
Abstract
Background
The ABC (ATP-binding cassette) gene superfamily is widespread across all living species. The majority of ABC genes encode ABC transporters, which are membrane-spanning proteins capable of transferring substrates across biological membranes by hydrolyzing ATP. Although ABC transporters have often been associated with resistance to drugs and toxic compounds, within the Arthropoda ABC gene families have only been characterized in detail in several insects and a crustacean. In this study, we report a genome-wide survey and expression analysis of the ABC gene superfamily in the spider mite, Tetranychus urticae, a chelicerate ~ 450 million years diverged from other Arthropod lineages. T. urticae is a major agricultural pest, and is among of the most polyphagous arthropod herbivores known. The species resists a staggering array of toxic plant secondary metabolites, and has developed resistance to all major classes of pesticides in use for its control.
Results
We identified 103 ABC genes in the T. urticae genome, the highest number discovered in a metazoan species to date. Within the T. urticae ABC gene set, all members of the eight currently described subfamilies (A to H) were detected. A phylogenetic analysis revealed that the high number of ABC genes in T. urticae is due primarily to lineage-specific expansions of ABC genes within the ABCC, ABCG and ABCH subfamilies. In particular, the ABCC subfamily harbors the highest number of T. urticae ABC genes (39). In a comparative genomic analysis, we found clear orthologous relationships between a subset of T. urticae ABC proteins and ABC proteins in both vertebrates and invertebrates known to be involved in fundamental cellular processes. These included members of the ABCB-half transporters, and the ABCD, ABCE and ABCF families. Furthermore, one-to-one orthologues could be distinguished between T. urticae proteins and human ABCC10, ABCG5 and ABCG8, the Drosophila melanogaster sulfonylurea receptor and ecdysone-regulated transporter E23. Finally, expression profiling revealed that ABC genes in the ABCC, ABCG ABCH subfamilies were differentially expressed in multi-pesticide resistant mite strains and/or in mites transferred to challenging (toxic) host plants.
Conclusions
In this study we present the first comprehensive analysis of ABC genes in a polyphagous arthropod herbivore. We demonstrate that the broad plant host range and high levels of pesticide resistance in T. urticae are associated with lineage-specific expansions of ABC genes, many of which respond transcriptionally to xenobiotic exposure. This ABC catalogue will serve as a basis for future biochemical and toxicological studies. Obtaining functional evidence that these ABC subfamilies contribute to xenobiotic tolerance should be the priority of future research.
Similar content being viewed by others

Background
ATP-binding cassette (ABC) proteins form one of the largest protein families that are present in all living organisms on earth. The majority of ABC proteins are membrane bound primary transporters, using ATP to translocate substrates across extra- and intracellular membranes. In addition, these ABC transporters are mostly uniporters, mediating the unidirectional translocation of a substrate [1–4]. The Major Facilitator Superfamily (MFS) is another large transporter family present in all living organisms, but as opposed to ABC transporters, it comprises secondary carriers that can be either uniporters, symporters or antiporters [5]. In most ABC proteins two types of domains can be distinguished, an ATP-binding domain (also named the nucleotide binding domain (NBD)) and a transmembrane domain (TMD). The highly conserved NBD contains three motifs: a Walker A and Walker B domain and the ABC signature (LSSG-motif). The NBD binds and hydrolyses ATP and provides energy to transport substrates. The TMD consists of five to six membrane spanning helices and provides the specificity for the substrate. Full transporters comprise two NBDs and two TMDs while half transporters have only one of each type and require homo- or heterodimerization to form a functional unit [1–4]. Based on the homology of their NBDs, ABC proteins have been divided into seven subfamilies, ABCA to ABCH [1, 6]. Interestingly, the ABCH subfamily was discovered during analysis of the Drosophila melanogaster genome and is present in all sequenced arthropod genomes to date and teleost fish, but not in mammals, plants or fungi [6–16].
In humans, ABC proteins mainly function in the membrane transport of substrates, including amino acids, sugars, lipids, inorganic ions, polysaccharides, metals, peptides, toxic metabolites and drugs [2, 4]. In addition to transporters, the human ABC protein superfamily also contains ion channels (CFTR), receptors (SUR1 and 2) and proteins involved in translation (human ABCE and ABCF1, 2 and 3) [1]. Mutations in ABC genes have been linked to several human disorders, like cystic fibrosis, adrenoleukodystrophy, sitosterolemia and diabetes [17, 18]. Furthermore, within the ABCB, C and G subfamilies, many genes code for proteins that contribute to resistance of cancer cells against chemotherapeutic agents: the multidrug resistance proteins or P-glycoproteins (MDR or P-gps, members of the ABCB subfamily), the multidrug resistance-associated proteins (MRP, members of the ABCC subfamily) and the breast cancer protein (BCRP or human ABCG2) [19, 20]. In insects, it has been shown that ABC transporters have functions that affect metabolism, development and resistance to xenobiotics including insecticides and plant secondary toxic compounds (allelochemicals) [14]. Some ABC transporters have specific functions that are well documented in arthropods. In D. melanogaster the ABCC transporter Mdr49 controls the export of a germ cell attractant [21]. D. melanogaster white, on the other hand, is a member of the ABCG subfamily and is involved in the uptake of pigment precursors in the developing eye [22]. Its orthologs in Bombyx mori (Bmwh3) and T. castaneum (TcABCG-9B) have similar functions, and w-3oeB. mori mutants and TcABCG-9B dsRNA injected adult beetles have white eyes [8, 23]. In the tobacco hornworm, Manduca sexta, orthologs of human P-gps are essential as they prevent the influx of nicotine across the blood brain barrier [24, 25]. Insect orthologs of human P-gps and MRPs have also been frequently linked to pesticide resistance [26–29]. Resistance to pesticides in insects is either related to reduced target-site sensitivity or sequestration/metabolism of the pesticide before it reaches the target site by quantitative or qualitative changes of genes involved in the detoxification process [30, 31]. These “detoxification” genes comprise members of the P450 mono-oxygenases (P450s), glutathione-S-transferases (GSTs), carboxyl/cholinesterases (CCEs) and also the less known ABC transporters. Although the ABC transporters have often been overlooked in studies that describe the detoxification toolkit in sequenced insect genomes [32–34], clear examples of their importance in detoxification have been documented. For example, Lanning et al. [28] showed that increased expression of human P-gp orthologues in H. virescens was associated with resistance to thiodicarb, and a mutation in the same ABCC member of four different lepidopteran species was recently associated with resistance to the Cry1A toxin [27, 29].
Complete and correctly annotated gene inventories are a prerequisite to study the biological role and evolutionary history of ABC genes. Among arthropods, detailed studies of ABC families have been published for members of several different insect orders [1, 8, 9, 12, 14, 16] and the crustacean Daphnia pulex[11]. In contrast, besides the identification of nine ABC genes in the mange mite Sarcoptes scabiei[35], there are no reported studies in the subphylum Chelicerata (spiders, scorpions, mites and ticks), one of the most diverse groups of terrestrial animals [36]. Recently, the first published draft genome sequence of a chelicerate, the two-spotted spider mite, Tetranychus urticae, was reported [37]. The spider mite is one of the most polyphagous herbivores known, and has been documented to feed on more than 1,100 plant species that belong to more than 140 different plant families, including many that produce toxic compounds [38, 39]. In addition, spider mites are major agricultural pests and are the “resistance champion” among arthropods as they have the most documented instances of resistance to diverse pesticides [31, 40]. The molecular mechanisms underlying spider mite resistance to xenobiotics (pesticides and allelochemicals) are less understood compared to insects [31, 41]. However, the availability of the draft genome sequence now provides unique information and tools for the study of the role of gene families involved in xenobiotic metabolism in spider mites [42–44]. Characterization of spider mite gene families associated with detoxification of xenobiotics, including ABC genes, is the first step towards a better understanding of how spider mites cope with these compounds [44]. An initial preliminary analysis of ABC genes in the spider mite genome focused solely on ABCB and ABCC subfamilies [37], and lacked a complete description of the phylogenetic relationships with other metazoan ABCs. In this study, we provide a detailed comparison of all ABC subfamilies (ABCA-ABCH) in T. urticae with those of the insect D. melanogaster, the crustacean D. pulex, the nematode Caenorhabditis elegans and the mammal Homo sapiens. Further, we show that expression levels of ABC genes change in pesticide resistant strains and when new and challenging plant host are encountered. Our analyses will facilitate biochemical and toxicological studies of the role of T. urticae ABC transporters in spider mite physiology, and in particular the extraordinary host range and pesticide resistance development.
Results and discussion
Identification of spider mite ABC genes
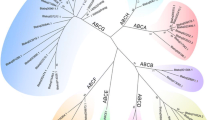
We identified 103 putative ABC genes in the genome of T. urticae (Table 1). To our knowledge, this is the largest number of ABC genes reported for any metazoan species so far [8, 9, 13, 45]. Of all organisms sequenced to date, only the protozoan ciliate Tetrahymena thermophila has more ABC genes [46]. A maximum likelihood phylogenetic analysis grouped the T. urticae ABC proteins into each of the eight known ABC subfamilies with high bootstrap support (Figure 1, Additional file 1). We identified 9, 4, 39, 2, 1, 3, 23 and 22 ABC proteins belonging to the ABCA, ABCB, ABCC, ABCD, ABCE, ABCF, ABCG and ABCH subfamilies, respectively (Table 2). Significant homology (E-value ≤ e-4) with one of the 103 putative T. urticae ABC genes was found at an additional 41 loci in the T. urticae genome. However, gene models at these loci, most of which had homology to the ABCC or ABCH subfamilies, lacked one or both vital domains (NBDs, TMD) of canonical ABC genes. These likely represent gene fragments or pseudogenized genes, and were excluded from detailed analysis (Additional file 2). From the 103 full-length T. urticae ABC genes, almost half (48) are located on only 5 genomic scaffolds (11, 9, 6, 11 and 10 ABC genes on scaffold 1, 3, 4, 9 and 11, respectively). In addition, a high complexity was observed within gene structures, with exon numbers ranging from 1 to 20 (Table 1). To determine the accurate evolutionary position of the 103 T. urticae ABC proteins, phylogenetic analyses, including full-length ABC protein sequences from the draft genomes of T. urticae and D. pulex and from the finished genomes of C. elegans, D. melanogaster and H. sapiens, were performed for each subfamily separately. The results of these analyses are discussed below.

Unrooted phylogenetic tree of N - terminal NBDs of 103 ABC proteins of T . urticae. Amino acid sequences of NBDs were aligned using MUSCLE [122] and subjected to a maximum likelihood analysis using Treefinder [124]. For amino acid alignment, amino acid substitution model and likelihood score of the constructed phylogenetic tree see Additional file 9 and Additional file 10. Numbers at important nodes represent the bootstrap values resulting from 1000 pseudoreplicates (LR-ELW). The scale bar represents 0.5 amino-acid substitutions per site. The different ABC protein subfamilies are indicated by shaded colors. T. urticae ABC protein sequences can be found in Additional file 12.
