
Abstract
Background
In recent years, genome-wide association studies have successfully uncovered single-nucleotide polymorphisms (SNPs) associated with complex traits such as diseases and quantitative phenotypes. These variations account for a small proportion of heritability. With the development of high throughput techniques, abundant submicroscopic structural variations have been found in organisms, of which the main variations are copy number variations (CNVs). Therefore, CNVs are increasingly recognized as an important and abundant source of genetic variation and phenotypic diversity.
Results
Analyses of CNVs in the genomes of three sheep breeds were performed using the Ovine SNP50 BeadChip array. A total of 238 CNV regions (CNVRs) were identified, including 219 losses, 13 gains, and six with both events (losses and gains), which cover 60.35 Mb of the sheep genomic sequence and correspond to 2.27% of the autosomal genome sequence. The length of the CNVRs on autosomes range from 13.66 kb to 1.30 Mb with a mean size of 253.57 kb, and 75 CNVRs events had a frequency > 3%. Among these CNVRs, 47 CNVRs identified by the PennCNV overlapped with the CNVpartition. Functional analysis indicated that most genes in the CNVRs were significantly enriched for involvement in the environmental response. Furthermore, 10 CNVRs were selected for validation and 6 CNVRs were further experimentally confirmed by qPCR. In addition, there were 57 CNVRs overlapped in our new dataset and other published ruminant CNV studies.
Conclusions
In this study, we firstly constructed a sheep CNV map based on the Ovine SNP50 array. Our results demonstrated the differences of two detection tools and integration of multiple algorithms can enhance the detection of sheep genomic structure variations. Furthermore, our findings would be of help for understanding the sheep genome and provide preliminary foundation for carrying out the CNVs association studies with economically important phenotypes of sheep in the future.
Similar content being viewed by others

Background
In recent years, genome-wide association studies (GWAS) have successfully uncovered single-nucleotide polymorphisms (SNPs) associated with complex diseases or traits [1]. With the rapid development of chip array-based genotyping techniques, thousands of genomic submicroscopic structural variations have been found in the human genome [2]. As a main genetic form of submicroscopic structural variation copy number variations (CNVs) are widely distributed in the human genome and influence gene expression, phenotypic variation and adaptation by disrupting genes and altering gene dosage [2–5]. Numerous studies showed that CNVs contributed to both disease susceptibility and phenotypic diversity [2, 5]. Now, CNV is increasingly considered to be an important and abundant source of genetic variation and phenotypic diversity [5, 6].
Investigations on CNVs have been successively carried out in human and other species [7–13]. In the domestic animals, there are involving in cattle [14–20], dog [21], chicken [22], pig [23, 24], goat [25], sheep [26] and rabbit [27]. As for sheep, Fontanesi et al. [26] provided a first comparative map of CNVs of the sheep genome referred to the cattle genome using a cross-species array comparative genome hybridization(aCGH). However, the cross-species analysis based on heterologous hybridization couldn’t identify all detectable CNVRs due to low homology between cattle probes and sheep DNA for some regions and doesn’t show the CNVR distributions on the sheep genome. In addition to CGH, another major platform commonly used to identify CNVs is the SNP array. In SNP array, intensity values of SNPs derived from each sample are used to detect CNVs in each individual. Comparison with two panels, CGH array has excellent performance in signal-to noise ratios, while the SNP array based approach is more convenient for high-throughput analyses and follow-up association studies [28]. With the development of high density SNP arrays, higher resolution of genomic regions can be achieved [29]. Moreover, due to their low cost and high-density, commercial SNP arrays have been widely used for CNV detection in domestic animals, and CNV mapping and functional studies have made important progress. However, there are no reports on CNV detection of sheep based on SNP array data.
In this study, we will investigate genome-wide CNV in three sheep populations. To pursue convincing results, we firstly employ the PennCNV program to analyze Ovine SNP50 genotyping data, and then use other algorithm program, cnvPartition, to validate CNVRs detected by PennCNV. To our knowledge, we will construct the first sheep CNV map based on SNP array data. This research will provide useful addition to the sheep CNV maps, and will provide potential genetic markers for further investigation on the roles of CNV in sheep productive traits and evolutionary adaptation.
