
Abstract
Background
Metal sulfide mineral dissolution during bioleaching and acid mine drainage (AMD) formation creates an environment that is inhospitable to most life. Despite dominance by a small number of bacteria, AMD microbial biofilm communities contain a notable variety of coexisting and closely related Euryarchaea, most of which have defied cultivation efforts. For this reason, we used metagenomics to analyze variation in gene content that may contribute to niche differentiation among co-occurring AMD archaea. Our analyses targeted members of the Thermoplasmatales and related archaea. These results greatly expand genomic information available for this archaeal order.
Results
We reconstructed near-complete genomes for uncultivated, relatively low abundance organisms A-, E-, and Gplasma, members of Thermoplasmatales order, and for a novel organism, Iplasma. Genomic analyses of these organisms, as well as Ferroplasma type I and II, reveal that all are facultative aerobic heterotrophs with the ability to use many of the same carbon substrates, including methanol. Most of the genomes share genes for toxic metal resistance and surface-layer production. Only Aplasma and Eplasma have a full suite of flagellar genes whereas all but the Ferroplasma spp. have genes for pili production. Cryogenic-electron microscopy (cryo-EM) and tomography (cryo-ET) strengthen these metagenomics-based ultrastructural predictions. Notably, only Aplasma, Gplasma and the Ferroplasma spp. have predicted iron oxidation genes and Eplasma and Iplasma lack most genes for cobalamin, valine, (iso)leucine and histidine synthesis.
Conclusion
The Thermoplasmatales AMD archaea share a large number of metabolic capabilities. All of the uncultivated organisms studied here (A-, E-, G-, and Iplasma) are metabolically very similar to characterized Ferroplasma spp., differentiating themselves mainly in their genetic capabilities for biosynthesis, motility, and possibly iron oxidation. These results indicate that subtle, but important genomic differences, coupled with unknown differences in gene expression, distinguish these organisms enough to allow for co-existence. Overall this study reveals shared features of organisms from the Thermoplasmatales lineage and provides new insights into the functioning of AMD communities.
Similar content being viewed by others


Background
Until recently, very few genomes of archaea had been sequenced. As of 2012 there were only 233 archaeal genomes in the NCBI database compared to 3843 bacterial genomes. In part because of this bias, much less is known about archaeal evolution and physiology than that of bacteria. Of the sequenced archaeal genomes, most come from isolates from disparate environments and therefore tell us little about how archaeal populations co-exist within environments. Notable exceptions include isolates and draft genomes from metagenomic sequencing projects in hypersaline [1] and hot springs environments [2–5] and genomes of different strains of one gut methanogen [6]. Metagenomics allows us to examine the genomes of closely related archaea in the same community and make inferences about physiological differences that allow them to coexist. Spatial and temporal distributions of populations may be related to differences in geochemical conditions, in nutrients, or in other resources that different strains and species can utilize. Finally, if the intention is to isolate organisms with particular metabolic capacities, metagenomic insights can aid in the determination of the vitamins, nutrients, cofactors, and environmental conditions necessary for the growth of potential isolates.
A number of archaea of the Euryarchaeal order Thermoplasmatales have been described. This order currently comprises five genera: Ferroplasma, Thermoplasma, Picrophilus, Thermogymnomonas, and Acidiplasma. All of the isolates from this order are obligate or facultative aerobes and extreme acidophiles that were isolated from acidic, high sulfur environments. However, there is some phenotypic variation within this clade. The Picrophilus spp. are characterized by a single cell membrane surrounded by a surface layer, whereas the species in the other Thermoplasmatales genera have no cell walls. The Thermoplasma spp., Picrophilus spp., and Thermogymnomonas acidicola are moderate thermophiles with temperature optima around 60°C, whereas the Ferroplasma spp. and Acidiplasma aeolicum are mesophiles with temperature optima around 40° and 45°C respectively [7–15]. All of the isolates from the Thermoplasmatales order except for Ferroplasma acidiphilum are heterotrophs. All of the Ferroplasma spp. and Acidiplasma sp. are Fe-oxidizers and grow anaerobically via Fe respiration, whereas the Thermoplasma spp. are capable of S0 respiration.
In this study, we compare the near-complete genomes of the two Ferroplasma acidarmanus types, the isolate Fer1 sequence and the environmental Fer2 sequence, with newly annotated genomes of related organisms that we call A-, E-, G-, and Iplasma (APL, EPL,GPL, and IPL; NCBI accession numbers are reported in the Availability of supporting data section) [16, 17]. These organisms coexist in biofilm communities sampled from within the Richmond Mine at Iron Mountain in Redding, California. Of these organisms, only Fer1 has been isolated [11]. Though some of the other genomes have been a part of previous metagenomic analyses [16–18], their gene content has not been fully examined. The gene annotations and microscopy reported here provide new insights into acid mine drainage (AMD) community function and genomic differentiation among these organisms that allows them to avoid competitive exclusion and thus co-occur.
Results and discussion
Phylogeny
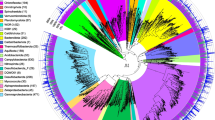
We previously published a phylogenetic tree of the 16S rRNA gene of the AMD plasmas [16, 17]. Here we improve upon that tree with the addition of a number of new taxa. This tree illustrates that the Richmond Mine AMD plasmas form the following clades: A-, B-, and Cplasma, E- with G-plasma, Dplasma with a number of environmental clones, I-plasma with a number of environmental clones, and the Ferroplasma spp. with Acidiplasma aeolicum. All of the 16S rRNA gene sequences, other than those of Fer1 and Fer2 (which have identical sequences), share less than 97% nucleotide identity. The Iplasma gene is the most divergent, and it is almost certainly not a member of the order Thermoplasmatales or the class Thermoplasmata (Figure 1, Additional file 1, Additional file 2). We found evidence for this classification in the phylogenetic analysis for both 16S rRNA and ribosomal protein S15 genes, where Iplasma groups outside of the Thermoplasmata clade (Figure 1 and Additional file 3) as observed previously [16, 17, 19, 20]. In the case of the 16S tree, Iplasma forms a monophyletic group with a number of environmental clones from acidic solfataric mud and acidic springs (Genbank) [21]. Because archaeal phylogeny is still unresolved, it is impossible to exactly determine the phylogeny of new taxa [22]. However, the branch length separating Iplasma and the Thermoplasmata organisms is greater than 0.25, supporting the separation of Iplasma into a new class of Euryarchaea. We previously suggested this in Justice et al., 2012 [20], but the current study provides much more extensive evidence for this classification. The monophyletic clustering of Eplasma and Gplasma and that of A-, B-, and C-, and Dplasma on the 16S rRNA tree suggests that they belong to new genera of Thermoplasmatales (Figure 1, Additional files 1, 2). This finding is further supported by similar amino acid identities of shared orthologs from A-, E-, and Gplasma to the other Thermoplasmatales archaea (Additional file 4).

16S rRNA tree indicating the possibility of a candidate class that includes Iplasma. Ferroplasma acidarmanus is Fer1 and Fer2. Bootstrap values are shown at branch splits. Gene start and stop positions and Genbank accession numbers are listed after organism names.
We examined a number of whole-genome measures of relatedness to further investigate evolutionary relationships. First, we identified the fraction of predicted orthologs in pairwise comparisons, and then determined their average amino acid identity. The normalization step involved dividing the number of orthologs by the average number of genes in the pair of genomes considered. Iplasma shares a lower percentage of orthologs, and a lower average amino acid identity with each of the other AMD plasma genomes than the other AMD plasma genomes share with each other (Additional files 4 and 5), consistent with a divergent phylogenetic placement. Fer1 vs. Fer2 has the highest amino acid identity (82%), as expected for closely related species. It was previously suggested that the genomes of Fer1 and Fer2 are different enough to merit classification as separate species based on analysis of recombination rates [23]. This result provides additional evidence supporting this claim, as Konstantinidis and Tiedje, 2005 found that approximately 95-96% amino acid identity corresponded to the 70% DNA-DNA hybridization species cut-off [24]. Eplasma and Gplasma are relatively closely related, as are Aplasma and Gplasma.
In addition to amino acid identity, we also looked at conserved gene order as a measure of evolutionary distance [16]. For each genome pair, we determined the number of syntenous orthologs and divided this by the number of shared orthologs. The Iplasma genome has the lowest synteny with the other AMD plasma genomes, Fer1 vs. Fer2 displays the highest synteny, followed by Eplasma vs. Gplasma (Additional file 6). The same trend holds true for another measure of synteny, the average length of syntenous blocks of genes in pairwise comparisons (Additional file 7). These whole-genome data support the tree topology and evolutionary distances assigned to the 16S rRNA genes in our phylogenetic analysis.
General genome features
Genome features of the AMD plasma organisms, including the number of tRNA synthetases and ribosomal genes, are summarized in Yelton et al., 2011 [16]. All of the genomes contain the full suite of tRNAs and most or all orthologous marker genes [16, 25], consistent with a high degree of genome completeness (Additional file 8). Important metabolic and structural features of each genome are listed and illustrated in Table 1 and Additional file 9.
Unique genomic island in G-plasma
A genomic island of potential importance was identified in the Gplasma genome. It consists of a block of nine genes that have virtually no orthologs in any of the other Thermoplasmatales genomes and is made up primarily of proteins of unknown function (Figure 2, Additional file 10). All nine of the proteins are represented in a whole community proteomic dataset reported previously [26], and three are among the most highly detected proteins of this organism in that dataset. The motifs and domains identified suggest that a number of these proteins are membrane associated, including a protein containing an AAA + FtsH ATPase domain (gene number 13327_0053) (found in a membrane-integrated metalloprotease [27]), a protein containing six transmembrane motifs and a signal peptide (13327_0056), and another with fourteen transmembrane motifs and a signal peptide (13327_0059). Additionally, three of these proteins include a rhodanese-like domain possibly involved in phosphatase or sulfurtransferase activity and another contains an armadillo repeat region, often used to bind large substrates such as peptides or nucleic acids (13327_0058).