The ABCA family comprises 9 full transporters (hereafter FTs) in T. urticae. In contrast to plants and some insects, no ABCA half transporters (hereafter HTs) were identified (Table 1) [9, 12, 47]. The T. urticae ABCA subfamily contains the largest T. urticae ABC protein, tetur25g01640 (2302 amino acids) (Table 1). ABCAs share a distinct set of characteristics across species: an extracellular loop between the first and second transmembrane helices (TMs), a conserved motif downstream of each NBD [48] and a conserved motif at the N-terminus (xLxxKN, [49]). All T. urticae ABCAs have a characteristic extracellular loop between the first and the second TMs of each TMD (see Additional file 3 for position of TMs). The conserved motif downstream of each NBD was, except for tetur25g01640, present in all T. urticae ABCAs, while the N-terminus conserved motif could only be found in a single T. urticae ABCA (tetur25g01640). Instead of xLxxKN, the remainder of T. urticae ABCAs harbor either a xMxxKD/S (7) or xLxxHR (1) N-terminal motif. A phylogenetic analysis of metazoan ABCAs is shown in Additional file 4. Six T. urticae ABCAs (tetur01g00580, tetur11g05030, tetur11g05040, tetur11g05200, tetur15g01990 and tetur30g01960) clustered together with high bootstrap support. These six ABCAs show high amino acid identity (62.3-94.1%, Additional file 5) and have identical exonic structure (Additional file 3), indicating they might have arisen by recent duplication events. Together with tetur01g15090, they form a sister-group with D. melanogaster CG31731, an ABCA reported to be down regulated in the salivary glands of an E93 mutant of D. melanogaster ([50]; the early ecdysone responsive gene E93 is a critical regulator of programmed cell death during D. melanogaster metamorphosis). D. melanogaster CG31731 and the seven T. urticae ABCA genes cluster together, albeit with moderate bootstrap support, with a group of C. elegans ABCA transporters. The latter contains Ced-7, which is involved in the engulfment of cell corpses during programmed cell death in C. elegans[51].
Further, tetur27g01890 and D. melanogaster CG34120 form a sister clade of human ABCA12 and ABCA13, while tetur25g01640, D. pulex Dappu1-312055 and Dappu1-312056 cluster with human ABCA1, ABCA2, ABCA4 and ABCA7 (Additional file 4). These human ABCAs contain conserved predicted N-glycosylation sites at N400, N1453 and N1637 of human ABCA1 [48]. In addition, it has been experimentally shown that D. melanogaster CG34120 is also glycosylated at an asparagine (N272, at NASFEEL motif of CG34120 [52]) aligning with one of these conserved sites (N400 of human ABCA1). The tetur27g01890 and tetur25g01640 proteins also have many predicted N-glycosylation sites (Table 1) of which at least one (N303 in tetur27g01890; N1705 in tetur25g01640) is shared with those conserved in human ABCA1, 2, 4, 7 and 12. In humans, these ABCAs have highly specialized roles in phospho- and sphingolipid export [48]. For example, human ABCA1 controls the initial steps leading to high-density lipoprotein (HDL) formation at the cell membrane and is crucial for reverse cholesterol transport from peripheral tissues to the liver [53]. Human ABCA12 works as an epidermal keratinocyte lipid transporter and a defective ABCA12 results in loss of the skin lipid barrier [54, 55]. Although we cannot assign such highly specific roles to the two T. urticae ABCA orthologues above, they may also be involved in lipid transport processes.
The ABCB subfamily consists of 2 FTs and 2 HTs in T. urticae (Table 1). A phylogenetic analysis of ABCB FTs revealed that transporters of each species in the analysis clustered into separate clades, confirming an earlier hypothesis by Sturm et al. that this subfamily has diversified through lineage-specific duplications [11] (Figure 2A). This diversification hypothesis is supported in mites by the fact that the T. urticae ABCB FTs, tetur11g04030 and tetur11g04040, have well-supported phylogenetic clustering, similar exon patterns (15 and 16 exons for tetur11g04030 and tetur11g04040, respectively (see Additional file 3)) and high amino acid identity (64.8%, see Additional file 5). T. urticae ABCB FTs form a sistergroup to a clade of C. elegans, H. sapiens and D. melanogaster ABCB FTs. The function of most members of this clade has been well documented in literature. Human ABCB FT, originally termed P-glycoproteins (P-gps) but now also known as multiple drug resistance (MDR) proteins, are among the best characterized ABC pumps and have been shown to be involved in transport of hydrophobic substrates including drugs, lipids, steroids, xenobiotics and peptides (Dean et al. 2001). The precise role of their orthologues in Drosophila has been a focus of recent study. D. melanogaster Mdr65 has been shown to function as an orthologue of human ABCB1/MDR1, a major ABC transporter of cytotoxic xenobiotics at the human blood–brain barrier, and is required for chemical protection of the fruitfly brain [56] while Mdr49 has been shown to be essential in germ cell migration [21]. Interestingly, arthropod ABCB FT orthologues have frequently been linked to pesticide resistance [14, 26]. For example, inhibition of a H. virescens orthologue of human ABCB1 by the P-gp inhibitor quinidine decreased the toxicity of thiodicarb by 12.5-fold in a resistant strain, compared to 1.8-fold in a susceptible strain [28]. Recently, it was found that pretreatment of D. melanogaster with the P-gp inhibitor verapamil reduced the toxicity of DDT by 10-fold in a resistant strain [57]. The involvement of P-gps in pesticide resistance is probably best documented for ivermectin resistance. This compound has been shown to be a substrate for both mammalian as insect-pgps and several cases of P-gp associated ivermectin resistance have been reported [26, 58–60].

Phylogenetic analysis of ABCB full and half transporters of five metazoan species: (A) ABCB full transporters, (B) ABCB half transporters. Full-length ABC proteins were aligned using MUSCLE [122] and subjected to a maximum likelihood analysis using Treefinder [124]. The resulting tree was midpoint rooted. For amino acid alignment, amino acid substitution model and likelihood score of the constructed phylogenetic tree see Additional file 9 and Additional file 10. Numbers at the branch point of each node represent the bootstrap value resulting from 1000 pseudoreplicates (LR-ELW). Species, abbreviations, and color codes are: Ce, C. elegans (purple); h, H. sapiens (green); Dm, D. melanogaster (red); Dp, D. pulex (yellow); and tetur, T. urticae (blue). The scale bar represents 0.2 and 0.5 amino-acid substitutions per site in Figure 2A and Figure 2B, respectively. Accession numbers of metazoan ABC protein sequences can be found in Additional file 11 while T. urticae ABC protein sequences can be found in Additional file 12.
A phylogenetic analysis of ABCB HTs revealed, as was also shown by Sturm et al. [11], clear orthologous relationships between ABCB HTs, suggesting they have evolutionary conserved roles in metazoan species (Figure 2B). In the case of T. urticae ABCB HTs, an orthologous relationship between tetur32g01330 and D. melanogaster CG7955, D. pulex Dappu1-347270, C. elegans ABTM-1 and human ABCB7 was found, while tetur17g02000 groups together with D. melanogaster CG1824, D. pulex Dappu1-347266, C. elegans Haf-6, and human ABCB8. As both tetur32g01330 and tetur17g02000 are predicted (data not shown) to have a mitochondrial targeting signal, these T. urticae transporters are most likely trafficked to the mitochondria, as has been demonstrated for their human orthologues (human ABCB7 and ABCB8) [61]. This suggests that tetur32g01330 and tetur17g02000 fulfill a similar role as their human orthologues. The human ABCB7 protein plays a crucial role in iron homeostasis in the cytoplasm and mutations in this gene have been linked to several diseases [61]. Recently, it was also shown that disruption of the C. elegans orthologue (ABTM-1) of human ABCB7 induced oxidative stress and premature cell death [62]. Furthermore, an orthologue (GenBank acc. No. AAEL006717) of human ABCB7 in the dengue vector, Aedes aegypti, was reported to be upregulated in an insecticide resistant strain [9, 63]. The function of human ABCB8 is not well understood, but it was shown to mediate resistance against the chemotherapeutic agent doxorubicin in melanoma cells [64, 65]. Also, Ichikawa et al. found that disruption of the mouse orthologue of human ABCB8 lead to cardiomyopathy and decreased mitochondrial iron export [66]. Orthologues of the remaining human mitochondrial transporters, ABCB6 and ABCB10, could be identified in D. pulex, D. melanogaster and C. elegans but were not found in T. urticae[1, 11] (Figure 2B). Interestingly, the localization of human ABCB6 is currently under debate, as some studies suggest that it is located in lysosomes [67]. Finally, similar to Sturm et al. [11], we did not identify arthropod orthologues of human ABCB HTs related to antigen processing (human ABCB2, ABCB3 and ABCB9) (Figure 2B).
The ABCC subfamily consists of 39 transporters in T. urticae. To our knowledge, this is the largest number of ABCC transporters reported in any metazoan species, including the flour beetle, T. castaneum, which also has an exceptionally large number of ABCCs [8, 9] (Table 2). ABCC proteins are FTs, bearing 2 TMDs and 2 NBDs, with diverse functions: ion transport, cell-surface receptor activity and translocation of a broad array of substrates like drugs, cyclic nucleotides, endogenous compounds and their glutathione conjugates and glutathione [19, 68–70]. Because of their ability to extrude drugs, many of these ABCCs are also termed multidrug resistance associated proteins (MRPs) [1]. MRPs can be categorized according to the presence or absence of a third N-terminal transmembrane-spanning domain (TMD0) . In humans, “long” MRPs like ABCC1, 2, 3, 6 and 10 (MRP1, 2, 3, 6 and 7, respectively) have such a TMD0 while “short” human MRPs like ABCC4, 5, 11 and 12 do not (MRP4, 5, 8 and 9, respectively) [71]. In addition to MRPs, the ABCC family also harbors the cystic fibrosis transmembrane conductance regulator (CFTR, human ABCC7) and sulfonylurea receptors (human ABCC8/SUR1 and ABCC9/SUR2) [1].
In our phylogenetic analysis, 23 T. urticae ABCCs clustered with D. melanogaster CG6214, D. pulex Dappu1-347281 and a group of human “long” MRPs (MRP1, 2, 3 and 6) (Figure 3). Twenty-two of the transporters (size ranging from 1481 to 1529 amino acids) from this T. urticae ABCC clade also have a TMD0, while we could not identify this additional transmembrane domain in tetur28g01950 (1287 amino acids) (Table 1), Additional file 3). Within this T. urticae ABCC clade, transporters clustered into 2 distinct groups. One group (“group 1”) consists of 8 T. urticae ABCC genes, each with 7 introns, and tetur04g04360, which has only 4 introns (see Additional file 3). Interestingly, tetur04g04360 is positioned at the basal node, indicating that intron gain events may have occurred in this group (Figure 3). The other group (“group 2”) consists of 14 T. urticae ABCC genes: 12 with 10 introns, tetur25g01780 with 11 introns and tetur28g01950 having only 6 introns (Additional file 3). The intron loss in tetur28g01950 can be directly linked to the lack of the TMD0 (see above, Additional file 3), while the nature of the intron gain event in tetur25g01780 is unclear (the extra intron contains stop codons in each frame, and is located in a non conserved region). Human MRP1, MRP2, MRP3 and MRP6 along with D. melanogaster CG6214 and D. pulex Dappu1-347281 form a sister clade of T. urticae ABCC groups 1 and 2 (Figure 3). These proteins have been extensively studied as transporters of natural product drugs like anthracyclines and plant alkaloids [72]. MRPs also share many substrates with human P-gps (see above) but, while P-gps transport drugs in their original form, MRPs mostly transport their glucuronate, sulfate and glutathione (GSH) conjugates. In the latter case, GSH is fused by GSTs with xenobiotics or their metabolites and finally transported out of the cell by MRPs [46, 72, 73]. In humans, GSTs from the alpha, mu and pi-class have been reported to act in synergy with MRPs [74]. Intriguingly, a clear expansion of GSTs in the T. urticae genome was found for the delta and mu GST subclasses, the latter of which is not present in insects and until recently was believed to be vertebrate specific [37, 75]. Future studies should point out if there is a coordinated action between the GSTs of these subfamilies and the many MRP orthologues of T. urticae.

