145. 10.1152/physrev.00017.2008.
Countryman RA, Orlowski JD, Brightwell JJ, Oskowitz AZ, Colombo PJ: CREB phosphorylation and c-Fos expression in the hippocampus of rats during acquisition and recall of a socially transmitted food preference. Hippocampus. 2005, 15: 56-67. 10.1002/hipo.20030.
Smith CA, Countryman RA, Sahuque LL, Colombo PJ: Time-courses of Fos expression in rat hippocampus and neocortex following acquisition and recall of a socially transmitted food preference. Neurobiol Learn Mem. 2007, 88: 65-74. 10.1016/j.nlm.2007.03.001.
Hall J, Thomas KL, Everitt BJ: Cellular imaging of zif268 expression in the hippocampus and amygdala during contextual and cued fear memory retrieval: selective activation of hippocampal CA1 neurons during the recall of contextual memories. J Neurosci. 2001, 21: 2186-2193.
Cunha C, Brambilla R, Thomas KL: A simple role for BDNF in learning and memory?. Front Mol Neurosci. 2010, 3: 1-
Babity JM, Armstrong JN, Plumier JC, Currie RW, Robertson HA: A novel seizure-induced synaptotagmin gene identified by differential display. Proc Natl Acad Sci U S A. 1997, 94: 2638-2641. 10.1073/pnas.94.6.2638.
Ferguson GD, Vician L, Herschman HR: Synaptotagmin IV. Mol Neurobiol. 2001, 23: 173-185. 10.1385/MN:23:2-3:173.
Paul V, Ekambaram P: Involvement of nitric oxide in learning & memory processes. Indian J Med Res. 2011, 133: 471-478.
Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, Li S, Yang H, Wang J, Wang J: De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20: 265-272. 10.1101/gr.097261.109.
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J: WEGO: a web tool for plotting GO annotations. Nucl Acids Res. 2006, 34: 293-297. 10.1093/nar/gkl031.
Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K, Wang J: SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009, 19: 1124-1132. 10.1101/gr.088013.108.
Morrissy AS, Morin RD, Delaney A, Zeng T, McDonald H, Jones S, Zhao Y, Hirst M, Marra MA: Next-generation tag sequencing for cancer gene expression profiling. Genome Res. 2009, 19: 1825-1835. 10.1101/gr.094482.109.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B: Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008, 5: 621-628. 10.1038/nmeth.1226.
Acknowledgments
This work was financially supported by the National Basic Research Program of China (2009CB118702, 2014CB138601), the Key Projects in the National Science & Technology Pillar Program during the Twelfth Five-year Plan Period (2012BAD25B04), the National Natural Science Foundation of China (31272641, 31172420), and the Fundamental Research Funds for the Central Universities (2010PY010, 2011PY030).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SH, JS and LL performed the de novo assembly and bioinformatical analysis. SH, YY and LC contributed to the fish and sample preparation. WH, CMQ and XLB contributed to the RT-PCR and SNP experiments. XFL gave technical advice and contributed to the study design. SH, XFL and YXT co-wrote the paper. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2013_5328_MOESM1_ESM.tiff
Additional file 1:Length distribution of All-Unigene. (> 200 bp, mean length = 506 bp, N50 = 611 bp, Max = 8,514 bp). (TIFF 112 KB)
12864_2013_5328_MOESM2_ESM.tiff
Additional file 2:Alignment of de novo assembled unigene (Unigene5395_All) with a Sanger-derived sequence. Multiple alignments were performed by ClustalW and DNAMAN. The hatched region shows the matched sequence, and coverage validation is 99.13%. (TIFF 289 KB)
12864_2013_5328_MOESM3_ESM.tiff
Additional file 3:Alignment of de novo assembled unigene (Unigene13401_All) with a Sanger-derived sequence. Multiple alignments were performed by ClustalW and DNAMAN. The hatched region shows the matched sequence, and coverage validation is 100%. (TIFF 516 KB)
12864_2013_5328_MOESM4_ESM.tiff
Additional file 4:Alignment of de novo assembled unigene (Unigene26407_All) with a Sanger-derived sequence. Multiple alignments were performed by ClustalW and DNAMAN. The hatched region shows the matched sequence, and coverage validation is 98.76%. (TIFF 868 KB)
12864_2013_5328_MOESM5_ESM.tiff
Additional file 5:Alignment of de novo assembled unigene (Unigene41802_All) with reference sequence deposited in NCBI. Insulin-like growth factor-1 (Genbank HM164110.1). Multiple alignments were performed by ClustalW and DNAMAN. The hatched region shows the matched sequence, and coverage validation is 99.56%. (TIFF 1012 KB)
12864_2013_5328_MOESM6_ESM.tiff
Additional file 6:Alignment of de novo assembled unigene (Unigene56717_All) with reference sequence deposited in NCBI. Insulin-like growth factor-2 (Genbank HM164111.1). Multiple alignments were performed by ClustalW and DNAMAN. The hatched region shows the matched sequence, and coverage validation is 98.04%. (TIFF 837 KB)
12864_2013_5328_MOESM7_ESM.tiff
Additional file 7:Alignment of de novo assembled unigene (Unigene58882_All) with reference sequence deposited in NCBI. Tropomyosin (Genbank JN165713.1). Multiple alignments were performed by ClustalW and DNAMAN. The hatched region shows the matched sequence, and coverage validation is 99.74%. (TIFF 634 KB)
12864_2013_5328_MOESM10_ESM.doc
Additional file 10:The differentially expressed genes involved in food preference determination of mandarin fish.(DOC 150 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
He, S., Liang, XF., Sun, J. et al. Insights into food preference in hybrid F1 of Siniperca chuatsi (♀) × Siniperca scherzeri (♂) mandarin fish through transcriptome analysis. BMC Genomics 14, 601 (2013). https://doi.org/10.1186/1471-2164-14-601
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-14-601