Cluster of unique genes in Gplasma. Arrows are proportional to the length of each gene and indicate its direction of transcription. The gene numbers are shown inside the arrows. All genes are from contig number 13327. Motif and domain-based annotations are shown above the arrows. Genes with no annotations are hypothetical proteins. Rhod indicates a rhodanese-like domain.
The absence of any orthologs to this block of hypothetical proteins in other Thermoplasmatales genomes is a strong indication that it may have been acquired by horizontal gene transfer. Many flanking genes have syntenous orthologs in other closely-related genomes. However, the lack of GC skew in the nucleotide signature of these genes suggests that the transfer event was not recent or that the donor had a similar GC content to Gplasma.
Cell wall biosynthesis and imaging
Thermoplasmatales cells are generally bounded by a single membrane, except for two Picrophilus species that have a single membrane surrounded by a surface-layer (S-layer) [13]. We characterized archaeal-rich biofilm communities via cryo-electron microscopy and identified surface layers on many single membrane bound cells (Figure 3, Additional file 11). Thus, we looked for the genes needed for surface layer structural proteins and their post-translational modifications (i.e., N-glycosylation). We found putative S-layer genes in all of the AMD plasma genomes (except Fer1) that are homologous with the predicted P. torridus S-layer genes (Additional file 12) [28], but found no homology to the predicted S-layer genes in their next closest relative, Acidiloprofundum boonei[29]. We also found genes potentially involved in archaeal S-layer protein N-glycosylation. Of particular interest were homologs to the AglD and AglB genes of Haloferax volcanii, which have been shown to be essential to S-layer protein N-glycosylation in that organism [30]. Many of the Iplasma S-layer-related genes occur in a cluster, and several have conserved gene order in distant relatives, including several enzymes that attach sugars to a dolichol that might serve as a membrane anchor for the formation of an oligosaccharide during N-glycosylation. The Iplasma genome contains a gene cluster syntenous with distant relatives that encodes all of the proteins in the ADP-L-glycero-β-D-manno-heptose (AGMH) biosynthesis pathway (Additional file 12). AGMH is attached to S-layer proteins in gram-positive bacteria [31–33], suggesting that this may be involved in S-layer glycosylation in Iplasma as well. Finally, in the same genomic region genes are found for the biosynthesis of GDP-L-fucose, a glycoprotein component, and dTDP-L-rhamnose, a lipopolysaccharide component, indicating that these may make up part of the AMD plasma S-layer polysaccharides.

Cryo-EM of surface-layer on an AMD plasma cell from the Richmond Mine. Insets show a higher magnification. Arrows point to putative surface-layer proteins. Panel A and panel B show evidence of proteinaceous surface layers in two different cells collected from the Richmond Mine AMD.
Energy metabolism (a) iron oxidation
Ferric iron produced by biotic iron oxidation drives metal sulfide mineral dissolution, and thus iron oxidation is one of the most important biochemical processes that occurs in acid mine drainage systems [34–36]. In order to assess which of the AMD plasmas were involved in this process, we looked for potential iron oxidation genes via BLASTP. Based on this analysis, Aplasma and Gplasma contain homologs to rusticyanin, a blue-copper protein implicated in iron oxidation in Acidithiobacillus ferrooxidans (Additional file 12) [37]. The Acidithiobacillus ferroxidans rusticyanin can complex with and reduce cytochrome c in that organism [38–41], is upregulated during growth on ferrous iron [40–47], and is believed to be essential to iron oxidation [48]. Allen et al.[49] inferred that a related blue-copper protein, sulfocyanin, is involved in iron oxidation in Ferroplasma spp. (e.g. Fer1), and Dopson et al. provided proteomic and spectrophotometric evidence that support this inference [50]. The Fer2 genome contains a sulfocyanin homolog, whereas E- and Iplasma do not appear to have a rusticyanin or a sulfocyanin gene, suggesting that they are not iron oxidizers.
Additional evidence for the function of these genes was found in their inferred protein structure. All of the AMD plasma blue-copper proteins (BCPs) contain the characteristic type I copper-binding site, consisting of two histidines, one cysteine, one methionine and a cupredoxin fold, identified by a 7 or 8-stranded β-barrel fold [51–53] (Additional file 13). However, the AMD plasma BCPs differ in their conservation of motifs identified by Vivekanandan Giri et al. in sulfocyanin and rusticyanin [54]. The Fer1 and Fer2 BCPs include one recognized sulfocyanin motif, FNFNGTS, as well as imperfect conservation of the motifs identified in both sulfocyanin and rusticyanin (Additional file 14). Conversely, the Aplasma and Gplasma blue-copper proteins do not contain any of the conserved sulfocyanin-specific motifs. Instead, they contain imperfect matches to the rusticyanin-specific motif. These results are consistent with the inferences made based on homology alone in that they suggest that Fer1 and Fer2 BCPs are sulfocyanins and that A- and Gplasma BCPs are rusticyanins.
Phylogenetic analysis was carried to confirm the original homology-based annotations of the AMD plasma BCPs and to look for evidence of horizontal gene transfer. The phylogenetic tree groups the Aplasma BCP gene with the rusticyanins, whereas the Fer1 and Fer2 genes group with the sulfocyanins (Additional file 15). Interestingly, the Gplasma gene is so divergent that it does not consistently group with the other iron-oxidation blue-copper proteins. Its divergence seems to stem from two more β-strands than most of the other rusticyanin-like proteins (Additional file 13). The tree also provides evidence for the horizontal transfer of both sulfocyanin and rusticyanin genes. Related rusticyanin-like genes are found in the Gammaproteobacteria and in a variety of Euryarchaea. Similarly, closely related sulfocyanin-like genes are found in Euryarchaea and Crenarchaea.
Tyson et al. hypothesized that the sulfocyanin found in the Fer1 genome forms part of an iron-oxidizing SoxM-like supercomplex, similar to the one involved in sulfur oxidation in Sulfolobus acidocaldarius[55–57]. The S. acidocaldarius SoxM supercomplex contains a BCP, a cytochrome b and a Rieske iron sulfur protein. In S. acidocaldarius the sulfocyanin functions much like the cytochrome c in the complex III/cytochrome bc complex used during iron oxidation (and aerobic respiration) in A. ferrooxidans[58]. The results presented here further support Tyson’s hypothesis in that both the cytochrome b and rieske Fe-S protein subunits of the hypothetical SoxM-like complex were identified in all AMD plasma genomes. None of the genomes contain homologs to any of the other genes in the A. ferrooxidans rus operon [42, 59, 60].
In general, the absence of blue-copper proteins suggests that E- and Iplasma lack the Fe-oxidation capability entirely, whereas the other AMD plasmas utilize two different pathways to carry out this metabolism. It is possible that E- and Iplasma do have blue-copper proteins in their genomes because gaps remain in their assemblies, but we took steps to rule out this possibility (see Methods section). Because Fe(II) is an abundant electron donor in the AMD environment, this observed genetic variation in Fe oxidation potential may be important in niche differentiation.
Energy metabolism (b) carbon monoxide dehydrogenase
The Iplasma, Fer1 and Fer2 genomes encode genes for a possible carbon monoxide dehydrogenase, (CODH) (Additional file 12), including genes for all three subunits of the CoxMLS complex. Recent research suggests that aerobic CO oxidation may be a widespread metabolism among bacteria [61]. Thus, it is a conceivable metabolism for organisms in AMD systems. In fact, it may be a good source of carbon or energy in the Richmond Mine, where up to 50 ppm of CO has been measured in the air (M. Jones, personal communication 2011).
A phylogenetic tree of the catalytic subunits of CODH indicates that all but one of the AMD plasma complexes is more closely related to the aerobic type than the anaerobic type (Additional file 16). The active site encoded by these genes also suggests that they are aerobic CODH proteins closely related to the form II CODH, which has the motif: AYRGAGR (Additional file 17) [61, 62]. This enzyme can be used to make CO2 either for C fixation or to make reducing equivalents. The AMD plasma genomes do not contain any of the genes for the known archaeal C fixation pathways. Based on these observations, we hypothesize that these CODH proteins are used solely to make electrons available for aerobic respiration. However, it is possible that they use a novel C fixation pathway that incorporates this CODH [63].
Interestingly, our CODH phylogenetic tree suggests that there is another AMD plasma gene that encodes a Ni-CODH, Fer2 scaffold 31 gene 47. Ni-CODHs are anaerobic and reduce CO2 to CO. This enzyme is generally involved in C fixation via the Wood-Ljungdahl pathway, the genes for which are not found in the AMD plasma genomes. Thus, this gene may be involved in a novel carbon fixation pathway in Fer2. Additional evidence for the annotation of this gene as a Ni-CODH is provided in its structural alignment with known Ni-CODH proteins (Additional file 18), and by the annotation of a neighbor gene as a Ni-CODH maturation factor (Additional file 12). As a whole, the genomic evidence suggests CO oxidation capacity among Fer1, Fer2, and Iplasma and a potential for CO reduction in Fer2.
Energy metabolism (c) aerobic respiration
Fer1 and T. acidophilum are known to be facultative anaerobes [11, 64–66], whereas T. volcanium and P. torridus are aerobes. Therefore, it is not surprising that all of the Richmond Mine AMD plasmas have the capacity for aerobic respiration and catabolism of organic compounds via two glucose catabolism pathways, pyruvate dehydrogenase, the TCA cycle and an aerobic electron transport chain (Additional file 12). Some AMD plasma genes in the aerobic electron transport chain have been observed in proteomic analyses as previously reported by Justice et al., 2012 [20].
The AMD plasmas’ electron transport chains are similar to that of other archaea in that they do not contain all of the subunits of the NADH ubiquinone-oxidoreductase complex [67]. All of the AMD plasmas except Aplasma are missing the NuoEFG subunits found in the bacterial type complex I and instead have the subunits found in the archaeal-type complex I, NuoABCDHIJKLMN. Fer2 is missing NuoIJKLM most likely because the genes for this complex are found at the end of an incomplete contig. Eplasma, Gplasma and Fer1 maintain the Nuo gene order found in a number of other archaea including, Halobacterium sp., Sulfolobus solfataricus, and T. acidophilum[68]. All contain succinate dehydrogenase complex genes (Additional file 12). In the case of A-, E-, and Gplasma, the complex is missing SdhD, and many of the SdhC genes have annotations with low confidence. This finding is congruent with previous research that shows that the genes for the membrane anchor subunits of the complex are poorly conserved in both bacteria and archaea, possibly due to low selective pressure [69]. As mentioned previously in section (v)(a), the AMD plasmas have genes homologous to several predicted archaeal complex III/cytochrome bc complex genes (Additional file 12).