Results
SNP genotyping
The genomic DNA of 329 individual samples from three sheep breeds (German Mutton sheep, Dorper and Sunite sheep) were genotyped using Illumina OvineSNP50 Genotyping BeadChip according to the manufacturer’s protocol, and the PennCNV (http://www.openbioinformatics.org/penncnv) software was used to identify the CNVs in the sheep genome (Table 1). According to the results of PennCNV, we defined the CNV call filtering criteria to exclude samples with low quality of signal intensity data. After applying the CNV quality control criteria detailed in the “Methods” section, 256 samples (157 German Mutton, 35 Dorper and 64 Sunite sheep) remained for further CNV analyses.
Genome-wide surveys of sheep CNVs
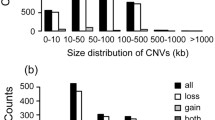
After filtering unreliable CNV calls, we discovered a total of 3624 CNV events (3416 losses and 208 gains), with an average number of 14.16 CNV events per individual. The average and median sizes of CNVs were 144.6 kb and 122.9 kb, respectively (Table 2). We found that approximately 58% of the CNVs ranged from 100 to 500 kb, and 25% ranged from 50 to 100 kb in size distribution (Figure 1A and Additional file 1: Table S1). Smaller CNVs (< 10 kb) were not identified in this study.

The size distribution of CNV. (A) The distribution of CNVs size in the three sheep breeds. (B) The distribution of CNVRs size in the three sheep breeds. Smaller CNVs (<10 kb) were not identified in this study.
Characteristics of CNVRs identified in sheep
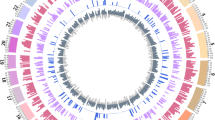
CNV regions (CNVRs) were determined by merging the overlapping CNVs identified in all samples, as reported previously [4]. A total of 238 autosomal CNVRs were identified, covering 60.35 Mb of the sheep genomic sequence and corresponding to 2.27% of the autosomal genome sequence (60.35 Mb/2655.6 Mb) and 2.17% of the whole sheep genome (60.35 Mb/2784.7 Mb). More information on CNVRs is also presented in Figure 2 and Additional file 1: Table S3.

Map of sheep CNVRs. Red, blue and green represented the Gain, Loss and both (Gain-Loss), respectively. A total of 238 CNV regions, including 219 Loss events, 13 Gain events, 6 both events(Gain-Loss); 60.35 Mb, ~2.27% of sheep autosomal genome.
These 238 CNVRs, ranging from 13.66 Kb to 1.30 Mb with mean and median sizes of 253.57 and 186.92 kb, respectively (Table 2), included 219 losses, 13 gains, and six with both events (gains and losses). Loss events were present approximately 16.8-fold more than gain events, similar to previously reports of other species [14, 16, 25, 26]. The majority (85%) of CNVRs ranged from 50 kb to 500 kb (Figure 1B and Additional file 1: Table S2). Furthermore, of the 238 CNVRs, 72 CNVRs were found in only one animal (Unique), whereas 166 CNVRs were found in ≥ two animals and breeds (multiple) [20] and 13 CNVRs had a frequency > 3%, 62 CNVRs had a frequency > 5%. (Table 2 and Additional file 2: Table S4). Specifically, the CNVR Ovis aries chromosome (OAR) 9:11627386–12923703 had the highest frequency (65%). In the report by McCarroll et al.[30], the copy number polymorphism (CNP) term was coined when describing 75 CNVRs as common CNVRs. In addition, as shown in Table 3, the numbers of CNVRs identified by PennCNV were large difference among three sheep breeds. In the German Mutton, we identified 172 CNVRs, which comprised 72.27% of the total CNVRs detected. In Sunite and Dorper breeds, 134 and 126 CNVRs were detected, corresponding to 56.30% and 52.9% of the total number, while 72 unique CNVRs, 29, 24 and 19 CNVRs were detected in German Mutton, Sunite and Dorper breeds, respectively.