Phylogenetic analysis of ABCC proteins of five metazoan species.
See the legend of Figure 2 for procedure and display details.
Most of the biochemical properties of human MRP1 have been confirmed in its D. melanogaster orthologue CG6214 [76, 77]. Recently, it was also found that exposure to the P450 mono-oxygenase inhibitor piperonylbutoxide and the antimetabolite/antifolate drug methotrexate alters the expression (27-fold and 1100-fold upregulation, respectively) of the D. melanogaster CG6214 gene in the Malpighian tubuli, which are organs known to play important roles in excretion and xenobiotic detoxification [78, 79]. The orthologue (PhABCC4) of CG6214 in the human body louse, Pediculus humanus, has also been reported to be upregulated after exposure to the pesticide ivermectin. Moreover, injection of PhABCC4 dsRNA in P. humanus female lice increased their sensitivity to ivermectin by 20-30% [60].
Fourteen spider mite ABCCs clustered with high bootstrap support in a group with 11 D. melanogaster ABCC proteins, human ABCC4/MRP4 and ABCC7 and D. pulex Dappu1-347288 (Figure 3). All fourteen T. urticae ABCCs show the structural properties of “short” MRPs (see above, Table 1), Additional file 3). Five of these T. urticae ABCCs group as a sister clade (“group 3”) of the 11 D. melanogaster ABCC proteins (Figure 3). Detailed physiological roles for most of the 11 D. melanogaster ABCC proteins are unknown. D. melanogaster CG10505 is regulated by heavy metals through the metal-responsive transcription factor 1 (MTF-1) and contributes to metal homeostasis [80]. D. melanogaster CG14709 controls responsiveness to O2 deprivation and might also be involved in oxidative stress response [81, 82] while 31% of embryos of D. melanogaster CG7627 mutants were unable to heal wounds 16 h postwounding [83]. Recently, it was also shown that D. melanogaster CG4562, which is highly expressed in the midgut, was upregulated in Cyp6g1 knockdown flies, indicating molecular crosstalk within a detoxification network [84]. Furthermore, a point mutation in lepidopteran homologs of this D. melanogaster ABCC group has been clearly linked with resistance against the B. thuringiensis Cry1A toxin [27, 29]. Another 9 T. urticae ABCCs (“group 4”) form, together with D. pulex Dappu1-347288 and the 11 D. melanogaster and five T. urticae ABCCs (“group 3”, see above) a sister clade of human ABCC4 and ABCC7 (Figure 3). The function of D. pulex Dappu1-347288 is not known, but human ABCC7/CFTR acts as a chloride channel, a unique function not found in any other ABC transporter [69]. In our analysis, CFTR clustered with human ABCC4/MRP4, which is its closest ABC paralog according to Jordan et al. [69]. Human MRP4 (and MRP5) have the ability to transport a range of endogenous molecules involved in cellular signaling, like cyclic nucleotides, eicosanoids and conjugated steroid hormones [19]. As a drug transporter, MRP4 also stands out for its broad substrate specificity, covering antiviral, antibiotic, cardiovascular and cytotoxic agents [85].
Interestingly, many transporter genes in T. urticae ABCC groups 1–4 form closely related sister groups and show high amino acid identity between their corresponding protein sequences (see Additional file 5). Together with their conserved exon pattern (see Additional file 3), this strongly suggests that multiple tandem duplications underlie the proliferation of these genes. This is in contrast to the crustacean D. pulex, which has only few ABCC genes [11], and more closely resembles what is generally observed for insects, where gene duplication of ABCC genes occurs frequently [8, 9, 12].
Furthermore, clear orthologous relationships were found for the two remaining T. urticae ABCCs: tetur03g07840 and tetur11g05990. Tetur03g07840 clustered as an orthologue of D. melanogaster CG7806, D. pulex Dappu1-347323 and human ABCC10/MRP7 (Figure 3). Similar to its orthologues, tetur03g07840 has a TMD0[11, 86] (Table 1), Additional file 3). The functions of D. melanogaster CG7806 and D. pulex Dappu1-34723 are not known. A growing understanding of the physiological role of human ABCC10/MRP7 is, on the other hand, beginning to emerge. Human MRP7 is distinct from other human ABCCs in that it shows little or no activity towards glutathione, sulfate conjugates and cyclic nucleotides, substrates that can be handled by other human MRPs (see above). Instead, human MRP7 is able to confer resistance to taxanes (which are diterpenes originally derived from plants of the genus Taxus and widely used as chemotherapy agents (e.g. docetaxel)) [86]. However, the presence of one-to-one orthologues in other metazoans, might indicate a more conserved function of this ABCC protein in this group of species.
Tetur11g05990 is located in the same clade as D. pulex, human and D. melanogaster sulfonylurea receptors (SURs). In contrast to vertebrates, the N-terminal SUR Interpro-motif (IPR000388) is not present in tetur11g05990 and other arthropod SURs (see OrthoDb group EOG531ZCW, [87]). However, the presence of a TMD0 typical for SURs and “long” MRPs [71], and the well-supported clustering with human ABCC8/SUR1 and ABCC9/SUR2 support the idea that tetur11g05990 is a SUR homologue. Four SUR subunits assemble into an octameric complex with four pore-forming subunits, characteristic for inwardly rectifying potassium (Kir) channels, to form ATP-sensitive potassium (KATP) channels [68]. Three orthologues of these pore- forming subunits were also found in the T. urticae genome (tetur17g01380, tetur24g01270 and tetur24g01280 having a BLASTx E-value of 1e-99, 4e-103 and 3e-103 with Ir (CG44159) of D. melanogaster), suggesting that a functional KATP channel can be formed in T. urticae. KATP channels are involved in multiple physiological processes, with roles in glucose homeostasis, ischemic protection and innate immunity [68, 88]. Intriguingly, in 2004 it was suggested that the SUR was the direct target of benzoylureas, a group of chitin synthesis inhibitors [89]. This was largely based on similar effects of glibenclamide, a well-known SUR inhibitor in humans and anti-diabetic drug, on the inhibition of chitin synthesis. However, it was later shown by Gangishetti et al. [90] that SUR is not expressed in the D. melanogaster epidermis, where chitin disruption is observed. Recently, based on genetic mapping of etoxazole resistance genes, it was suggested that the action of chitin synthesis inhibitors is mediated by a direct interaction with chitin synthase, a processive glycosyl transferase [43]. The lack of a role for SUR in chitin production, transport or metabolism is further confirmed by recent studies, where it was shown that the SUR receptor is dispensable for chitin synthesis in D. melanogaster[91], and RNAi knockdown of its orthologue in T. castaneum did not result into a phenotype [8]. Elucidating the role of SUR in T. urticae will therefore require additional studies.
Finally, no orthologues of human ABCC5, 11 and 12 were identified in T. urticae, although three orthologues were found in the genome of D. pulex (Figure 3), confirming earlier findings by Sturm et al. [11]. Surprisingly, a single nucleotide polymorphism in human ABCC11 was identified as the determinant of the human earwax type [92]. However, the potential roles of related transporters in other organisms (such as D. pulex) are not clear.
The ABCD subfamily harbors HTs that in humans are located in the peroxisome where they are involved in the import of long and branched chain acyl-coA into this organelle [93]. The T. urticae genome has 2 ABCD genes, tetur05g06640 and tetur35g01360 (Table 1). This number of ABCD genes equals those found in insects [9] while 3, 4 and 5 are found in the genomes of D. pulex, H. sapiens and C. elegans, respectively (Table 1). T. urticae ABCDs carry the EAA-like motif between TM4 and TM5 and the loop1 motif (L105, R108 and T109 (S. cerevisiae numbering)), both considered to be essential for canonical ABCD function [94]. The clear orthologous relationships we identified between T. urticae ABCDs and other metazoan ABCDs (Additional file 6) suggests that the function of T. urticae ABCDs is likely to be conserved with those in other metazoans.
The ABCE and F proteins are characterized by two linked NBDs, but lack TMDs and thus are involved in biological processes other than transport. The ABCE protein is essential in all eukaryotes examined to date and is one of the most conserved proteins known [95]. Human ABCE1 was first discovered as an inhibitor of RNase L [96], but was later found to have a more fundamental role in ribosome biogenesis and translation regulation [95]. In line with all eukaryotes to date, we found one ABCE protein (tetur30g01400) in T. urticae (Table 1) and Table 2) that has high amino acid identity (77.1%) with D. melanogaster ABCE1 (pixie) (Additional file 5). Similar to human ABCE, ABCF1 is involved in translation regulation, but probably does not play a role in ribosome biogenesis [97]. In most eukaryotes 3 ABCF genes are found, and T. urticae conforms to this expectation (Liu et al. [9], Table 1) and Table 2). The essential role of ABCE and ABCF genes was recently shown in the flour beetle, T. castaneum, where RNAi–mediated knockdown of members of the ABCE and F families resulted in 100% mortality in penultimate larvae [8].
A phylogenetic analysis of ABCE and ABCF proteins was performed together (Additional file 7). Tetur30g01400 grouped with metazoan ABCE1 orthologues, while each T. urticae ABCF (tetur20g02610, tetur29g00620 and tetur32g00940) clustered into well-supported separate clades with its metazoan orthologues, C. elegans F42A10.1 excluded (Additional file 7). The ABCE and ABCF subfamilies are highly conserved, and T. urticae ABCE1 and ABCFs probably have analogous roles as their orthologues in other metazoans.
The ABCG transporter family is present in most metazoan species, fungi and plants such as Arabidopsis. For metazoan species, only ABCG HTs have been reported to date, while in plants and fungi also ABCG FTs are present [1, 15, 47]. In humans, ABCG HTs are primarily implicated in transport of endogenous and dietary lipids, while the human ABCG2 functions as a multidrug efflux pump [18, 98]. Within the T. urticae genome we identified 23 ABCGs, all having a typical reverse domain organization (the NBD is localized to the N-terminal side of the TMD) (Table 1), see Additional file 3). A similar number of ABCGs has also been found in D. pulex, and is the highest reported among metazoan species [11] (Table 2). According to Sturm et al. [11], the high number of ABCG genes in D. pulex and D. melanogaster genomes is due to extensive lineage specific duplications. Our phylogenetic analysis confirms this hypothesis not only for these two arthropod species but also for T. urticae, with twenty out of 23 ABCGs grouping into one of the two T. urticae specific clades (Figure 4).

Phylogenetic analysis of ABCG proteins of five metazoan species.
See the legend of Figure 2 for procedure and display details.