Archaeal-type aerobic terminal oxidases include cytochrome c oxidases (CCOs) and cytochrome bd oxidases. Genes for the cytochrome bd complex are found in P. torridus, T. acidophilum and T. volcanium[70]. All of the AMD plasma genomes contain the two genes for this complex. They also all contain the two essential genes for the archaeal heme-copper oxidase/CCO complex (subunit I and II) [70], and we confirm that subunit II contains the Cu-binding motif generally found in CCOs [71] (Additional file 19). Like the other CCO genes in B. subtilis and E. coli, the two cytochrome c genes in the AMD plasmas occur in a gene cluster with a protoheme IX farnesyltransferase, required for synthesis of the heme type used in aa(3) type CCOs [72]. The subunit II gene shares a high amino acid identity with several oxidases of this type, further indicating an aa(3) type CCO (Additional file 20).
Archaea use A-type ATP synthases to generate ATP from an electrochemical gradient. All of the AMD archaeal genomes contain the AhaABCDEFIK genes that comprise this complex in Methanosarcina mazei, although they are missing an ortholog to AhaG. All but Eplasma and Iplasma contain a putative AhaH gene. AhaG is also absent in T. acidophilum, indicating that it may not be necessary for ATP synthesis in these organisms.
Energy metabolism (d) alternative electron acceptors
In addition to aerobic respiratory capabilities, some Thermoplasmatales organisms are able to respire anaerobically [66]. Anaerobic reduction of S0 or sulfur ions could allow archaea in AMD systems to survive under anoxic conditions deep inside floating biofilms or in sunken biofilms and sediment, where many sulfur compounds are present [73]. The Iplasma genome contains several genes that are homologous to asrA and asrB, known sulfite reduction protein genes (13606_0515 and 13606_0514). These proteins comprise two of the three subunits of the AsrABC dissimilatory sulfite reductase complex found in Salmonella typhimurium[74]. However, the Iplasma genome does not contain the AsrC subunit, which contains the siroheme-binding motif and thus is thought to contain the active site for sulfite reduction. As the Asr proteins are not well characterized in many organisms, it is possible that these genes are misannotated. Synteny-based annotation ties these two genes to an adjacent FdhF formate dehydrogenase alpha subunit gene, indicating a possible involvement of these genes in formate dehydrogenase activity. In fact, one of these genes is structurally related to the HycB hydrogenase 3 Fe-S protein formate dehydrogenase subunit based on CBLAST against the NCBI protein structure database. Additional protein modeling suggests that one of the proteins in Iplasma could be a subunit of the formate dehydrogenase complex (Yelton, Zemla, and Thelen; unpublished observation). Thus, we suggest that these two proteins are functionally related to formate dehydrogenase in Iplasma.
Interestingly, the Iplasma genome contains homologs to all of the genes overexpressed under anaerobic conditions for T. volcanium as well as all of the genes overexpressed or over-transcribed under anaerobic conditions for T. acidophilum (except for their predicted sulfur respiration gene Ta1129) in two previous studies [75, 76] (Additional file 21). The other AMD archaea also share most, but not all, of these genes. Although there is no direct genomic evidence for anaerobic respiration, novel anaerobic respiratory pathways are possible. In fact, there is evidence that Fer1 can grow via anaerobic Fe(III) reduction [64], and enrichment cultures of Fer1 and Aplasma reduce iron [20].
Energy metabolism (e) heterotrophy
Chemolithoautotrophy is a common lifestyle in AMD communities (e.g., of Leptospirillum spp.) [77]. However, the Thermoplasmatales archaea are mostly heterotrophs (only F. acidiphilum has been shown to have any autotrophic capability [10]). The AMD plasma genomes encode genes for a wide variety of heterotrophic metabolisms, both aerobic and anaerobic. The AMD plasmas have the genes necessary for energy generation via catabolism of organic compounds, including fatty acids, sugars, starch, and glycogen, but not refractory organic matter such as cellulose (Additional file 12).
All of the AMD plasmas have genes for sugar and polysaccharide catabolism, including glucoamylase genes required to break down starch and alpha-amylase genes for glycogen catabolism into glucose and dextrin. They have the conventional Embden-Meyerhoff (EM) glycolytic pathway (Additional file 12). Moreover, they also have the genes for the non-phosphorylative Entner-Doudoroff (NPED) pathway for glucose degradation also found in a number of (hyper)thermophilic archaea, including T. acidophilum, P. torridus, S. solfataricus, Sulfolobus acidocaldarius, Sulfolobus tokodai and Thermoproteus tenax[78–81]. The AMD plasma genomes contain homologs to all of the genes in this pathway, including a homolog to the proven P. torridus KDG aldolase [82]. Thus, the AMD plasmas are similar to their Thermoplasmatales relatives, all of which have genes homologous to those of both the EM and NPED pathways. Previously published proteomic data indicates that all of the AMD plasma organisms express some of the genes in these two pathways [20].
Another potential carbon source for the AMD plasmas is lipids from lysed cells. All of the AMD plasma genomes contain a full set of homologs to the genes for the aerobic fatty acid oxidation pathway from E. coli (Additional file 12). Because many of the proteins in this pathway are acyl-CoA dehydrogenases, which are known to have undergone frequent gene duplication and horizontal transfer events [83], it is difficult to discern which role each gene plays in fatty acid degradation. However the number of β-oxidation-related annotations suggests that the AMD plasmas are capable of fatty acid breakdown, and many of the proteins from this pathway have been identified by proteomics [20].
Interestingly, the AMD plasmas have the genetic capacity to catabolize one-carbon compounds such as methanol. All except for Gplasma have several genes for subunits of a formate dehydrogenase. These genes were previously discussed by Yelton et al. [16], and a number are found in gene clusters with biosynthesis genes for their specific molybdopterin cofactor. We find that a formate hydrogen lyase complex gene cluster is evident in the Fer1 genome, as previously noted by Cárdenas et al.[63], but we also find a cluster of orthologous genes in Eplasma and Gplasma. It is possible that Fer1 is capable of the chimeric pathway of carbon fixation involving the formate hydrogen lyase described by Cárdenas et al.[84] (See section (vi) for further discussion of the putative group 4 hydrogenase hycE gene in this cluster). Eplasma also has the genes necessary for this pathway, but all of the other AMD plasma genomes are missing either the formate hydrogen lyase genes or the formate dehydrogenase subunit genes. Thus, we surmise that the AMD plasma formate dehydrogenases are primarily involved in an oxidative pathway for methanol methylotrophy (i.e., methanol degradation to formaldehyde, formaldehyde to formate, and formate oxidation to CO2). The AMD plasmas have homologs to all of the enzymes in this pathway, including the enzyme used by all thermotolerant methanol-oxidizing bacteria, a NAD-linked methanol dehydrogenase [85] (Additional file 12). Among the AMD plasmas, only Iplasma appears to have the genes necessary for the ribulose monophosphate cycle, which is commonly used for carbon assimilation from formaldehyde [85]. None of the genomes contain the genes necessary for the other known formaldehyde assimilation pathway, the serine cycle. As Fer1 has been shown to produce methanethiol during cysteine degradation [86], any methanol in the AMD biofilm may be a product of methanethiol catabolism.
Energy metabolism (f) fermentation and the use of fermentation products
AMD archaea are typically more abundant in thick, mature AMD biofilms [87] where they may encounter anoxic microenvironments [73]. Thus, we looked for potential fermentation genes in their genomes. They all have the genes for fermentation of pyruvate to acetate found in Pyrococcus furiosus and a number of other anaerobic fermentative and aerobic archaea [88–91] (Additional file 12). This pathway is unique in that it converts acetyl-CoA to acetate in only one step, with an ADP-forming acetyl-CoA synthetase. It is the only phosphorylating step of pyruvate fermentation via the NPED pathway. Previously this enzyme had been detected in hyperthermophilic and mesophilic archaea as well as some eukaryotes [91]. In anaerobic archaea this enzyme is involved in fermentation, whereas in aerobic archaea it makes acetate that is then catabolized via aerobic respiration [92]. The AMD plasmas have the genes necessary for fermentation to acetate under anaerobic conditions and for acetate respiration under aerobic conditions via an acetate-CoA ligase or the reversal of the direction of the acetate-CoA synthetase.
Putative hydrogenase 4 genes
Several AMD plasma genomes contain a number of genes that group with the putative group 4 hydrogenases according to phylogenetic analysis (Additional file 22). A group 4 hydrogenase complex and formate dehydrogenase comprise the formate hydrogen lyase that catalyzes non-syntrophic growth on formate and production of H2 in hyperthermophilic archaea (Thermococcus onnurineus) [93, 94]. The putative group 4 hydrogenases, though closely related to the group 4 hydrogenases, lack the two conserved hydrogen and Ni-binding motifs that are thought to be necessary for H2 formation [94, 95], possibly indicating some other function.
Toxic metal resistance
The Richmond Mine solutions contain extremely high (mM) concentrations of arsenic, cadmium, copper, and zinc [96]. Genomic evidence indicates that the AMD plasmas utilize multiple strategies to protect themselves from these elements, such as oxidation/reduction to less toxic forms and efflux (Additional file 12) [8, 97]. All of the AMD plasmas have at least two genes from the arsenic resistance (arsRABC) operon. Only Gplasma has all of the genes in the operon, but Fer1 has previously been shown to have resistance to both arsenate and arsenite, despite lacking the arsenate reductase [97]. All of the AMD plasmas except for Fer2 have two of the genes in the mercury resistance operon (merTPCAD), merA and merP (mercuric reductase and the mercuric ion-binding protein, respectively). All of the genomes also contain some putative copper resistance genes in the copABCD operon or the copYBZ loci, identified previously in Fer1 [98]. Specifically they all have homologs to copB. This gene has been shown to be involved in copper sequestration as a copper resistance strategy in Pseudomonas syringae[99]. The heavy metal transporter genes found in the AMD plasma genomes group into two different clades in a phylogenetic tree of metal resistance P-type ATPases. All of the genomes except for that of Iplasma contain two types of metal resistance transporters according to this phylogenetic analysis, a Cu/Ag transporter related to copA or copBZ and a Zn/Cd transporter related to cadA.