Construction of copy number variation map
We constructed the map of CNVR distribution on the chromosomes based on the sheep whole genome SNP genotyping chips (Figure 2). The results indicated that the CNVs among sheep breeds were non-randomly distributed among the different chromosomes, and that the percentage of CNVRs in the chromosomes varies from 0.46–5.08% (Figure 2 and Additional file 1: Table S3), with the top three highest percentages of CNVRs located on chromosomes 16, 26 and 9 (5.08%, 4.94%, and 4.11%, respectively). This might be due to bias derived by the analysis of the Ovine SNP50 BeadChip. Chromosome 1 had the greatest number and largest length of CNVRs, whereas chromosome 14 had the smallest length and number of CNVRs. The most enriched chromosomes for CNVRs in sheep were chromosomes 1, 2 and 5.
To verify the CNVs detected by PennCNV, we also used the CNVpartition program implemented in Illumina GenomeStudio to analyze the data and detect CNVs. After applying the quality filtering criteria, we identified 179 individuals with CNVs, and 41 CNVRs were determined by merging overlapping CNVs identified across all samples. It should be noted that all 179 individuals were encompassed by 256 individuals passed the quality filtering criteria of PennCNV. The results of CNVRs distributed on chromosomes were similar to those of PennCNV (Additional file 1: Table S3), and the OAR16 showed the greatest enrichment for CNV (18.45%), which was consistent with the result of PennCNV program. But, chromosomes with more five CNVRs were OAR3 and OAR7. The different results might be due to the different algorithms between PennCNV and CNVpartition. In a comparison of the results from the PennCNV and CNVpartition programs, we found 47 CNVRs identified by the PennCNV program (19.7%) that overlapped with the CNVpartition data, and the ‘losses’ and ‘gains’ of 47 CNVRs were consistent with the CNVpartition data. Some CNVRs identified by the CNVpartition analysis were larger in size, resulting in overlapping of multiple CNVRs identified by PennCNV, thus, we segmented these large CNVRs in the CNVpartition results. In total, we obtained 52 CNVRs by CNVpartition analysis (see Additional file 3: Figure S1).
CNV validation by quantitative PCR
We performed quantitative real-time PCR experiments to evaluate the accuracy of the copy number assignments. Ten putative CNVRs were selected for CNV validation, these ten CNVRs represent different predicted status of copy numbers (i.e., gain and loss) and CNVR frequencies varied from 0.30 to 15.20%, of which four contained known sheep RefSeq genes in the UCSC database (see Additional file 4: Table S5); the remaining six CNVRs were selected randomly. We performed 60 qPCR assays in 49 animals, out of the 60 qPCR assays, 16 were in agreement with prediction by PennCNV. When counting the CNVRs, 6 out of the 10 CNVRs confirmed the existence of copy number variations in at least one qPCR assay, whereas primer pairs in four other regions did not work (Additional file 4: Table S5).
Functional enrichment analysis of CNVRs
We found five sheep RefSeq genes partially or entirely encompassing four CNVRs. To further analyze the gene content of the 238 CNVRs, we used a BLASTN search for homologous human and cattle sequences using the UCSC table browser tool. There were 1043 RefSeq homologous human genes located within or partially overlapping with 127 CNVRs of the 238 CNVRs and similarly, there were 270 RefSeq homologous cattle genes located within or partially overlapping with 106 CNVRs of the 238 CNVRs (Additional file 5: Table S6). To assess the functional annotation of these CNVRs, we conducted gene ontology analysis (GO) using the Database for Annotation, Visualization and Integrated Discovery (DAVID) functional annotation tool. We retrieved 1313 gene symbols (homologous genes) to load into the DAVID tool. We selected ‘human’ as the background, and the 563 corresponding human gene IDs were identified in DAVID. GO analysis indicated that the genes overrepresented in DAVID were involved in olfactory receptor activity, sensory perception of smell, sensory perception of chemical stimulus, sensory perception, cognition, neurological system processes, G-protein coupled receptor protein signaling pathway, cell surface receptor linked signal transduction, plasma membrane as well as integral and intrinsic membrane components, protein polymerization and microtubule-based movement. In addition to the above gene functions, the DAVID pathway results showed genes involved in olfactory transduction and pathogenic Escherichia coli (Table 4). We also found that a number of CNVRs overlapped with known disease-related genes in the DGV database. A total of 341 disease-related genes reported in DGV were located either wholly or partially within CNVRs in our results (see the Additional file 6: Table S7).