One clade (bottom of tree, Figure 4) consists of 11 T. urticae ABCGs, each having a maximum of 1 intron (see Additional file 3). Another T. urticae specific ABCG clade comprises 9 transporters of which 7 are located next to each other on scaffold 9. These seven ABCGs show high amino acid identity (between 50.8 and 91.7%) and have a conserved exon pattern (9 exons), indicating a common origin by successive tandem duplication events. Together with tetur06g05430 and tetur02g11270 they form a well-supported sister clade of D. melanogaster white and its D. pulex orthologues. Interestingly, no orthologues of D. melanogaster ABCGs brown and scarlet were found in T. urticae, while only one D. pulex orthologue of scarlet could be identified (Figure 4, [11]). About a century ago, the discovery of D. melanogaster white mutants with a remarkable eye-color phenotype marked the beginning of Drosophila genetics. As a consequence, D. melanogaster white is one of the most intensively studied fruit fly genes [99]. D. melanogaster white dimerises with either D. melanogaster scarlet or brown to form a transporter involved in the uptake of pigment precursors (guanine and tryptophan) in cells of developing compound and simple eyes [22]. T. urticae has, in contrast to D. pulex and D. melanogaster, no compound eyes and only four simple eyes (ocelli) [100]. Although no T. urticae orthologues of scarlet or brown were identified, dimerisation between the nine T. urticae co-orthologues of D. melanogaster white might result in a transporter capable of translocating pigment precursors into the cells of the spider mite ocelli (which have red pigment, as in D. melanogaster). However, these transporters might also have other functions besides transporting pigment precursors, as in other species roles have been documented in courtship behavior [101, 102], transport of biogenic amines [103] and uptake of uric acid [104] as was shown for D. melanogaster white and/or its B. mori orthologue.
In the middle of the ABCG phylogenetic tree, tetur01g16280 clustered with human ABCG8, D. melanogaster CG31121 and D. pulex Dappu1-258299, while tetur01g16290 clustered with human ABCG5, D. melanogaster CG11069, and D. pulex Dappu1-300887. C. elegans orthologues of human ABCG5/8 could not be identified (Figure 4). Similar to human ABCG5/8, D. melanogaster CG31121/CG11069[105] and D. pulex Dappu1-258299/Dappu1-300887[106], tetur01g16280 and tetur01g16290 are found juxtaposed in a head to head orientation. Annilo et al. [7] have suggested an evolutionary constraint on the separation of these genes, probably for the maintenance of shared regulatory regions. In humans, ABCG5 and ABCG8 are both glycoproteins and obligate heterodimers that limit intestinal absorption and promote biliary excretion of neutral sterols [107]. Both tetur01g16280 and tetur01g16290 have at least one well-predicted glycosylation site (Table 1). Together with their head-to-head arrangement and the well-supported clustering with human ABCG8 and 5, it seems likely that these T. urticae ABCGs have similar functions as their human counterparts.
A clear orthologous relationship was found between tetur17g02510, D. melanogaster CG3327 and D. pulex Dappu1-347416. D. melanogaster CG3327, also known as E23 (Early gene at 23), is a 20-OH ecdysone (20E) induced ABC transporter that is capable of regulating 20E responses during metamorphosis, probably by removing 20E from cells [108]. Recently, Broehan et al. [8] showed through RNAi-mediated knockdown experiments and expression profiling that the T. castaneum orthologue of E23 (TcABCG-8A) appears to serve a similar function in metamorphosis. In addition, it is also believed that E23 controls the circadian clock in adult flies through ecdysone-mediated expression of the clock gene vrille[109]. Interestingly, it was shown that not only the B. mori orthologue of E23 (BmABC010557) but also four other midgut-specific B. mori ABCG genes (BmABC005226, BmABC005203, BmABC005202 and BmABC010555) could be induced by 20E [9]. As T. urticae uses a different molting hormone (ponasterone A instead of 20E) compared to arthropods [37], future experiments are required to establish if tetur17g02510 has a similar function as its insect counterparts, and more specifically whether it can be induced by ponasterone A.
T. urticae orthologues of human ABCG1 and 4 were not identified in the phylogenetic analysis of ABCG transporters, and only one was found in D. melanogaster (Atet) and D. pulex (Dappu1-34744) (Figure 4). The function of human ABCG4 is not well understood, while it is proposed that human ABCG1 functions in conjunction with human ABCA1 and is involved in cholesterol homeostasis [18, 98]. The D. melanogaster orthologue of human ABCG1 (Atet) has been poorly characterized, but is expressed in the trachea [110]. Finally, no clear orthologues of human ABCG2 were identified (Figure 4). This transporter is the most thoroughly characterized human ABCG and is capable of transporting an array of substrates, including anticancer drugs [18, 19, 98]. Because of this feature, it has been proposed that arthropod ABCGs could be involved in pesticide resistance [14]. However, to the best of our knowledge there has only been two studies that correlated increased arthropod ABCG expression levels with resistance. In both reports, however, no functional evidence was obtained [13, 111]. In Fungi on the other hand, several cases of ABCG FTs involved in fungicide resistance, have been reported [112].
The ABCH subfamily was first discovered in D. melanogaster and is lacking in mammals, plants or fungi [1, 15, 47]. In addition to arthropods ([1, 8, 9, 11], this study), members of this subfamily have also been reported in teleost fish [6, 7, 10]. Most insects have only 3 ABCH genes, while 15 and 22 are present in D. pulex and T. urticae, respectively ([9, 13], Table 2). Liu et al. [9] suggested that all insect ABCHs diversified from a common ancestral copy. According to our phylogenetic analysis, this insect ancestor seems not to be shared with T. urticae ABCH proteins (Figure 5). Tetranychus ABCHs clustered, similar to D. pulex ABCHs (see also [11]), into a distinct clade, indicating that the diversity of the ABCH family in T. urticae has been due to lineage specific duplications. Interestingly, 16 of the 22 T. urticae ABCHs appear to be intronless (Additional file 3). Although ABCHs have the same structural organization as metazoan ABCGs (HTs, NBD at N-terminal side of TMD), their physiological functions have remained enigmatic. In the zebrafish, D. rerio, ABCH1 has highest expression in brain, gills and kidney followed by lower expression in intestine, gonads, skeletal muscle and liver [10]. D. melanogaster ABCHs are enriched in the adult crop and hindgut [113] and at least one of them (CG9990) is glycosylated as shown by mass spectrometry of N-glycosylated peptides [52]. An RNAi screen of D. melanogaster genes revealed that an RNAi line that silences CG9990 is lethal [114, 115]. In addition, microarray analysis demonstrated an almost two-fold upregulation of a D. melanogaster ABCH (CG33970) after cold hardening of adult fruit flies [116]. In the diamondback moth Plutella xylostella, it was recently found that an ABCH transporter (Px014955, [117]) was the most up-regulated ABC gene in two resistant strains [13]. The most groundbreaking finding about insect ABCH function was just recently reported by the excellent study of Broehan et al. [8]. RNAi-mediated knockdown of an ABCH gene (TcABCH-9C) in T. castaneum larvae resulted in dessication and 100% mortality. Injection of TcABCH-9C dsRNA into adults also drastically reduced the number of eggs laid and all eggs failed to hatch. Furthermore, cryosections of TcABCH-9C dsRNA injected larvae stained with Nile Red (a fluorescent dye that stains lipids) revealed a lack of lipids in the epicuticle. Based on these results, the authors suggested that TcABCH-9C functions as a transporter of lipids to the cuticle and is required for the formation of a waterproof barrier in the epicuticle.

Phylogenetic analysis of ABCH proteins of five metazoan species.
See the legend of Figure 2 for procedure and display details.
Expression profiling of ABC genes
We assessed expression of ABC genes across development in the T. urticae London reference strain, as well as in London after transfer from a benign host (bean, Phaseolus vulgaris) to two more challenging hosts (Arabidopsis thaliana and tomato, Solanum lycopersicum; [37]). For the developmental and host transfer experiments, we used existing RNA-seq reads, but we recalculated gene expression using newly described or corrected ABC gene models curated as part of this study. We further examined previously published microarray data to assess the expression profiles of ABC genes in two spider mite strains, MR-VP and MAR-AB, that are resistant to multiple pesticides [42].
As assessed by RNA-seq expression quantification, the majority of ABC genes were found to be expressed – 88 of the 103 full length T. urticae ABC genes had an RPKM of >1 in at least one of the spider mite life stages or on one of the plant hosts (Figure 6). In contrast, nearly all T. urticae ABC fragments or pseudogenes were not expressed (Additional file 8). Most full-length T. urticae ABC genes for which we detected no expression across development or on different hosts belonged to either ABCA, C or G subfamilies, many of which are tandem duplicated genes ((Table 1), Figure 6). In C. elegans, tandem duplicated genes were shown to be subfunctionalized, with strong stage or tissue dependent expression [118]. Whether T. urticae ABC genes that lacked expression support in the existing data are expressed at low levels, in highly restricted expression domains, or alternatively are expressed under specific environmental conditions (i.e., host plants not included in this analysis), remains to be determined.

Heat map of expression values (RPKM values) of 103 T . urticae ABC genes of mites on different host plants (bean , tomato and Arabidopsis) and from four different life stages (embryo , larvae , nymph and adult). A color legend with corresponding RPKM values is shown at the top of the figure. ABC genes were considered as being expressed when they had an RPKM of >1 in at least one of the spider mite life stages or on one of the plant hosts.
Many T. urticae ABC genes were broadly expressed, and more than half (57 genes, or 55% of the total) were expressed across all developmental stages analyzed (embryos, larvae, nymphs, and adults). However, the expression of many ABCC and ABCH genes was restricted (or at their highest levels) in larvae and nymphs; overall, embryos and adult females had the highest number of non-expressed ABCs (40 and 36 ABC genes in the adult and embryos, respectively (Figure 6)). Furthermore, tetur 30g01400 and tetur 20g02610, members of the ABCE and ABCF subfamilies respectively, showed very high expression in all stages (Figure 6), coinciding with their presumed conserved role in translation regulation (see above). Similar high expression of the T. castaneum ABCE gene was reported in all developmental stages examined [8]. Finally, within the other ABC subfamilies, tetur 27g01890 (ABCA), tetur 11g04030 (ABCB), tetur 04g04360 (ABCC), tetur 05g06640 (ABCD), tetur 02g11270 (ABCG) and tetur 28g00870 (ABCH) had the highest average expression across developmental stages (Figure 6).
Upon host transfer, 22 and 28 ABC genes were differentially expressed in strain London mites transferred from bean to Arabidopsis and tomato plants, respectively (Figure 7; fold change ≥ 2, FDR < 0.05 as calculated for all T. urticae genes, see Methods). We found that 73% (16/22) of the differentially expressed ABC genes in mites transferred to Arabidopsis were also differentially expressed on tomato, and belonged to three subfamilies, ABCC (4), -G (5) and -H (7). Surprisingly, ABC genes differentially expressed in two multi-pesticide resistant T. urticae strains as detected with expression microarrays [42] belonged to the same ABC subfamilies as identified with RNA-seq data in the host transfer experiment (Table 3). The ABCC subfamily has been frequently linked with xenobiotic detoxification in arthropods (see above). On the other hand, arthropod members of the ABCG and H subfamilies have only very recently been reported to be associated with detoxification of xenobiotic compounds (see discussions for the ABCG and ABCH families). The differential expression of members from these ABC subfamilies (G and H) upon host transfer/exposure to xenobiotics should be further (functionally) validated in future studies.

Full length differentially expressed (fold change ≥ 2, FDR adjusted p-value < 0.05) ABC genes in mites after host plant change from bean to either Arabidopsis or tomato. Subfamilies are color-coded as follows: ABCA, red; ABCB, yellow; ABCC, green; ABCE, blue; ABCG, purple; and ABCH, brown. Genes found to be significant in only one of the pairwise comparisons have had their fold change values assigned to zero for the non-significant comparison. Fold change values of differentially expressed ABC genes can be found in Additional file 13.