Biosynthesis
Because the AMD plasmas live in dense biofilms, they could potentially benefit from biomolecules (cofactors, amino acids, etc.) provided by other organisms .We previously demonstrated a lack of genes for de novo cobalamin biosynthesis in A-, E-, G-, and Iplasma [16]. Here we examined the AMD plasma genomes for other biosynthetic pathways.
Biosynthesis (a) glyoxylate shunt
Only Eplasma has the genes for the glyoxylate shunt, a pathway closely related to the TCA cycle that allows the use of organic compounds that are degraded to acetyl-CoA (i.e. fatty acids) for biosynthesis (Additional file 12). One of the proteins encoded in this pathway, the malate synthase, has been detected in proteomic analyses [20].
Biosynthesis (b) amino acid synthesis
The Thermoplasmatales archaea exhibit differential abilities to synthesize amino acids, suggesting that some of them rely more heavily on organic compound uptake than others. The genomes of E-, G- and Iplasma do not contain most of the histidine synthesis pathway genes. Eplasma and Iplasma also lack many of the genes necessary for the valine and (iso)leucine synthesis pathway (Additional file 12). They are also among the subset of organisms that do not make their own cobalamin [16]. This group of organisms may rely on amino acid and cobalamin scavenging to avoid the energetic costs of de novo synthesis.
Biosynthesis (c) trehalose biosynthesis
Compatible solutes allow organisms to maintain osmotic balance under high salt conditions or to protect against heat shock and cold shock [100]. A number of archaea make organic solutes for this purpose. T. acidophilum and a number of Sulfolobales archaea have been shown to produce trehalose as a compatible solute. In these organisms it has also been suggested that it is used to thermostabilize macromolecules and as a carbon storage molecule [100]. All of the AMD plasmas except for Iplasma have the genes necessary for trehalose biosynthesis from maltose (Additional file 12). The monophyletic group of A-, E-, and Gplasma also has the genetic potential for trehalose synthesis from glycogen.
Motility
Motility can provide a competitive advantage for archaea in aquatic environments by allowing them to colonize new sites and move across environmental gradients. To determine potential for motility, we looked for flagellar, chemotaxis and pili genes in the AMD plasma genomes.
Both the A- and Gplasma genomes contain the full flagella flaBCDEFGHIJ operon found in Methanococcus voltae[101–103] and Halobacterium salinarum[104] (Additional file 12). Thus, these organisms are predicted to be motile, yet they lack identifiable chemotaxis genes.
No flagellar genes are found in the other AMD plasma genomes, suggesting differences in motility. We used cryo-EM to confirm the existence of flagella on cells inferred to be archaea based on the presence of a single cell membrane (Figure 4). We found flagella-like structures with diameters of about 10–14 nm, similar in width to the flagella of T. volcanium[105]. The structures are also thicker than the pili observed in similar AMD plasmas or in bacteria [106]. A high-electron density area can be seen inside the cytoplasm immediately adjacent to the flagella that may be part of the associated protein motor complex.

Cryo-electron microscopy of AMD plasma cells. Panel A and panel B show evidence of flagella on two different cells collected from the Richmond Mine AMD. Arrows point to flagella. The box surrounds a potential motor protein complex.
In addition to flagellar assembly genes, a number of the AMD plasma genomes contain genes for Type II secretion or Type IV pili that are used in twitching motility or possibly conjugation or attachment to the biofilm or other surfaces. All of the genomes except for Fer1 and Fer2 contain some of these genes, and in Eplasma, Gplasma, and Iplasma they are in a cluster with conserved gene order among the AMD plasmas (Additional file 23). Cryo-EM confirms the existence of pili, and shows attachment of the pili from the original cell to other cells (Figure 5, Additional file 24).

Cryo-electron microscopy of AMD plasma cells with putative pili. Panel A and panel B show evidence of pili on two different cells collected from the Richmond Mine AMD. Arrows point to pili. Vesicle-like structures are delineated by a single membrane layer around an ovoid shape in each cell’s cytoplasm.
Vesicle-like cavities
Cryo-EM imaging demonstrates that a number of the AMD plasma cells harbor low electron-density inclusions within what appears to be a lipid membrane (Figure 5). These are similar in appearance to the gas vesicles that some extreme halophiles use for buoyancy [107], although those vesicles are enclosed in a proteinaceous membrane. We did not find genomic evidence of gas vesicle formation in the AMD plasmas by performing BLASTP searches of their genomes against the gas vesicle protein (gvp) genes of Haloarchaea[108]. Novel vesicle formation genes are expected and we speculate that these are liquid vesicles because their apparent lipid membrane would be gas-permeable.
Conclusions
The metagenomic and phylogenetic analyses presented here reveal evolutionary, metabolic and cell structural differences among uncultivated archaea that occur in AMD biofilm communities. We recognize Iplasma as a representative of a phylogenetically distinct class and provide both ribosomal RNA gene-based and genomic evidence supporting this conclusion. We present evidence for two new genera of the Thermoplasmatales order (one comprising E- and Gplasma and another including A-, B-, C-, and Dplasma). Based on genome content, it appears that all of the AMD plasmas have the capacity to grow both aerobically and anaerobically. However, their differing genetic potentials for biosynthesis of cofactors and amino acid precursors may allow the coexisting AMD plasmas to take advantage of microniches that occur in structurally differentiated biofilms [87]. Similarly, differences in motility may allow some AMD plasmas to colonize new sites or move along physicochemical gradients. We report new types of blue-copper proteins that future work may show are involved in iron oxidation and may further differentiate the AMD plasmas. Comparative genomic analyses also provide new information about organisms in the Thermoplasmatales clade, indicating the importance of methylotrophy, carbon monoxide oxidation, and other heterotrophic metabolisms to the AMD plasmas and demonstrating the existence of S-layer proteins outside of the Picrophilus genus.
Methods
DNA sequencing and assembly
The new genomes presented here are composite assemblies of DNA extracted from a number of biofilm samples from the Richmond Mine, Iron Mountain, CA. Sample collection, DNA extraction, sequencing, genome assembly, and automated annotation were described previously [16, 55, 109, 110], though current assemblies of Aplasma and Gplasma have been updated with recently acquired Illumina sequencing. All of the genomes were automatically assembled using velvet [111] and then manually curated, using the Consed software [112] to correct misassemblies and join contigs across gaps. Assembly data were published in Yelton, et al., 2011 [16].
Gene annotation
In addition to the automated annotation pipeline for the genomes described [16], we used a synteny-based method to improve the annotations of poorly annotated genes. This method was described previously [16], and provides either specific or general functional annotations based on gene context in distantly related genomes.
We manually curated all annotations that are specifically cited in this paper in the following manner. Genes were aligned against the Interpro and nr databases with a BLASTP algorithm. Genes were then annotated if they had a TIGR or Pfam domain hit that predicted a specific function with an e-value of at least 1 × 10-10 and coverage of more than 70% of the protein. Genes were given a “putative” annotation if they met the previous criteria except they had an e-value between 1 × 10-4 and 1 × 10-10 and matched 50-70% of the protein, or if their domain-based hits provided only general functional information. In these cases, additional evidence from hits from the nr database was used if possible to provide a specific functional annotation. Genes were given a “probable” annotation if they had annotated hits in the nr database with greater than 30% amino acid identity over 70% of the length of the gene. For incomplete metabolic and structural pathways, BLASTP searches were carried out against the entire Richmond Mine metagenomic database. Missing genes were searched for based on the amino acid sequence of their closest relative. In the case where significant hits were uncovered, maximum-likelihood amino acid trees were used to place these genes within the AMD plasma group of archaea and this placement was used to associate the genes with a specific AMD plasma genome or outside the group altogether.
Phylogenetic analyses
Phylogenetic analyses of certain genes were used to help place them in evolutionary context (e.g. 16S rRNA, blue-copper proteins). In these cases, the genes were aligned using the MAFFT alignment tool and default parameters [113, 114]. The alignment was then manually corrected if needed. For protein trees, the completed alignment was used to make a phylogenetic tree with the FastTree [115, 116] maximum likelihood-based tree software. In the case of the 16S rRNA gene, the phylogenetic tree was made using RaxML for improved accuracy based on the taxonomy of isolate organisms [117]. Support values were calculated for each branch split via the Shimodaira-Hasegawa test provided by the –boot option set to 1000 bootstraps for FastTree trees and using the rapid bootstrap for the RaxML tree.
Cryo-EM specimen preparation
For cryo-EM, aliquots of 5 μl were taken directly from the fresh biofilm samples and placed onto lacey carbon grids (Ted Pella 01881) that were pre-treated by glow-discharge. For cryo-ET, samples were deposited onto support grids pre-loaded with 10 nm colloidal gold particles. The Formvar support was not removed from the lacey carbon. The grids were manually blotted and plunged into liquid ethane by a compressed air piston, then stored in liquid nitrogen.
Electron tomography imaging
Images were acquired on a JEOL–3100 electron microscope equipped with a FEG electron source operating at 300 kV, an Omega energy filter, a Gatan 795 2K×2K CCD camera, and cryo-transfer stage. The stage was cooled to 80 K with liquid nitrogen. For more information on imaging and analysis see Additional file 25.
Availability of supporting data
The data sets supporting the results of this article are available in the NCBI repository.
Aplasma: This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession ACXK00000000. The version described in this paper is version ACXK02000000. Eplasma: This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession ACXL00000000. The version described in this paper is version ACXL02000000. Gplasma: This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession ATDV00000000. The version described in this paper is version ATDV01000000. FER1: This isolate genome has been deposited at DDBJ/EMBL/GenBank under the accession AMD_IFERC00001. FER2: This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession ATDU00000000. The version described in this paper is version ATDU01000000. Iplasma: This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession ACXM00000000. The version described in this paper is version ACXM02000000. Additional data sets supporting the results of this article are included within the article and its additional files.
References
Narasingarao P, Podell S, Ugalde JA, Brochier-Armanet C, Emerson JB, Brocks JJ, Heidelberg KB, Banfield JF, Allen EE: De novo metagenomic assembly reveals abundant novel major lineage of Archaea in hypersaline microbial communities. ISME J. 2012, 6 (1): 81-93. 10.1038/ismej.2011.78.
Guo L, Brugger K, Liu C, Shah SA, Zheng HJ, Zhu YQ, Wang SY, Lillestol RK, Chen LM, Frank J, et al: Genome analyses of Icelandic strains of Sulfolobus islandicus, model organisms for genetic and virus-host interaction studies. J Bacteriol. 2011, 193 (7): 1672-1680. 10.1128/JB.01487-10.
Reno ML, Held NL, Fields CJ, Burke PV, Whitaker RJ: Biogeography of the Sulfolobus islandicus pan-genome. Proc Natl Acad Sci USA. 2009, 106 (21): 8605-8610. 10.1073/pnas.0808945106.
Whitaker RJ, Grogan DW, Taylor JW: Geographic barriers isolate endemic populations of hyperthermophilic archaea. Science. 2003, 301 (5635): 976-978. 10.1126/science.1086909.
Inskeep WP, Rusch DB, Jay ZJ, Herrgard MJ, Kozubal MA, Richardson TH, Macur RE, Hamamura N, Jennings RD, Fouke BW, et al: Metagenomes from high-temperature chemotrophic systems reveal geochemical controls on microbial community structure and function. PLoS One. 2010, 5 (3): e9773-10.1371/journal.pone.0009773.
Hansen EE, Lozupone CA, Rey FE, Wu M, Guruge JL, Narra A, Goodfellow J, Zaneveld JR, McDonald DT, Goodrich JA, et al: Pan-genome of the dominant human gut-associated archaeon, Methanobrevibacter smithii, studied in twins. Proc Natl Acad Sci USA. 2011, 108: 4599-4606. 10.1073/pnas.1000071108.
Huber H, Stetter KO: From Thermoplasmatales. Prokaryotes: A Handbook on the Biology of Bacteria, Vol 3. Edited by: Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E. 2006, 233 Spring Street, New York, NY 10013, United States: Springer, 101-112. 3
Dopson M, Baker-Austin C, Hind A, Bowman JP, Bond PL: Characterization of Ferroplasma isolates and Ferroplasma acidarmanus sp nov., extreme acidophiles from acid mine drainage and industrial bioleaching environments. Appl Environ Microbiol. 2004, 70 (4): 2079-2088. 10.1128/AEM.70.4.2079-2088.2004.
Zhou H, Zhang R, Hu P, Zeng W, Xie Y, Wu C, Qiu G: Isolation and characterization of Ferroplasma thermophilum sp. nov., a novel extremely acidophilic, moderately thermophilic archaeon and its role in bioleaching of chalcopyrite. J Appl Microbiol. 2008, 105 (2): 591-601. 10.1111/j.1365-2672.2008.03807.x.
Golyshina OV, Pivovarova TA, Karavaiko GI, Kondrat’eva TF, Moore ERB, Abraham WR, Lunsdorf H, Timmis KN, Yakimov MM, Golyshin PN: Ferroplasma acidiphilum gen. nov., sp nov., an acidophilic, autotrophic, ferrous-iron-oxidizing, cell-wall-lacking, mesophilic member of the Ferroplasmaceae fam. nov., comprising a distinct lineage of the Archaea. Int J Syst Evol Microbiol. 2000, 50: 997-1006. 10.1099/00207713-50-3-997.
Edwards KJ, Bond PL, Gihring TM, Banfield JF: An archaeal iron-oxidizing extreme acidophile important in acid mine drainage. Science. 2000, 287 (5459): 1796-1799. 10.1126/science.287.5459.1796.
Darland G, Brock TD, Samsonof W, Conti SF: Thermophilic, acidophilic mycoplasma isolated from a coal refuse pile. Science. 1970, 170 (3965): 1416-1418. 10.1126/science.170.3965.1416.
Schleper C, Puhler G, Klenk HP, Zillig W: Picrophilus oshimae and Picrophilus torridus fam nov, gen nov, sp nov, two species of hyperacidophilic, thermophilic, heterotrophic, aerobic archaea. Int J Syst Bacteriol. 1996, 46 (3): 814-816. 10.1099/00207713-46-3-814.
Hawkes RB, Franzmann PD, O’Hara G, Plumb JJ: Ferroplasma cupricumulans sp nov., a novel moderately thermophilic, acidophilic archaeon isolated from an industrial-scale chalcocite bioleach heap. Extremophiles. 2006, 10 (6): 525-530. 10.1007/s00792-006-0527-y.
Itoh T, Yoshikawa N, Takashina T: Thermogymnomonas acidicola gen. nov., sp nov., a novel thermoacidophilic, cell wall-less archaeon in the order Thermoplasmatales, isolated from a solfataric soil in Hakone, Japan. Int J Syst Evol Microbiol. 2007, 57: 2557-2561. 10.1099/ijs.0.65203-0.
Yelton AP, Thomas BC, Simmons SL, Wilmes P, Zemla A, Thelen MP, Justice N, Banfield JF: A semi-quantitative, synteny-based method to improve functional predictions for hypothetical and poorly annotated bacterial and archaeal genes. PLoS Comp Biol. 2011, 7 (10): e1002230-10.1371/journal.pcbi.1002230.
Dick GJ, Andersson AF, Baker BJ, Simmons SL, Yelton AP, Banfield JF: Community-wide analysis of microbial genome sequence signatures. Genome Biol. 2009, 10 (8): 50-
Jones DS, Albrecht HL, Dawson KS, Schaperdoth I, Freeman KH, Pi YD, Pearson A, Macalady JL: Community genomic analysis of an extremely acidophilic sulfur-oxidizing biofilm. ISME J. 2012, 6 (1): 158-170. 10.1038/ismej.2011.75.
Durbin AM, Teske A: Archaea in organic-lean and organic-rich marine subsurface sediments: an environmental gradient reflected in distinct phylogenetic lineages. Front Microbiol. 2012, 3: 168-
Justice N, Pan C, Mueller R, Spaulding SE, Shah V, Sun CS, Yelton AP, Miller CS, Thomas BC, Shah M, VerBerkmoes N, Hettich R, Banfield JF: Heterotrophic archaea contribute to carbon cycling in low-pH, suboxic biofilm communities. Appl Environ Microbiol. 2012, 78: 8321-8330. 10.1128/AEM.01938-12.
Kato S, Itoh T, Yamagishi A: Archaeal diversity in a terrestrial acidic spring field revealed by a novel PCR primer targeting archaeal 16S rRNA genes. FEMS Microbiol Lett. 2011, 319 (1): 34-43. 10.1111/j.1574-6968.2011.02267.x.
Pace NR: Mapping the tree of life: Progress and prospects. Microbiol Mol Biol Rev. 2009, 73 (4): 565-576. 10.1128/MMBR.00033-09.
Eppley JM, Tyson GW, Getz WM, Banfield JF: Genetic exchange across a species boundary in the archaeal genus Ferroplasma. Genetics. 2007, 177 (1): 407-416. 10.1534/genetics.107.072892.
Konstantinidis KT, Tiedje JM: Towards a genome-based taxonomy for prokaryotes. J Bacteriol. 2005, 187 (18): 6258-6264. 10.1128/JB.187.18.6258-6264.2005.
Raes J, Korbel JO, Lercher MJ, von Mering C, Bork P: Prediction of effective genome size in metagenomic samples. Genome Biol. 2007, 8 (1): R10-10.1186/gb-2007-8-1-r10.
Denef VJ, Kalnejais LH, Mueller RS, Wilmes P, Baker BJ, Thomas BC, VerBerkmoes NC, Hettich RL, Banfield JF: Proteogenomic basis for ecological divergence of closely related bacteria in natural acidophilic microbial communities. Proc Natl Acad Sci USA. 2010, 107 (6): 2383-2390. 10.1073/pnas.0907041107.
Niwa H, Tsuchiya D, Makyio H, Yoshida M, Morikawa K: Hexameric ring structure of the ATPase domain of the membrane-integrated metalloprotease FtsH from Thermus thermophilus HB8. Structure. 2002, 10 (10): 1415-1423. 10.1016/S0969-2126(02)00855-9.
Futterer O, Angelov A, Liesegang H, Gottschalk G, Schleper C, Schepers B, Dock C, Antranikian G, Liebl W: Genome sequence of Picrophilus torridus and its implications for life around pH 0. Proc Natl Acad Sci USA. 2004, 101 (24): 9091-9096. 10.1073/pnas.0401356101.
Reysenbach AL, Flores GE: Electron microscopy encounters with unusual thermophiles helps direct genomic analysis of Aciduliprofundum boonei. Geobiology. 2008, 6 (3): 331-336. 10.1111/j.1472-4669.2008.00152.x.
Abu-Qarn M, Yurist-Doutsch S, Giordano A, Trauner A, Morris HR, Hitchen P, Medalia O, Dell A, Eichler J: Haloferax volcanii AglB and AglD are involved in N-glycosylation of the S-layer glycoprotein and proper assembly of the surface layer. J Mol Biol. 2007, 374 (5): 1224-1236. 10.1016/j.jmb.2007.10.042.
Kosma P, Wugeditsch T, Christian R, Zayni S, Messner P: Glycan structure of a heptose-containing S-layer glycoprotein of Bacillus thermoaerophilus. Glycobiology. 1995, 5 (8): 791-796. 10.1093/glycob/5.8.791.
Wugeditsch T, Zachara NE, Puchberger M, Kosma P, Gooley AA, Messner P: Structural heterogeneity in the core oligosaccharide of the S-layer glycoprotein from Aneurinibacillus thermoaerophilus DSM 10155. Glycobiology. 1999, 9 (8): 787-795. 10.1093/glycob/9.8.787.