It is worth noting that the differentially expressed T. urticae ABCC, -G and -H genes mentioned above were not among the most highly differentially expressed genes in these experiments, both in expression level and fold change. In total, 893 and 977 differentially expressed genes were identified in the two multi-resistant strains [42]. Likewise, 2,502 and 3,951 differentially expressed genes were detected in mites when fed on Arabidopsis and tomato in comparison with bean, respectively [37]. The Major Facilitator Superfamily, another large and widespread transporter family [5], showed overall a more pronounced response both in the number of genes differentially expressed, and in the fold change values of differentially expressed genes in both resistant strains and after host plant change [37, 42]. Thus, despite the exceptional number of ABC transporters in the T. urticae genome, other transporters and non-transporter proteins also play key roles in the detoxification of xenobiotics [37, 42].
Conclusions
The spider mite T. urticae is among the most polyphagous pests worldwide and is notorious for its ability to develop resistance against numerous pesticides. One of the prerequisites to study xenobiotic metabolism (pesticides and plant secondary metabolites) in this species is to inventory genes related to detoxification. Here, we provide a survey of the ABC gene superfamily, whose members have frequently been reported to play roles in detoxification, either by directly transporting toxicants out of cells, or after conjugation with glutathione. We identified 103 ABC genes (distributed over eight subfamilies, ABCA-H) in the genome of the spider mite T. urticae. To date, this is the largest number of ABC genes reported in any metazoan species. The large number is mainly due to lineage-specific expansions in subfamilies C, G and H. Of particular note, most of the differentially expressed ABC genes in acaricide resistant strains and after introduction of mites to challenging host plants belong to these expanded ABC subfamilies. This hints at their potential role in detoxification and may explain their retention after duplication in the mite genome. However, obtaining functional evidence that members of these ABC subfamilies contribute to xenobiotic tolerance should be the priority of further research.
Due to the lineage specific expansions in the ABCC, G, and H families, inferring the function of specific T. urticae ABC family members based on phylogenetic relationships is not straightforward. Nevertheless, we found clear orthologous relationships between some of the T. urticae ABC proteins and human ABCC10, ABCG5 and ABCG8, the D. melanogaster sulfonylurea receptor and the ecdysone-regulated transporter E23. Furthermore, we found a high conservation between T. urticae ABC proteins and members of the ABCB-half transporters and ABCD, -E, and –F subfamilies, which are known to be involved in fundamental processes. To conclude, this study provides the first thorough ABC gene analysis of a polyphagous arthropod herbivore and represents a useful resource for future biochemical and toxicological studies on the role of ABC transporters in the extremely broad host range and development of pesticide resistance of T. urticae.
Methods
Annotation and phylogeny of ABC transporters
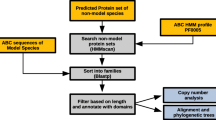
ABCs were identified in a similar way as for D. pulex[11]. Briefly, tBLASTn searches [119] were performed on the T. urticae genome sequence assembly (version July 2012, available at http://bioinformatics.psb.ugent.be/orcae/overview/Tetur[120]) using the highly conserved nucleotide binding domain (NBD) of D. melanogaster ABC proteins as queries. One search was carried out per subfamily, using the sequence of the NBD of a representative D. melanogaster protein (A: CG1718; B: CG3879 (Mdr49); C: CG9270; D: CG12703; E: CG5651; F: CG9330; G: white; H: CG9990). If the D. melanogaster transporter had two NBDs, the N-terminal domain was used. All hits with an E-value less than e-4 were withdrawn for analysis and gene models were refined or created on the basis of homology and RNA-seq support [120]. The NBDs from those T. urticae gene models encoding complete ABCs (i.e. not lacking one or both vital domains (TMD and NBD)) were extracted using the ScanProsite facility [121] and the Prosite profile PS50893. T. urticae ABC protein NBDs were aligned with NBDs of D. melanogaster and human ABC transporters using MUSCLE [122]. Model selection was done with Prottest 2.4 [123]. According to the Akaike information criterion LG+F+G was optimal for phylogenetic analysis (see Additional file 9 and Additional file 10 for protein alignment and likelihood scores, respectively). A maximum likelihood phylogenetic analysis of T. urticae, D. melanogaster and human ABC protein NBDs, bootstrapping with 1000 pseudoreplicates, was performed using Treefinder [124] to confirm the position of T. urticae ABCs within ABC classes (A-G). A similar phylogenetic analysis, restricted to N-terminal NBDs of T. urticae, was also performed (optimal model according to the Akaike Information Criterion: LG+I+G+F, see Additional file 9 and Additional file 10 for protein alignment and likelihood score, respectively). Similar to previous studies, in the phylogenetic analysis using T. urticae, D. melanogaster and human ABC protein NBDs C-terminal NBDs of the ABCC subfamily clustered together with NBDs of the ABCB subfamily [9, 45, 125]. The subfamily assignment was further confirmed by BLASTp analyses of the manually corrected models on the NCBI website. We adopted the guidelines set forth by the human genome organization nomenclature committee (HGNC) for naming the T. urticae ABC proteins. Separate phylogenetic analyses on full ABC protein sequences of T. urticae, D. pulex, C. elegans, D. melanogaster and H. sapiens ABCs were also carried out for each subfamily, using the same methodology as above (for ABC protein alignments see Additional file 9, for model choice and likelihood scores see Additional file 10). According to previous studies [7, 11], this approach facilitates bioinformatics analyses and results in a more meaningful degree of resolution in phylogenetic analysis. Finally, in order to detect ABC pseudogenes/fragments not containing ABC NBDs, all protein sequences of complete ABCs were used as query in tBLASTn-searches against the T. urticae genome. Phylogenetic trees were visualized and edited using MEGA5 [126] and CorelDraw X3 (Corel Inc.), respectively.
Sequence similarity, transmembrane prediction and gene structure of T. urticaeABC proteins
ABC protein sequence similarities and identities were calculated using MatGAT 2.03 [127] using default settings (BLOSUM50 matrix, gap opening and extending gap penalty set to 12 and 2, respectively). Transmembrane domains of T. urticae ABCs were predicted using the SCAMPI prediction server [128]. Subcellular localization was predicted using TargetP 1.0 [129] . Gene structures of T. urticae ABCs were visualized using the coordinates of each T. urticae ABC transporter (available at [120]) and the fancyGene visualization software [130]. N-glycosylation sites were predicted with NetNGlyc1.0 server [131]. Only N-glycosylation sites with a “potential” score > 0.5 and with a jury agreement (“++”-sign or higher) were included in analyses. O-glycosylation sites were predicted using NetOGlyc 3.1 server [132]. If the G-score was higher than 0.5 the residue was considered to be O-glycosylated. The number of O-glycoslated sites (glycosylated serines and threonines) is shown in Table 1).
Expression profiling of ABC genes
Expression profiling of ABC genes was assessed using microarray expression data of two multi-pesticide resistant strains (MR-VP and MAR-AB) [42] and a previously published RNA-seq dataset [37]. The RNA-seq dataset consists of replicated RNA-seq libraries of spider mites feeding on different host plants (bean, tomato and Arabidopsis) and a single RNA-seq library for different developmental stages of spider mites (embryo, larvae, nymph and adult). Experimental details can be found in Grbić et al. [37] and the RNA-seq data are available via Gene Expression Omnibus under reference GSE32342. To ensure the best possible alignment of RNA-seq reads to our manually curated ABC transporter gene models, we re-mapped the RNA-seq reads to the spider mite genome with an updated annotation (Nov 15, 2012 release; [120]). Read alignments and expression quantification were performed after Grbić et al. [37]. For host transfer experiments, differential gene expression was assessed with the DESeq R package [133] as previously described [37]. For the microarray experiment, differentially expressed genes were assessed as reported earlier [42]. For both the host transfer experiment (RNA-seq) and expression profiling with multi-pesticide resistant strains (microarrays), ABC genes with a fold change higher than two and a FDR adjusted p-value less than 0.05 were considered as differentially expressed.
Abbreviations
- ABC:
-
ATP-binding cassette
- FDR:
-
False discovery rate
- FT:
-
Full transporter
- GST:
-
Glutathione S-transferase
- HT:
-
Half transporter
- MDR:
-
Multidrug resistance protein
- MRP:
-
Multidrug resistance associated protein
- P-gp:
-
P-glycoprotein
- RPKM:
-
Reads per kilobase per million mapped reads
- SUR:
-
Sulfonylurea receptor.
References
Dean M, Rzhetsky A, Allikmets R: The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11: 1156-1166. 10.1101/gr.GR-1649R.
Higgins CF: ABC transporters - from microorganisms to man. Annu Rev Cell Biol. 1992, 8: 67-113. 10.1146/annurev.cb.08.110192.000435.
Hollenstein K, Dawson RJP, Locher KP: Structure and mechanism of ABC transporter proteins. Curr Opin Struct Biol. 2007, 17: 412-418. 10.1016/j.sbi.2007.07.003.
Rees DC, Johnson E, Lewinson O: ABC transporters: the power to change. Nat Rev Mol Cell Biol. 2009, 10: 218-227. 10.1038/nrm2646.
Pao SS, Paulsen IT, Saier MH: Major facilitator superfamily. Microbiol Mol Biol Rev. 1998, 62: 1-34.
Dean M, Annilo T: Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annual Review of Genomics and Human Genetics Vol 6. Edited by: Page DS, Chakravarti A. 2005, California: Palo Alto: Annual Review of Genomics and Human Genetics, 123-142.
Annilo T, Chen ZQ, Shulenin S, Costantino J, Thomas L, Lou H, Stefanov S, Dean M: Evolution of the vertebrate ABC gene family: Analysis of gene birth and death. Genomics. 2006, 88: 1-11. 10.1016/j.ygeno.2006.03.001.
Broehan G, Kroeger T, Lorenzen M, Merzendorfer H: Functional analysis of the ATP-binding cassette (ABC) transporter gene family of Tribolium castaneum. BMC Genomics. 2013, 14: 6-10.1186/1471-2164-14-6.
Liu S, Zhou S, Tian L, Guo E, Luan Y, Zhang J, Li S: Genome-wide identification and characterization of ATP-binding cassette transporters in the silkworm, Bombyx mori. BMC Genomics. 2011, 12: 491-10.1186/1471-2164-12-491.
Popovic M, Zaja R, Loncar J, Smital T: A novel ABC transporter: the first insight into zebrafish (Danio rerio) ABCH1. Mar Environ Res. 2010, 69: S11-S13.
Sturm A, Cunningham P, Dean M: The ABC transporter gene family of Daphnia pulex. BMC Genomics. 2009, 10: 170-10.1186/1471-2164-10-170.
Xie X, Cheng T, Wang G, Duan J, Niu W, Xia Q: Genome-wide analysis of the ATP-binding cassette (ABC) transporter gene family in the silkworm, Bombyx mori. Mol Biol Rep. 2012, 39: 7281-7291. 10.1007/s11033-012-1558-3.
You M, Yue Z, He W, Yang X, Yang G, Xie M, Zhan D, Baxter SW, Vasseur L, Gurr GM, et al: A heterozygous moth genome provides insights into herbivory and detoxification. Nat Genet. 2013, 45: 220-225. 10.1038/ng.2524.
Labbé R, Caveney S, Donly C: Genetic analysis of the xenobiotic resistance-associated ABC gene subfamilies of the Lepidoptera. Insect Mol Biol. 2011, 20: 243-256. 10.1111/j.1365-2583.2010.01064.x.
Kovalchuk A, Driessen AJM: Phylogenetic analysis of fungal ABC transporters. BMC Genomics. 2010, 11: 177-10.1186/1471-2164-11-177.
Roth CW, Holm I, Graille M, Dehoux P, Rzhetsky A, Wincker P, Weissenbach J, Brey PT: Identification of the Anopheles gambiae ATP-binding cassette transporter superfamily genes. Mol Cells. 2003, 15: 150-158.
Borst P, Elferink RO: Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002, 71: 537-592. 10.1146/annurev.biochem.71.102301.093055.
Tarr PT, Tarling EJ, Bojanic DD, Edwards PA, Baldan A: Emerging new paradigms for ABCG transporters. Biochim Biophys Acta Mol Cell Biol Lipids. 2009, 1791: 584-593. 10.1016/j.bbalip.2009.01.007.
Fukuda Y, Schuetz JD: ABC transporters and their role in nucleoside and nucleotide drug resistance. Biochem Pharmacol. 2012, 83: 1073-1083. 10.1016/j.bcp.2011.12.042.
Schinkel AH, Jonker JW: Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Delivery Rev. 2003, 55: 3-29. 10.1016/S0169-409X(02)00169-2.
Ricardo S, Lehmann R: An ABC transporter controls export of a Drosophila germ cell attractant. Science. 2009, 323: 943-946. 10.1126/science.1166239.
Mackenzie SM, Brooker MR, Gill TR, Cox GB, Howells AJ, Ewart GD: Mutations in the white gene of Drosophila melanogaster affecting ABC transporters that determine eye colouration. Biochim Biophys Acta Biomembranes. 1999, 1419: 173-185. 10.1016/S0005-2736(99)00064-4.
Komoto N, Quan G-X, Sezutsu H, Tamura T: A single-base deletion in an ABC transporter gene causes white eyes, white eggs, and translucent larval skin in the silkworm w-3(oe) mutant. Insect Biochem Mol Biol. 2009, 39: 152-156. 10.1016/j.ibmb.2008.10.003.
Gaertner LS, Murray CL, Morris CE: Transepithelial transport of nicotine and vinblastine in isolated malpighian tubules of the tobacco hornworm (Manduca sexta) suggests a P-glycoprotein-like mechanism. J Exp Biol. 1998, 201: 2637-2645.
Murray CL, Quaglia M, Arnason JT, Morris CE: A putative nicotine pump at the metabolic blood–brain-barrier of the tobacco hornworm. J Neurobiol. 1994, 25: 23-34. 10.1002/neu.480250103.
Buss DS, Callaghan A: Interaction of pesticides with p-glycoprotein and other ABC proteins: A survey of the possible importance to insecticide, herbicide and fungicide resistance. Pestic Biochem Physiol. 2008, 90: 141-153. 10.1016/j.pestbp.2007.12.001.
Heckel DG: Learning the ABCs of Bt: ABC transporters and insect resistance to Bacillus thuringiensis provide clues to a crucial step in toxin mode of action. Pestic Biochem Physiol. 2012, 104: 103-110. 10.1016/j.pestbp.2012.05.007.
Lanning CL, Fine RL, Corcoran JJ, Ayad HM, Rose RL, AbouDonia MB: Tobacco budworm P-glycoprotein: biochemical characterization and its involvement in pesticide resistance. Biochim Biophys Acta Gen Subj. 1996, 1291: 155-162. 10.1016/0304-4165(96)00060-8.
Baxter SW, Badenes-Perez FR, Morrison A, Vogel H, Crickmore N, Kain W, Wang P, Heckel DG, Jiggins CD: Parallel evolution of Bacillus thuringiensis toxin resistance in Lepidoptera. Genetics. 2011, 189: 675-U814. 10.1534/genetics.111.130971.
Li X, Schuler M, Berenbaum M: Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu Rev Entomol. 2007, 52: 231-253. 10.1146/annurev.ento.51.110104.151104.
Van Leeuwen T, Vontas J, Tsagkarakou A, Dermauw W, Tirry L: Acaricide resistance mechanisms in the two-spotted spider mite Tetranychus urticae and other important Acari: A review. Insect Biochem Mol Biol. 2010, 40: 563-572. 10.1016/j.ibmb.2010.05.008.
Claudianos C, Ranson H, Johnson RM, Biswas S, Schuler MA, Berenbaum MR, Feyereisen R, Oakeshott JG: A deficit of detoxification enzymes: pesticide sensitivity and environmental response in the honeybee. Insect Mol Biol. 2006, 15: 615-636. 10.1111/j.1365-2583.2006.00672.x.
Oakeshott JG, Johnson RM, Berenbaum MR, Ranson H, Cristino AS, Claudianos C: Metabolic enzymes associated with xenobiotic and chemosensory responses in Nasonia vitripennis. Insect Mol Biol. 2010, 19: 147-163.
Ramsey JS, Rider DS, Walsh TK, De Vos M, Gordon KHJ, Ponnala L, Macmil SL, Roe BA, Jander G: Comparative analysis of detoxification enzymes in Acyrthosiphon pisum and Myzus persicae. Insect Mol Biol. 2010, 19: 155-164.
Mounsey KE, Holt DC, McCarthy J, Walton SF: Identification of ABC transporters in Sarcoptes scabiei. Parasitology. 2006, 132: 883-892. 10.1017/S0031182005009716.
Walter DE, Proctor HC: Mites: Ecology, Evolution and Behaviour. 1999, London: CABI Publishing
Grbić M, Van Leeuwen T, Clark RM, Rombauts S, Rouzé P, Grbić V, Osborne EJ, Dermauw W, Ngoc PCT, Ortego F, et al: The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature. 2011, 479: 487-492. 10.1038/nature10640.
Jeppson LR, Keifer HH, Baker EW: Mites injurious to Economic Plants. 1975, Berkeley, Los Angeles: University of California Press
Spider Mites Web: a comprehensive database for the Tetranychidae. http://www.montpellier.inra.fr/CBGP/spmweb,
Arthropod Pesticide Resistance Database. http://www.pesticideresistance.org,
Yang XM, Buschman LL, Zhu KY, Margolies DC: Susceptibility and detoxifying enzyme activity in two spider mite species (Acari : Tetranychidae) after selection with three insecticides. J Econ Entomol. 2002, 95: 399-406. 10.1603/0022-0493-95.2.399.
Dermauw W, Wybouw N, Rombauts S, Menten B, Vontas J, Grbić M, Clark RM, Feyereisen R, Van Leeuwen T: A link between host plant adaptation and pesticide resistance in the polyphagous spider mite Tetranychus urticae. Proc Natl Acad Sci USA. 2013, 110: E113-E122. 10.1073/pnas.1213214110.
Van Leeuwen T, Demaeght P, Osborne EJ, Dermauw W, Gohlke S, Nauen R, Grbic M, Tirry L, Merzendorfer H, Clark RM: Population bulk segregant mapping uncovers resistance mutations and the mode of action of a chitin synthesis inhibitor in arthropods. Proc Natl Acad Sci USA. 2012, 109: 4407-4412. 10.1073/pnas.1200068109.
Van Leeuwen T, Dermauw W, Grbic M, Tirry L, Feyereisen R: Spider mite control and resistance management: does a genome help?. Pest Manag Sci. 2013, 69: 156-159. 10.1002/ps.3335.
Sheps JA, Ralph S, Zhao ZY, Baillie DL, Ling V: The ABC transporter gene family of Caenorhabditis elegans has implications for the evolutionary dynamics of multidrug resistance in eukaryotes. Genome Biol. 2004, 5: R15-10.1186/gb-2004-5-3-r15.
Xiong J, Feng L, Yuan D, Fu C, Miao W: Genome-wide identification and evolution of ATP-binding cassette transporters in the ciliate Tetrahymena thermophila: A case of functional divergence in a multigene family. BMC Evol Biol. 2010, 10: 330-10.1186/1471-2148-10-330.
Verrier PJ, Bird D, Buria B, Dassa E, Forestier C, Geisler M, Klein M, Kolukisaoglu U, Lee Y, Martinoia E, et al: Plant ABC proteins - a unified nomenclature and updated inventory. Trends Plant Sci. 2008, 13: 151-159. 10.1016/j.tplants.2008.02.001.
Peelman F, Labeur C, Vanloo B, Roosbeek S, Devaud C, Duverger N, Denefle P, Rosier M, Vandekerckhove J, Rosseneu M: Characterization of the ABCA transporter subfamily: Identification of prokaryotic and eukaryotic members, phylogeny and topology. J Mol Biol. 2003, 325: 259-274. 10.1016/S0022-2836(02)01105-1.
Beers MF, Hawkins A, Shuman H, Zhao M, Newitt JL, Maguire JA, Ding W, Mulugeta S: A novel conserved targeting motif found in ABCA transporters mediates trafficking to early post-Golgi compartments. J Lipid Res. 2011, 52: 1471-1482. 10.1194/jlr.M013284.
Dutta S: PhD thesis. Genetic regulation of autophagic cell death in Drosophila melanogaster. 2008, College Park: University of Maryland, Department Molecular and Cell Biology
Wu YC, Horvitz HR: The C. elegans cell corpse engulfment gene ced-7 encodes a protein similar to ABC transporters. Cell. 1998, 93: 951-960. 10.1016/S0092-8674(00)81201-5.
Baycin-Hizal D, Tian Y, Akan I, Jacobson E, Clark D, Chu J, Palter K, Zhang H, Betenbaugh MJ: GlycoFly: a database of Drosophila N-linked glycoproteins identified using SPEG-MS techniques. J Proteome Res. 2011, 10: 2777-2784. 10.1021/pr200004t.
Zarubica A, Trompier D, Chimini G: ABCA1, from pathology to membrane function. Pflueg Arch Eur J Physiol. 2007, 453: 569-579. 10.1007/s00424-006-0108-z.
Akiyama M, Sugiyama-Nakagiri Y, Sakai K, McMillan JR, Goto M, Arita K, Tsuji-Abe Y, Tabata N, Matsuoka K, Sasaki R, et al: Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest. 2005, 115: 1777-1784. 10.1172/JCI24834.
Albrecht C, Viturro E: The ABCA subfamily - gene and protein structures, functions and associated hereditary diseases. Pflueg Arch Eur J Physiol. 2007, 453: 581-589. 10.1007/s00424-006-0047-8.
Mayer F, Mayer N, Chinn L, Pinsonneault RL, Kroetz D, Bainton RJ: Evolutionary conservation of vertebrate blood–brain barrier chemoprotective mechanisms in Drosophila. J Neurosci. 2009, 29: 3538-3550. 10.1523/JNEUROSCI.5564-08.2009.
Strycharz JP: Polygenic resistance in the highly DDT-resistant 91-R strain of Drosophila melanogaster involves decreased penetration, increased metabolism and direct excretion of DDT. 2010, Department of Veterinary and Animal Sciences: M.S. University of Massachussets
Lespine A, Ménez C, Bourguinat C, Prichard RK: P-glycoproteins and other multidrug resistance transporters in the pharmacology of anthelmintics: Prospects for reversing transport-dependent anthelmintic resistance. Int J Parasitol Drugs Drug Resist. 2012, 2: 58-75.