Valvano MA, Messner P, Kosma P: Novel pathways for biosynthesis of nucleotide-activated glycero-manno-heptose precursors of bacterial glycoproteins and cell surface polysaccharides. Microbiology-Sgm. 2002, 148: 1979-1989.
Johnson DB, Hallberg KB: The microbiology of acidic mine waters. Res Microbiol. 2003, 154 (7): 466-473. 10.1016/S0923-2508(03)00114-1.
Fowler TA, Holmes PR, Crundwell FK: Mechanism of pyrite dissolution in the presence of Thiobacillus ferrooxidans. Appl Environ Microbiol. 1999, 65 (7): 2987-2993.
Rawlings DE: From characteristics and adaptability of iron- and sulfur-oxidizing microorganisms used for the recovery of metals from minerals and their concentrates. Microb Cell Fact. 2005, 4: 1-10.1186/1475-2859-4-1.
Cobley JG, Haddock BA: Respiratory chain of Thiobacillus ferrooxidans: Reduction of cytochromes by Fe2+ and preliminary characterization of rusticyanin, a novel blue copper protein. FEBS Lett. 1975, 60 (1): 29-33. 10.1016/0014-5793(75)80411-X.
Giudici-Orticoni MT, Guerlesquin F, Bruschi M, Nitschke W: Interaction-induced redox switch in the electron transfer complex rusticyanin-cytochrome c(4). J Biol Chem. 1999, 274 (43): 30365-30369. 10.1074/jbc.274.43.30365.
Castelle C, Guiral M, Malarte G, Ledgham F, Leroy G, Brugna M, Giudici-Orticoni M-T: A new iron-oxidizing/O-2-reducing supercomplex spanning both inner and outer membranes, isolated from the extreme acidophile Acidithiobacillus ferrooxidans. J Biol Chem. 2008, 283 (38): 25803-25811. 10.1074/jbc.M802496200.
Giudici-Orticoni MT, Leroy G, Nitschke W, Bruschi M: Characterization of a new dihemic c(4)-type cytochrome isolated from Thiobacillus ferrooxidans. Biochemistry (Mosc). 2000, 39 (24): 7205-7211. 10.1021/bi992846p.
Malarte G, Leroy G, Lojou E, Abergel C, Bruschi M, Giudici-Orticoni MT: Insight into molecular stability and physiological properties of the diheme cytochrome CYC41 from the acidophilic bacterium Acidithiobacillus ferrooxidans. Biochemistry (Mosc). 2005, 44 (17): 6471-6481. 10.1021/bi048425b.
Yarzabal A, Appia-Ayme C, Ratouchniak J, Bonnefoy V: Regulation of the expression of the Acidithiobacillus ferrooxidans rus operon encoding two cytochromes c, a cytochrome oxidase and rusticyanin. Microbiology. 2004, 150: 2113-2123. 10.1099/mic.0.26966-0.
Ramirez P, Guiliani N, Valenzuela L, Beard S, Jerez CA: Differential protein expression during growth of Acidithiobacillus ferrooxidans on ferrous iron, sulfur compounds, or metal sulfides. Appl Environ Microbiol. 2004, 70 (8): 4491-4498. 10.1128/AEM.70.8.4491-4498.2004.
Quatrini R, Appia-Ayme C, Denis Y, Ratouchniak J, Veloso F, Valdes J, Lefimil C, Silver S, Roberto F, Orellana O, et al: Insights into the iron and sulfur energetic metabolism of Acidithiobacillus ferrooxidans by microarray transcriptome profiling. Hydrometallurgy. 2006, 83 (1–4): 263-272.
Kucera J, Bouchal P, Lochman J, Potesil D, Janiczek O, Zdrahal Z, Mandl M: Ferrous iron oxidation by sulfur-oxidizing Acidithiobacillus ferrooxidans and analysis of the process at the levels of transcription and protein synthesis. Antonie Van Leeuwenhoek International Journal of General and Molecular Microbiology. 2013, 103 (4): 905-919. 10.1007/s10482-012-9872-2.
Bouchal P, Zdrahal Z, Helanova S, Janiczek O, Hallberg KB, Mandl M: Proteomic and bioinformatic analysis of iron- and sulfur-oxidizing Acidithiobacillus ferrooxidans using immobilized pH gradients and mass spectrometry. Proteomics. 2006, 6 (15): 4278-4285. 10.1002/pmic.200500719.
Carlos C, Reis FC, Vicentini R, Madureira DJ, Ottoboni LMM: The rus operon genes are differentially regulated when Acidithiobacillus ferrooxidans LR is kept in contact with metal sulfides. Curr Microbiol. 2008, 57 (4): 375-380. 10.1007/s00284-008-9208-7.
Holmes DS, Bonnefoy V: Genetic and bioinformatic insights into iron and sulfur oxidation mechanisms of bioleaching organisms. Biomining. Edited by: Rawlings DE, Johnson DB. 2007, Berlin Heidelberg: Springer-Verlag, 281-307.
Allen EE, Tyson GW, Whitaker RJ, Detter JC, Richardson PM, Banfield JF: Genome dynamics in a natural archaeal population. Proc Natl Acad Sci USA. 2007, 104 (6): 1883-1888. 10.1073/pnas.0604851104.
Dopson M, Baker-Austin C, Bond PL: Analysis of differential protein expression during growth states of Ferroplasma strains and insights into electron transport for iron oxidation. Microbiology. 2005, 151: 4127-4137. 10.1099/mic.0.28362-0.
Ambler RP: Blue copper proteins as honorary cytochromes: The structure and evolution of blue copper proteins. J Chem Soc Pak. 1999, 21 (3): 213-228.
Redinbo MR, Yeates TO, Merchant S: Plastocyanin: Structural and functional analysis. J Bioenerg Biomembr. 1994, 26 (1): 49-66. 10.1007/BF00763219.
Gough J, Chothia C: The linked conservation of structure and function in a family of high diversity: The monomeric cupredoxins. Structure. 2004, 12 (6): 917-925. 10.1016/j.str.2004.03.029.
Giri AV, Anishetty S, Gautam P: Functionally specified protein signatures distinctive for each of the different blue copper proteins. BMC Bioinforma. 2004, 5: 127-10.1186/1471-2105-5-127.
Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF: Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004, 428 (6978): 37-43. 10.1038/nature02340.
Komorowski L, Schafer G: Sulfocyanin and subunit II, two copper proteins with novel features, provide new insight into the archaeal SoxM oxidase supercomplex. FEBS Lett. 2001, 487 (3): 351-355. 10.1016/S0014-5793(00)02343-7.
Komorowski L, Verheyen W, Schaefer G: The archaeal respiratory supercomplex SoxM from S. acidocaldarius combines features of quinole and cytochrome c oxidases. Biol Chem. 2002, 383 (11): 1791-1799.
Elbehti A, Nitschke W, Tron P, Michel C, Lemesle-Meunier D: Redox components of cytochrome bc-type enzymes in acidophilic prokaryotes I. Characterization of the cytochrome bc(1)-type complex of the acidophilic ferrous ion-oxidizing bacterium Thiobacillus ferrooxidans. J Biol Chem. 1999, 274 (24): 16760-16765. 10.1074/jbc.274.24.16760.
Appia-Ayme C, Guiliani N, Ratouchniak J, Bonnefoy V: Characterization of an operon encoding two c-type cytochromes, an aa(3)-type cytochrome oxidase, and rusticyanin in Thiobacillus ferrooxidans ATCC 33020. Appl Environ Microbiol. 1999, 65 (11): 4781-4787.
Yarzabal A, Duquesne K, Bonnefoy V: Rusticyanin gene expression of Acidithiobacillus ferrooxidans ATCC 33020 in sulfur- and in ferrous iron media. Hydrometallurgy. 2003, 71 (1–2): 107-114.
King GA: Molecular and culture-based analyses of aerobic carbon monoxide oxidizer diversity. Appl Environ Microbiol. 2003, 69 (12): 7257-7265. 10.1128/AEM.69.12.7257-7265.2003.
Cunliffe M: Correlating carbon monoxide oxidation with cox genes in the abundant marine Roseobacter clade. ISME J. 2011, 5 (4): 685-691. 10.1038/ismej.2010.170.
Cardenas JP, Martinez V, Covarrubias P, Holmes DS, Quatrini R: Predicted CO/CO(2) fixation in Ferroplasma spp. via a novel chimaeric pathway. Biohydrometallurgy: A meeting point between microbial ecology, metal recovery processes and environmental remediation, Volume 71–73. Edited by: Donati ER, Viera MR, Tavani EL, Giaveno MA, Lavalle TL, Chiacchiarini PA. 2009, 219-222.
Dopson M, Baker-Austin C, Bond P: Towards determining details of anaerobic growth coupled to ferric iron reduction by the acidophilic archaeon ‘Ferroplasma acidarmanus’ Fer1. Extremophiles. 2007, 11 (1): 159-168. 10.1007/s00792-006-0029-y.
Ruepp A, Graml W, Santos-Martinez ML, Koretle KK, Volker C, Mewes HW, Frishman D, Stocker S, Lupas AN, Baumeister W: The genome sequence of the thermoacidophilic scavenger Thermoplasma acidophilum. Nature. 2000, 407 (6803): 508-513. 10.1038/35035069.
Segerer A, Langworthy TA, Stetter KO: Thermoplasma acidophilum and Thermoplasma volcanium sp nov from solfatara fields. Syst Appl Microbiol. 1988, 10 (2): 161-171. 10.1016/S0723-2020(88)80031-6.
Kletzin A: General characteristics and important model organisms, vol. 1. Washington. 2007, D.C.: ASM Press
Omelchenko MV, Makarova KS, Wolf YI, Rogozin IB, Koonin EV: Evolution of mosaic operons by horizontal gene transfer and gene displacement in situ. Genome Biol. 2003, 4 (9): R55-10.1186/gb-2003-4-9-r55.
Lemos RS, Fernandes AS, Pereira MM, Gomes CM, Teixeira M: Quinol : fumarate oxidoreductases and succinate : quinone oxidoreductases: phylogenetic relationships, metal centres and membrane attachment. Biochim Biophys Acta. 2002, 1553 (1–2): 158-170.