Pohl PC, Klafke GM, Carvalho DD, Martins JR, Daffre S, IdS V, Masuda A: ABC transporter efflux pumps: A defense mechanism against ivermectin in Rhipicephalus (Boophilus) microplus. Int J Parasitol. 2011, 41: 1323-1333. 10.1016/j.ijpara.2011.08.004.
Yoon KS, Strycharz JP, Baek JH, Sun W, Kim JH, Kang JS, Pittendrigh BR, Lee SH, Clark JM: Brief exposures of human body lice to sublethal amounts of ivermectin over-transcribes detoxification genes involved in tolerance. Insect Mol Biol. 2011, 20: 687-699. 10.1111/j.1365-2583.2011.01097.x.
Zutz A, Gompf S, Schaegger H, Tampe R: Mitochondrial ABC proteins in health and disease. Biochim Biophys Acta Bioenerg. 2009, 1787: 681-690. 10.1016/j.bbabio.2009.02.009.
Gonzalez-Cabo P, Bolinches-Amoros A, Cabello J, Ros S, Moreno S, Baylis HA, Palau F, Vazquez-Manrique RP: Disruption of the ATP-binding cassette B7 (ABTM-1/ABCB7) induces oxidative stress and premature cell death in Caenorhabditis elegans. J Biol Chem. 2011, 286: 21304-21314. 10.1074/jbc.M110.211201.
Bariami V, Jones CM, Poupardin R, Vontas J, Ranson H: Gene amplification, ABC transporters and cytochrome P450s: unraveling the molecular basis of pyrethroid resistance in the dengue vector, Aedes aegypti. PLoS Negl Trop Dis. 2012, 6: e1692-10.1371/journal.pntd.0001692.
Elliott AM, Ai-Hajj MA: ABCB8 Mediates doxorubicin resistance in melanoma cells by protecting the mitochondrial genome. Mol Canc Res. 2009, 7: 79-87. 10.1158/1541-7786.MCR-08-0235.
Sachrajda I, Ratajewski M: Mithramycin A suppresses expression of the human melanoma-associated gene ABCB8. Mol Genet Genomics. 2011, 285: 57-65. 10.1007/s00438-010-0586-8.
Ichikawa Y, Bayeva M, Ghanefar M, Potini V, Sun L, Mutharasan RK, Wu R, Khechaduri A, Naik TJ, Ardehali H: Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proc Natl Acad Sci USA. 2012, 109: 4152-4157.
Kiss K, Brozik A, Kucsma N, Toth A, Gera M, Berry L, Vallentin A, Vial H, Vidal M, Szakacs G: Shifting the paradigm: the putative mitochondrial protein ABCB6 resides in the lysosomes of cells and in the plasma membrane of erythrocytes. PLoS One. 2012, 7: e37378-10.1371/journal.pone.0037378.
Akrouh A, Halcomb SE, Nichols CG, Sala-Rabanal M: Molecular biology of K-ATP channels and implications for health and disease. IUBMB Life. 2009, 61: 971-978. 10.1002/iub.246.
Jordan IK, Kota KC, Cui G, Thompson CH, McCarty NA: Evolutionary and functional divergence between the cystic fibrosis transmembrane conductance regulator and related ATP-binding cassette transporters. Proc Natl Acad Sci USA. 2008, 105: 18865-18870. 10.1073/pnas.0806306105.
Kruh GD, Belinsky MG: The MRP family of drug efflux pumps. Oncogene. 2003, 22: 7537-7552. 10.1038/sj.onc.1206953.
Deeley RG, Westlake C, Cole SPC: Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol Rev. 2006, 86: 849-899. 10.1152/physrev.00035.2005.
Borst P, Evers R, Kool M, Wijnholds J: A family of drug transporters: The multidrug resistance-associated proteins. J Natl Canc Inst. 2000, 92: 1295-1302. 10.1093/jnci/92.16.1295.
Ishikawa T: The ATP-dependent glutathione s-conjugate export pump. Trends Biochem Sci. 1992, 17: 463-469. 10.1016/0968-0004(92)90489-V.
Sau A, Tregno FP, Valentino F, Federici G, Caccuri AM: Glutathione transferases and development of new principles to overcome drug resistance. Arch Biochem Biophys. 2010, 500: 116-122. 10.1016/j.abb.2010.05.012.
Reddy BPN, Prasad GBKS, Raghavendra K: In silico analysis of glutathione S-transferase supergene family revealed hitherto unreported insect specific delta- and epsilon-GSTs and mammalian specific mu-GSTs in Ixodes scapularis (Acari: Ixodidae). Comput Biol Chem. 2011, 35: 114-120. 10.1016/j.compbiolchem.2011.03.004.
Szeri F, Ilias A, Pomozi V, Robinow S, Bakos E, Varadi A: The high turnover Drosophila multidrug resistance-associated protein shares the biochemical features of its human orthologues. Biochim Biophys Acta Biomembranes. 2009, 1788: 402-409. 10.1016/j.bbamem.2008.11.007.
Tarnay JN, Szeri F, Ilias A, Annilo T, Sung C, Le Saux O, Varadi A, Dean M, Boyd CD, Robinow S: The dMRP/CG6214 gene of Drosophila is evolutionarily and functionally related to the human multidrug resistance-associated protein family. Insect Mol Biol. 2004, 13: 539-548. 10.1111/j.0962-1075.2004.00512.x.
Chahine S, O'Donnell MJ: Physiological and molecular characterization of methotrexate transport by Malpighian tubules of adult Drosophila melanogaster. J Insect Physiol. 2009, 55: 927-935. 10.1016/j.jinsphys.2009.06.005.
Chahine S, O'Donnell MJ: Interactions between detoxification mechanisms and excretion in Malpighian tubules of Drosophila melanogaster. J Exp Biol. 2011, 214: 462-468. 10.1242/jeb.048884.
Yepiskoposyan H, Egli D, Fergestad T, Selvaraj A, Treiber C, Multhaup G, Georgiev O, Schaffner W: Transcriptome response to heavy metal stress in Drosophila reveals a new zinc transporter that confers resistance to zinc. Nucleic Acids Res. 2006, 34: 4866-4877. 10.1093/nar/gkl606.
Huang H, Haddad GG: Drosophila dMRP4 regulates responsiveness to O-2 deprivation and development under hypoxia. Physiol Genomics. 2007, 29: 260-266. 10.1152/physiolgenomics.00166.2006.
Monnier V, Girardot F, Cheret C, Andres O, Tricoire H: Modulation of oxidative stress resistance in Drosophila melanogaster by gene overexpression. Genesis. 2002, 34: 76-79. 10.1002/gene.10130.
Campos I, Geiger JA, Santos AC, Carlos V, Jacinto A: Genetic screen in Drosophila melanogaster uncovers a novel set of genes required for embryonic epithelial repair. Genetics. 2010, 184: 129-U239. 10.1534/genetics.109.110288.
Shah S, Yarrow C, Dunning R, Cheek B, Vass S, Windass J, Hadfield S: Insecticide detoxification indicator strains as tools for enhancing chemical discovery screens. Pest Manag Sci. 2012, 68: 38-48. 10.1002/ps.2218.
Russel FGM, Koenderink JB, Masereeuw R: Multidrug resistance protein 4 (MRP4/ABCC4): a versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol Sci. 2008, 29: 200-207. 10.1016/j.tips.2008.01.006.
Hopper-Borge E, Xu X, Shen T, Shi Z, Chen Z-S, Kruh GD: Human Multidrug Resistance Protein 7 (ABCC10) is a resistance factor for nucleoside analogues and epothilone B. Cancer Res. 2009, 69: 178-184. 10.1158/0008-5472.CAN-08-1420.
Waterhouse RM, Zdobnov EM, Tegenfeldt F, Li J, Kriventseva EV: OrthoDB: the hierarchical catalog of eukaryotic orthologs in 2011. Nucleic Acids Res. 2011, 39: D283-D288. 10.1093/nar/gkq930.
Eleftherianos I, Won S, Chtarbanova S, Squiban B, Ocorr K, Bodmer R, Beutler B, Hoffmann JA, Imler J-L: ATP-sensitive potassium channel (K-ATP)-dependent regulation of cardiotropic viral infections. Proc Natl Acad Sci USA. 2011, 108: 12024-12029. 10.1073/pnas.1108926108.
Abo-Elghar GE, Fujiyoshi P, Matsumura F: Significance of the sulfonylurea receptor (SUR) as the target of diflubenzuron in chitin synthesis inhibition in Drosophila melanogaster and Blattella germanica. Insect Biochem Mol Biol. 2004, 34: 743-752. 10.1016/j.ibmb.2004.03.009.
Gangishetti U, Breitenbach S, Zander M, Saheb SK, Mueller U, Schwarz H, Moussian B: Effects of benzoylphenylurea on chitin synthesis and orientation in the cuticle of the Drosophila larva. Eur J Cell Biol. 2009, 88: 167-180. 10.1016/j.ejcb.2008.09.002.
Meyer F, Flötenmeyer M, Moussian B: The sulfonylurea receptor SUR is dispensable for chitin synthesis in Drosophila melanogaster embryos. Pest Manag Sci. 2012, 10.1002/ps.3476
Yoshiura K, Kinoshita A, Ishida T, Ninokata A, Ishikawa T, Kaname T, Bannai M, Tokunaga K, Sonoda S, Komaki R, et al: A SNP in the ABCC11 gene is the determinant of human earwax type. Nat Genet. 2006, 38: 324-330. 10.1038/ng1733.
Morita M, Imanaka T: Peroxisomal ABC transporters: Structure, function and role in disease. Biochim Biophys Acta Mol Basis Dis. 1822, 2012: 1387-1396.
Shani N, Sapag A, Valle D: Characterization and analysis of conserved motifs in a peroxisomal ATP-binding cassette transporter. J Biol Chem. 1996, 271: 8725-8730. 10.1074/jbc.271.15.8725.
Barthelme D, Dinkelaker S, Albers S-V, Londei P, Ermler U, Tampe R: Ribosome recycling depends on a mechanistic link between the FeS cluster domain and a conformational switch of the twin-ATPase ABCE1. Proc Natl Acad Sci USA. 2011, 108: 3228-3233. 10.1073/pnas.1015953108.
Zhou AM, Hassel BA, Silverman RH: Expression cloning of 2-5a-dependent RNAse - a uniquely regulated mediator of interferon action. Cell. 1993, 72: 753-765. 10.1016/0092-8674(93)90403-D.
Paytubi S, Wang X, Lam YW, Izquierdo L, Hunter MJ, Jan E, Hundal HS, Proud CG: ABC50 promotes translation initiation in mammalian cells. J Biol Chem. 2009, 284: 24061-24073. 10.1074/jbc.M109.031625.
Kerr ID, Haider AJ, Gelissen IC: The ABCG family of membrane-associated transporters: you don't have to be big to be mighty. Br J Pharmacol. 2011, 164: 1767-1779. 10.1111/j.1476-5381.2010.01177.x.
Green MM: The ''genesis of the white-eyed mutant'' in Drosophila melanogaster: A reappraisal. Genetics. 1996, 142: 329-331.
Mills LR: Structure of visual system of 2-spotted spider-mite, Tetranychus urticae. J Insect Physiol. 1974, 20: 795-808. 10.1016/0022-1910(74)90171-1.
Zhang SD, Odenwald WF: Misexpression of the white (omega) gene triggers male-made courtship in Drosophila. Proc Natl Acad Sci USA. 1995, 92: 5525-5529. 10.1073/pnas.92.12.5525.
Anaka M, MacDonald CD, Barkova E, Simon K, Rostom R, Godoy RA, Haigh AJ, Meinertzhagen IA, Lloyd V: The white gene of Drosophila melanogaster encodes a protein with a role in courtship behavior. J Neurogenet. 2008, 22: 243-276. 10.1080/01677060802309629.
Borycz J, Borycz JA, Kubow A, Lloyd V, Meinertzhagen IA: Drosophila ABC transporter mutants white, brown and scarlet have altered contents and distribution of biogenic amines in the brain. J Exp Biol. 2008, 211: 3454-3466. 10.1242/jeb.021162.
Tatematsu K-i, Yamamoto K, Uchino K, Narukawa J, Iizuka T, Banno Y, Katsuma S, Shimada T, Tamura T, Sezutsu H, Daimon T: Positional cloning of silkworm white egg 2 (w-2) locus shows functional conservation and diversification of ABC transporters for pigmentation in insects. Genes Cells. 2011, 16: 331-342. 10.1111/j.1365-2443.2011.01490.x.
McQuilton P, St Pierre SE, Thurmond J, FlyBase C: FlyBase 101-the basics of navigating FlyBase. Nucleic Acids Res. 2012, 40: D706-D714. 10.1093/nar/gkr1030.
wFleaBase - Daphnia Water Flea Genome Database. http://wfleabase.org/,
Graf GA, Yu LQ, Li WP, Gerard R, Tuma PL, Cohen JC, Hobbs HH: ABCG5 and ABCG8 are obligate heterodimers for protein trafficking and biliary cholesterol excretion. J Biol Chem. 2003, 278: 48275-48282. 10.1074/jbc.M310223200.
Hock T, Cottrill T, Keegan J, Garza D: The E23 early gene of Drosophila encodes an ecdysone-inducible ATP-binding cassette transporter capable of repressing ecdysone-mediated gene activation. Proc Natl Acad Sci USA. 2000, 97: 9519-9524. 10.1073/pnas.160271797.
Itoh TQ, Tanimura T, Matsumoto A: Membrane-bound transporter controls the circadian transcription of clock genes in Drosophila. Genes Cells. 2011, 16: 1159-1167. 10.1111/j.1365-2443.2011.01559.x.
Kuwana H, ShimizuNishikawa K, Iwahana H, Yamamoto D: Molecular cloning and characterization of the ABC transporter expressed in Trachea (ATET) gene from Drosophila melanogaster. Biochim Biophys Acta Gene Struct Expr. 1996, 1309: 47-52. 10.1016/S0167-4781(96)00137-6.
Jones CM, Toé HK, Sanou A, Namountougou M, Hughes A, et al: Additional selection for insecticide resistance in urban malaria vectors: DDT Resistance in Anopheles arabiensis from Bobo-Dioulasso, Burkina Faso. PLoS One. 2012, 7: e45995-10.1371/journal.pone.0045995.
Lamping E, Baret PV, Holmes AR, Monk BC, Goffeau A, Cannon RD: Fungal PDR transporters: Phylogeny, topology, motifs and function. Fungal Genet Biol. 2010, 47: 127-142. 10.1016/j.fgb.2009.10.007.
Robinson SW, Herzyk P, Dow JAT, Leader DP: FlyAtlas: database of gene expression in the tissues of Drosophila melanogaster. Nucleic Acids Res. 2013, 41: D744-D750. 10.1093/nar/gks1141.
Mummery-Widmer JL, Yamazaki M, Stoeger T, Novatchkova M, Bhalerao S, Chen D, Dietzl G, Dickson BJ, Knoblich JA: Genome-wide analysis of Notch signalling in Drosophila by transgenic RNAi. Nature. 2009, 458: 987-U959. 10.1038/nature07936.
Zhang S, Feany MB, Saraswati S, Littleton JT, Perrimon N: Inactivation of Drosophila Huntingtin affects long-term adult functioning and the pathogenesis of a Huntington's disease model. Dis Model Mech. 2009, 2: 247-266. 10.1242/dmm.000653.
Qin W, Neal SJ, Robertson RM, Westwood JT, Walker VK: Cold hardening and transcriptional change in Drosophila melanogaster. Insect Mol Biol. 2005, 14: 607-613. 10.1111/j.1365-2583.2005.00589.x.
Diamond back moth Genome database. http://iae.fafu.edu.cn/DBM,
Zhao ZY, Sheps JA, Ling V, Fang LL, Baillie DL: Expression analysis of ABC transporters reveals differential functions of tandemly duplicated genes in Caenorhabditis elegans. J Mol Biol. 2004, 344: 409-417. 10.1016/j.jmb.2004.09.052.
Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
ORCAE. Online Resource for Communnity Annotation of Eukaryots. http://bioinformatics.psb.ugent.be/orcae/overview/Tetur,
ScanProsite tool. http://prosite.expasy.org/scanprosite,
Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32: 1792-1797. 10.1093/nar/gkh340.
Abascal F, Zardoya R, Posada D: ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005, 21: 2104-2105. 10.1093/bioinformatics/bti263.
Jobb G, Von Haeseler A, Strimmer K: TREEFINDER: a powerful graphical analysis environment for molecular phylogenetics. BMC Evol Biol. 2004, 4: 18-10.1186/1471-2148-4-18.
Allikmets R, Gerrard B, Hutchinson A, Dean M: Characterization of the human ABC superfamily: Isolation and mapping of 21 new genes using the expressed sequence tags database. Hum Mol Genet. 1996, 5: 1649-1655. 10.1093/hmg/5.10.1649.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol. 2011, 28: 2731-2739. 10.1093/molbev/msr121.
Campanella JJ, Bitincka L, Smalley J: MatGAT: An application that generates similarity/identity matrices using protein or DNA sequences. BMC Bioinforma. 2003, 4: 29-10.1186/1471-2105-4-29.
Bernsel A, Viklund H, Falk J, Lindahl E, Von Heijne G, Elofsson A: Prediction of membrane-protein topology from first principles. Proc Natl Acad Sci USA. 2008, 105: 7177-7181. 10.1073/pnas.0711151105.
Emanuelsson O, Brunak S, Von Heijne G, Nielsen H: Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007, 2: 953-971. 10.1038/nprot.2007.131.
Rambaldi D, Ciccarelli FD: FancyGene: dynamic visualization of gene structures and protein domain architectures on genomic loci. Bioinformatics. 2009, 25: 2281-2282. 10.1093/bioinformatics/btp381.
NetNGlyc server 1.0. http://www.cbs.dtu.dk/services/NetNGlyc/,
Julenius K, Molgaard A, Gupta R, Brunak S: Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology. 2005, 15: 153-164.
Anders S, Huber W: Differential expression analysis for sequence count data. Genome Biol. 2010, 11: R106-10.1186/gb-2010-11-10-r106.
Acknowledgements
TVL is a post-doctoral fellow of the Fund for Scientific Research Flanders (FWO). This work was supported by FWO grant 3G061011 (to TVL) and 3G009312 (to TVL and LT), a Ghent University Special Research Fund grant 01J13711 (to TVL and LT), the Government of Canada through Genome Canada and the Ontario Genomics Institute OGI-046 (to MG), a University of Utah Funding Incentive Seed Grant (to RMC), and a National Institutes of Health Genetics Training Grant T32 GM07464 (to EJO).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
WD and TVL designed research. WD and EJO analyzed data. WD and TVL wrote the manuscript, with input from RMC, EJO and LT. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2013_5057_MOESM1_ESM.pdf
Additional file 1: Midpoint rooted maximum likelihood phylogenetic tree of ABC NBDs of D. melanogaster , H. sapiens and T. urticae . For amino acid alignment, amino acid substitution model and likelihood score of the constructed phylogenetic tree see Additional file 9 and Additional file 10. Main nodes were collapsed to create a better overview of the phylogenetic relationships between the different ABC subfamilies. Numbers at the branch point of each node represent the bootstrap value resulting from 1000 pseudoreplicates (LR-ELW). The scale bar represents 0.5 amino-acid substitutions per site. For accession numbers of metazoan ABC protein sequences see Additional file 11 while T. urticae ABC protein sequences can be found in Additional file 12. (PDF 7 KB)
12864_2013_5057_MOESM3_ESM.pdf
Additional file 3: Exon-intron pattern of 103 T. urticae ABC genes. Exons are depicted as light grey boxes while strandlines represent introns. Within exons red boxes represent NBDs, while small dark grey boxes represent transmembrane helices (TM). Solid arrows indicate direction of transcription. (PDF 2 MB)
12864_2013_5057_MOESM4_ESM.pdf
Additional file 4: Phylogenetic analysis of ABCA proteins of five metazoan species, derived according to the procedure in the Figure 2 legend.(PDF 18 KB)
12864_2013_5057_MOESM5_ESM.xlsx
Additional file 5: Identity/similarity matrices between T. urticae, D. pulex, D. melanogaster and H. sapiens ABC proteins. In the upper triangle identity values are shown, while in the lower triangle similarity values are presented. (XLSX 147 KB)
12864_2013_5057_MOESM6_ESM.pdf
Additional file 6: Phylogenetic analysis of ABCD proteins of five metazoan species, derived according to the procedure in the Figure 2 legend.(PDF 10 KB)
12864_2013_5057_MOESM7_ESM.pdf
Additional file 7: Phylogenetic analysis of ABCE and ABCF proteins of five metazoan species, derived according to the procedure in the Figure 2 legend.(PDF 12 KB)
12864_2013_5057_MOESM8_ESM.pdf
Additional file 8: Heat plot of mean expression values (rpkm) of T. urticae ABC fragments from mites on different host plants (bean, tomato and Arabidopsis ) and of expression values of T. urticae ABC fragments from four different life stages (embryo, larvae, nymph and adult).(PDF 307 KB)
12864_2013_5057_MOESM9_ESM.rar
Additional file 9: Alignment of N-terminal NBDs of T. urticae ABC proteins (ABC_N-term_NBD_Tetranychus.fas), N- and C-terminal NBDs of metazoan ABC proteins (ABC_NBD_Metazoa_simple.fas) and of full length metazoan ABCA, -B, C, D, E, F, G and H protein sequences (ABCA-H.fas) (bundled in a .rar file) (examination of the files in this .rar file requires the BioEdit program (http://www.mbio.ncsu.edu/bioedit/bioedit.html)). (RAR 244 KB)
12864_2013_5057_MOESM10_ESM.xlsx
Additional file 10: Amino acid substitution models used for maximum likelihood phylogenetic analyses and likelihood scores of constructed phylogenetic trees.(XLSX 10 KB)
12864_2013_5057_MOESM12_ESM.fas
Additional file 12: 103 T. urticae ABC protein sequences (.fasta) (examination of this file requires the BioEdit program (http://www.mbio.ncsu.edu/bioedit/bioedit.html)). (FAS 116 KB)
12864_2013_5057_MOESM13_ESM.xlsx
Additional file 13: Fold change values of differentially expressed ABC genes of mites after host plant change to either Arabidopsis or tomato.(XLSX 19 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Dermauw, W., Osborne, E.J., Clark, R.M. et al. A burst of ABC genes in the genome of the polyphagous spider mite Tetranychus urticae. BMC Genomics 14, 317 (2013). https://doi.org/10.1186/1471-2164-14-317
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-14-317