Schroder I, De Vries S: Archaea: new models for prokaryotic biology, vol. 1. 2008, Norfolk, UK: Caister Academic Press, 4-5.
Paumann M, Lubura B, Regelsberger G, Feichtinger M, Kollensberger G, Jakopitsch C, Furtmuller PG, Peschek GA, Obinger C: Soluble Cu(A) domain of cyanobacterial cytochrome c oxidase. J Biol Chem. 2004, 279 (11): 10293-10303.
Garciahorsman JA, Barquera B, Rumbley J, Ma JX, Gennis RB: The superfamily of heme-copper respiratory oxidases. J Bacteriol. 1994, 176 (18): 5587-5600.
Ma S, Banfield JF: Micron-scale Fe(2+)/Fe(3+), intermediate sulfur species and O(2) gradients across the biofilm-solution-sediment interface control biofilm organization. Geochim Cosmochim Acta. 2011, 75 (12): 3568-3580. 10.1016/j.gca.2011.03.035.
Huang CJ, Barrett EL: Sequence analysis and expression of the Salmonella typhimurium asr operon encoding production of hydrogen sulfide from sulfite. J Bacteriol. 1991, 173 (4): 1544-1553.
Kawashima T, Yokoyama K, Higuchi S, Suzuki M: Identification of proteins present in the archaeon Thermoplasma volcanium cultured in aerobic or anaerobic conditions. Proc Jpn Acad Ser B Phys Biol Sci. 2005, 81 (6): 204-219. 10.2183/pjab.81.204.
Sun N, Pan CP, Nickell S, Mann M, Baumeister W, Nagy I: Quantitative proteome and transcriptome analysis of the archaeon Thermoplasma acidophilum cultured under aerobic and anaerobic conditions. J Proteome Res. 2010, 9 (9): 4839-4850. 10.1021/pr100567u.
Golovacheva RS, Golyshina OV, Karavaiko GI, Dorofeev AG, Pivovarova TA, Chernykh NA: A new iron-oxidizing bacterium, Leptospirillum thermoferrooxidans sp. nov. Microbiology. 1992, 61 (6): 744-750.
Reher M, Schonheit P: Glyceraldehyde dehydrogenases from the thermoacidophilic euryarchaeota Picrophilus torridus and Thermoplasma acidophilum, key enzymes of the non-phosphorylative Entner-Doudoroff pathway, constitute a novel enzyme family within the aldehyde dehydrogenase superfamily. FEBS Lett. 2006, 580 (5): 1198-1204. 10.1016/j.febslet.2006.01.029.
Budgen N, Danson MJ: Metabolism of glucose via a modified Entner-Doudoroff pathway in the thermoacidophilic archaebacterium Thermoplasma acidophilum. FEBS Lett. 1986, 196 (2): 207-210. 10.1016/0014-5793(86)80247-2.
Derosa M, Gambacorta A, Nicolaus B, Giardina P, Poerio E, Buonocore V: Glucose-metabolism in the extreme thermoacidophilic archaebacterium Sulfolobus solfataricus. Biochem J. 1984, 224 (2): 407-414.
Siebers B, Tjaden B, Michalke K, Dorr C, Ahmed H, Zaparty M, Gordon P, Sensen CW, Zibat A, Klenk HP, et al: Reconstruction of the central carbohydrate metabolism of Thermoproteus tenax by use of genomic and biochemical data. J Bacteriol. 2004, 186 (7): 2179-2194. 10.1128/JB.186.7.2179-2194.2004.
Reher M, Fuhrer T, Bott M, Schoenheit P: The nonphosphorylative Entner-Doudoroff pathway in the thermoacidophilic euryarchaeon Picrophilus torridus involves a novel 2-keto-3-deoxygluconate-specific aldolase. J Bacteriol. 2010, 192 (4): 964-974. 10.1128/JB.01281-09.
Swigonova Z, Mohsen AW, Vockley J: Acyl-CoA dehydrogenases: Dynamic history of protein family evolution. J Mol Evol. 2009, 69 (2): 176-193. 10.1007/s00239-009-9263-0.
Cardenas E, Wu W, Leigh M, Carley J, Carroll S, Gentry T, Luo J, Watson D, Gu B, Ginder-Vogel M: Microbial communities in contaminated sediments, associated with bioremediation of uranium to submicromolar levels. Appl Environ Microbiol. 2008, 74: 3718-3729. 10.1128/AEM.02308-07.
Lidstrom ME: Aerobic methylotrophic prokaryotes, vol. 2. 2006, New York, NY, USA: Springer, 3-5.
Baumler DJ, Hung KF, Jeong KC, Kaspar CW: Production of methanethiol and volatile sulfur compounds by the archaeon “Ferroplasma acidarmanus”. Extremophiles. 2007, 11 (6): 841-851. 10.1007/s00792-007-0108-8.
Wilmes P, Remis JP, Hwang M, Auer M, Thelen MP, Banfield JF: Natural acidophilic biofilm communities reflect distinct organismal and functional organization. ISME J. 2009, 3 (2): 266-270. 10.1038/ismej.2008.90.
Schafer T, Selig M, Schonheit P: Acetyl-Coa synthetase (ADP forming) in archaea, a novel enzyme involved in acetate formation and ATP synthesis. Arch Microbiol. 1993, 159 (1): 72-83. 10.1007/BF00244267.
Brasen C, Schonheit P: Unusual ADP-forming acetyl-coenzyme A synthetases from the mesophilic halophilic euryarchaeon Haloarcula marismortui and from the hyperthermophilic crenarchaeon Pyrobaculum aerophilum. Arch Microbiol. 2004, 182 (4): 277-287. 10.1007/s00203-004-0702-4.
Sakuraba H, Ohshima T: Novel energy metabolism in anaerobic hyperthermophilic archaea: A modified Embden-Meyerhof pathway. J Biosci Bioeng. 2002, 93 (5): 441-448.
Siebers B, Schonheit P: Unusual pathways and enzymes of central carbohydrate metabolism in Archaea. Curr Opin Microbiol. 2005, 8 (6): 695-705. 10.1016/j.mib.2005.10.014.
Brasen C, Schonheit P: Regulation of acetate and acetyl-CoA converting enzymes during growth on acetate and/or glucose in the halophilic archaeon Haloarcula marismortui. FEMS Microbiol Lett. 2004, 241 (1): 21-26. 10.1016/j.femsle.2004.09.033.
Kim YJ, Lee HS, Kim ES, Bae SS, Lim JK, Matsumi R, Lebedinsky AV, Sokolova TG, Kozhevnikova DA, Cha SS, et al: Formate-driven growth coupled with H2 production. Nature. 2010, 467 (7313): 352-U137. 10.1038/nature09375.
Lim JK, Kang SG, Lebedinsky AV, Lee JH, Lee HS: Identification of a novel class of membrane-bound NiFe-hydrogenases in Thermococcus onnurineus NA1 by in silico analysis. Appl Environ Microbiol. 2010, 76 (18): 6286-6289. 10.1128/AEM.00123-10.
Wu LF, Mandrand MA: Microbial hydrogenases: Primary structure, classification, signatures and phylogeny. FEMS Microbiol Rev. 1993, 104 (3–4): 243-270.
Edwards KJ, Gihring TM, Schrenk MO, Hamers RJ, Banfield JF: Microbial populations and distribution at an extreme acid mine drainage environment: A study using fluorescent in-situ hybridization [abstract]. Abstr Gen Meet Am Soc Microbiol. 1998, 98: 382-
Gihring TM, Bond PL, Peters SC, Banfield JF: Arsenic resistance in the archaeon “Ferroplasma acidarmanus”: new insights into the structure and evolution of the ars genes. Extremophiles. 2003, 7 (2): 123-130.
Baker-Austin C, Dopson M, Wexler M, Sawers RG, Bond PL: Molecular insight into extreme copper resistance in the extremophilic archaeon ‘Ferroplasma acidarmanus’ Fer1. Microbiology. 2005, 151: 2637-2646. 10.1099/mic.0.28076-0.
Cooksey DA: Molecular mechanisms of copper resistance and accumulation in bacteria. FEMS Microbiol Rev. 1994, 14 (4): 381-386. 10.1111/j.1574-6976.1994.tb00112.x.
Martins LO, Huber R, Huber H, Stetter KO, Da Costa MS, Santos H: Organic solutes in hyperthermophilic archaea. Appl Environ Microbiol. 1997, 63 (3): 896-902.
Jarrell KF, Bayley DP, Florian V, Klein A: Isolation and characterization of insertional mutations in flagellin genes in the archaeon Methanococcus voltae. Mol Microbiol. 1996, 20 (3): 657-666. 10.1046/j.1365-2958.1996.5371058.x.
Thomas NA, Mueller S, Klein A, Jarrell KF: Mutants in flaI and flaJ of the archaeon Methanococcus voltae are deficient in flagellum assembly. Mol Microbiol. 2002, 46 (3): 879-887. 10.1046/j.1365-2958.2002.03220.x.
Thomas NA, Pawson CT, Jarrell KF: Insertional inactivation of the flaH gene in the archaeon Methanococcus voltae results in non-flagellated cells. Mol Genet Genomics. 2001, 265 (4): 596-603. 10.1007/s004380100451.
Patenge N, Berendes A, Engelhardt H, Schuster SC, Oesterhelt D: The fla gene cluster is involved in the biogenesis of flagella in Halobacterium salinarum. Mol Microbiol. 2001, 41 (3): 653-663. 10.1046/j.1365-2958.2001.02542.x.
Faguy DM, Bayley DP, Kostyukova AS, Thomas NA, Jarrell KF: Isolation and characterization of flagella and flagellin proteins from the thermoacidophilic archaea Thermoplasma volcanium and Sulfolobus shibatae. J Bacteriol. 1996, 178 (3): 902-905.
Wang YA, Yu X, Silverman PM, Harris RL, Egelman EH: The Structure of F-Pili. J Mol Biol. 2009, 385 (1): 22-29. 10.1016/j.jmb.2008.10.054.
Walsby AE: Gas vesicles. Annu Rev Plant Physiol Plant Mol Biol. 1975, 26: 427-439. 10.1146/annurev.pp.26.060175.002235.
Chu LJ, Chen MC, Setter J, Tsai YS, Yang HY, Fang XF, Ting YS, Shaffer SA, Taylor GK, von Haller PD, et al: New Structural Proteins of Halobacterium salinarum Gas Vesicle Revealed by Comparative Proteomics Analysis. J Proteome Res. 2011, 10 (3): 1170-1178. 10.1021/pr1009383.
Baker BJ, Comolli LR, Dick GJ, Hauser LJ, Hyatt D, Dill BD, Land ML, VerBerkmoes NC, Hettich RL, Banfield JF: Enigmatic, ultrasmall, uncultivated Archaea. Proc Natl Acad Sci USA. 2010, 107 (19): 8806-8811. 10.1073/pnas.0914470107.
Goltsman DSA, Denef VJ, Singer SW, VerBerkmoes NC, Lefsrud M, Mueller RS, Dick GJ, Sun CL, Wheeler KE, Zemla A, et al: Community genomic and proteomic analyses of chemoautotrophic iron-oxidizing “Leptospirillum rubarum” (Group II) and “Leptospirillum ferrodiazotrophum” (Group III) bacteria in acid mine drainage biofilms. Appl Environ Microbiol. 2009, 75 (13): 4599-4615. 10.1128/AEM.02943-08.
Zerbino DR, Birney E: Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18 (5): 821-829. 10.1101/gr.074492.107.
Gordon D, Abajian C, Green P: Consed: A graphical tool for sequence finishing. Genome Res. 1998, 8 (3): 195-202. 10.1101/gr.8.3.195.
Katoh K, Toh H: Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008, 9 (4): 286-298. 10.1093/bib/bbn013.
Katoh K, Misawa K, Kuma K, Miyata T: MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30 (14): 3059-3066. 10.1093/nar/gkf436.
Price MN, Dehal PS, Arkin AP: FastTree 2-Approximately maximum-likelihood trees for large alignments. PLoS One. 2010, 5 (3): e9490-10.1371/journal.pone.0009490.
Price MN, Dehal PS, Arkin AP: FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009, 26 (7): 1641-1650. 10.1093/molbev/msp077.
Stamatakis A: RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006, 22 (21): 2688-2690. 10.1093/bioinformatics/btl446.
Lin K, Simossis VA, Taylor WR, Heringa J: A simple and fast secondary structure prediction method using hidden neural networks. Bioinformatics. 2005, 21 (2): 152-159. 10.1093/bioinformatics/bth487.
Drennan CL, Heo JY, Sintchak MD, Schreiter E, Ludden PW: Life on carbon monoxide: X-ray structure of Rhodospirillum rubrum Ni-Fe-S carbon monoxide dehydrogenase. Proc Natl Acad Sci USA. 2001, 98 (21): 11973-11978. 10.1073/pnas.211429998.
Acknowledgements
We thank Mr. Ted Arman (President, Iron Mountain Mines), Mr. Rudy Carver, and Mr. Richard Sugarek for site access and other assistance. This work was supported by DOE Genomics: GTL project Grant No. DE-FG02-05ER64134 (Office of Science). APY acknowledges NSF Graduate Research Fellowship Program support. LRC also acknowledges support by the Director, Office of Science, Office of Biological and Environmental Research, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Genome assemblies were carried out by AY, VD, and JB BT called and annotated genes in these assemblies. AY manually curated these gene annotations and applied the synteny-based annotation method to provide more specific annotations. LC performed the cryo-EM. NJ provided insight into metabolic comparisons. AY did the comparisons between the genomes. CC did the protein structural alignments. The manuscript was written by AY and JB. All authors read and approved the manuscript.
Electronic supplementary material
12864_2013_5234_MOESM1_ESM.xls
Additional file 1: Percent nucleotide identity of 16S rRNA genes in the AMD plasmas relative to one another.(XLS 176 KB)
12864_2013_5234_MOESM2_ESM.xls
Additional file 2: 16S rRNA nucleotide identity for AMD Thermoplasmatales organisms and close relatives. Note that all of the organisms in the first column except for Aciduliprofundum boonei are classified as Thermoplasmatales. (XLS 146 KB)
12864_2013_5234_MOESM6_ESM.xls
Additional file 6: Gene order conservation between the AMD plasma genomes. Synt/Orth indicates the number of syntenous orthologs divided by the total number of orthologs. (XLS 20 KB)
12864_2013_5234_MOESM7_ESM.xls
Additional file 7: Average length of syntenous blocks of genes between the AMD plasma genomes. Synt Block indicates the average number of genes of syntenous blocks of genes in each pairwise comparison. (XLS 20 KB)
12864_2013_5234_MOESM8_ESM.xls
Additional file 8: Estimate of genome completeness based on orthologous marker gene homologs. Note that genome estimates of 100% are not exact. These genomes still contain gaps between contigs. (XLS 30 KB)
12864_2013_5234_MOESM9_ESM.jpeg
{kind=link}
Additional file 9: Metabolic and structural features of the AMD plasma organisms. The surface layer proteins are pink. Pili are blue. Flagella are brown. The electron transport chain is yellow. The metal resistance proteins are blue. The archaeal type ATP synthase is yellow. Sulfocyanin is yellow and rusticyanin is blue. (JPEG 10 MB)
12864_2013_5234_MOESM10_ESM.xls
Additional file 10: Cluster of unique genes in Gplasma. PUF indicates a protein of unknown function. Bold font indicates gene numbers for proteins detected in proteomic data. (XLS 22 KB)
12864_2013_5234_MOESM12_ESM.xls
Additional file 12: Genes of metabolic and structural importance in the AMD plasma genomes. * indicates a putative annotation. ** indicates a probable annotation. *** indicates a possible annotation. Gray indicates additional evidence of function via synteny analysis. Bold font indicates gene numbers for proteins detected in proteomic data. “split” indicates a split gene. “fusion” indicates a fused gene. (XLS 72 KB)
12864_2013_5234_MOESM13_ESM.jpeg
{kind=link}
Additional file 13: Structural alignment of blue copper proteins. β-Strands (cupredoxin fold) predicted by YASPIN [118] are highlighted (cyan for β-strand 1, yellow and light green for β-strand 2, pink for β-strand 3, dark blue for β-strand 4, dark green for β-strand 5, purple for β-strand 6 and red for β-strand 7). Amicyanin from Paracoccus denitrificans [GenBank: CAA39199] and Plastocyanin from Synechococcus elongatus GenBank: ABB57 [118] serve as references. Red circles indicate copper-binding ligands. Residues highlighted by light grey correspond to additional β-strands and those in bold orange correspond to α-helices. Sulfocyanin-specific motifs are boxed in red. Black arrows indicate copper-binding ligands. Additional loops are indicated at the bottom of the alignment by a light orange line. (JPEG 1 MB)
12864_2013_5234_MOESM15_ESM.jpeg
{kind=link}
Additional file 15: AMD plasma blue-copper protein tree. bcp indicates a blue-copper protein of unknown function. (JPEG 2 MB)
{kind=link}
12864_2013_5234_MOESM17_ESM.jpeg
{kind=link}
Additional file 17: Active site alignment of aerobic CODH catalytic subunit genes. The red box indicates the active site residues. H. pseudoflava is Hydrogenophaga pseudoflava, O. carboxidovorans is Oligotropha carboxidovorans, M. loti is Mesorhizobium loti, B. japonicum is Bradyrhizobium japonicum, and B. fungorum is Burkholderia fungorum. (JPEG 244 KB)
12864_2013_5234_MOESM18_ESM.jpeg
{kind=link}
Additional file 18: Ni-CODH catalytic subunit alignment. Genes in this alignment are the Ni-CODH catalytic subunits from R. rubrum (CooS, PDB:1JQK), M. thermoacetica (AcsA, PDB:1MJG) and Fer2 (fer2_31_0047). fer2_31_0047’s secondary structure was predicted by YASPIN [118]. β-strands are shown in green and α-helices are highlighted in cyan. Residues belonging to the D-cluster are boxed in yellow (Cys41 and Cys49). Ligands of the B-cluster are boxed in black (Cys50, Cys53, Cys58 and Cys72). Catalytic residues binding the Ni-Fe-S cluster from C-cluster are boxed in purple (His265, Cys300, Cys338, Cys451, Cys481, and Cys531) and catalyze the oxidation of carbon. His95 and Lys568 (boxed in dark red) are non-coordinating residues conserved in Ni-CODHs and have been suggested to be involved in facilitating the reaction [119]. Residue numbering is from the R. rubrum Ni-CODH. (JPEG 2 MB)
12864_2013_5234_MOESM19_ESM.jpeg
{kind=link}
Additional file 19: Cytochrome c oxidase subunit II alignment. * indicates the copper-binding motif found in other cytochrome c oxidase proteins. S. acidocaldarius is Sulfolobus acidocaldarius, A. pernix is Aeropyrum pernix, P. oguniense is Pyrobaculum oguniense, T. thermophilus is Thermus thermophilus, P. denitrificans is Paracoccus denitrificans. (JPEG 445 KB)
12864_2013_5234_MOESM20_ESM.xls
Additional file 20: Amino acid identity of AMD plasma cytochrome c oxidase subunit II genes with closely related genes.(XLS 20 KB)
12864_2013_5234_MOESM22_ESM.jpeg
{kind=link}
Additional file 22: AMD plasma putative hydrogenase 4 gene tree. Accession numbers are to the left of the species names. (JPEG 831 KB)
12864_2013_5234_MOESM23_ESM.xls
Additional file 23: Pili genes in the AMD plasmas. * indicates a putative annotation. ** indicates a probable annotation. *** indicates a possible annotation. Gray indicates additional evidence of function via synteny analysis. “split” indicates a split gene. Bold font indicates gene numbers for proteins detected in proteomic data. (XLS 18 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Yelton, A.P., Comolli, L.R., Justice, N.B. et al. Comparative genomics in acid mine drainage biofilm communities reveals metabolic and structural differentiation of co-occurring archaea. BMC Genomics 14, 485 (2013). https://doi.org/10.1186/1471-2164-14-485
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-14-485