Comparison with other ruminant CNV studies
To compare CNVRs identified by SNP platform in sheep genome which were overlapped with CNVRs reported in other ruminants, we migrated 238 CNVRs from ISGC Ovis aries 1.0 to Btau_4.0 using UCSC liftOver tool [31] and 230 CNVRs were obtained on Btau_4.0 assembly. And then we compared these CNVRs with those previous CNV studies (Additional file 7: Table S8), including four experiments that two were carried out in sheep, goat and two in cattle using aCGH platform [15, 20, 25, 26] and other two in cattle using the Illumina BovineSNP50 BeadChip [14, 16]. Comparing sheep CNVRs detected by us and Fontanesi et al. [26], we found three CNVRs overlapped. In addition, there were found two sheep CNVRs matched goat CNVRs identified with aCGH panel [25]. Comparing sheep CNVRs with four other cattle CNVRs reported by Liu et al. [20]. Fadista et al. [15]. Hou et al. [16] and Bea et al. [14], we obtained 54 of 230 CNVRs mapped on Btau_4.0 assembly overlapped with cattle CNVRs with aCGH panel or SNP panel.
Discussion
In recent years, CNVs have been increasingly recognized as an important source of genetic variation and phenotypic diversity [4, 5, 30]. A number of CNVs associated with diseases have been found in human genetic researches, involving autoimmune disorders, schizophrenia, autism, as well as infectious and cardiovascular diseases [32, 33]. In addition, CNVs have been shown to play a role in pharmacokinetics in terms of drug efficacy and toxicity [34]. Therefore, the systematic analysis and characterization of CNVs improves our understanding of genetic variation and is an important tool for deciphering the role of CNV in the heritability of complex traits. In the past few years, with the development of high-density genotyping arrays, detection of CNVs using SNP genotyping arrays has become a cost-effective and efficient approach. Fine-scale CNV maps for human and other species have been constructed and refined [18, 35, 36].
In this study, we used whole genome genotyping based on Ovine SNP50 BeadChip arrays to identify CNVs in the sheep genome. After genotyping 329 samples using the Illumina platform from three sheep breeds, the signal intensity (LRR) and allelic intensity (BAF) ratio of all samples were exported using the Illumina GenomeStudio software (Illumina Inc., San Diego, CA, USA). In order to obtain a higher accuracy, quality control and CNV detection optimization was conducted using the PennCNV software when generating CNV calls [16]. To identify reliable CNV data, other studies have used different criteria for filtering unreliable CNV calls. Jakobsson et al. restricted analyses to CNVs with a minimum of 10 markers per CNV to minimize the number of false positives [37]. In our study using PennCNV analysis, we tested the impact of genomic waves on CNV calls and the filtering parameters with or without the –gcmodel option set. Compared with the results from the PennCNV using the –gcmodel option, more CNVs were identified without the –gcmodel option (5418 CNVs) than with the –gcmodel option (5008 CNVs; data not shown). This result was similar to that reported by Hou et al.[16]. These discordant calls were likely due to false positives called from the differentiating signal intensities caused by ‘genomic waves’. This further demonstrated that genomic waves have a significant effect on CNV analysis. We also used the ‘filter_cnv.pl’ program to perform sample-level quality control (see Materials and Methods). After this step, 73 further samples were excluded because their intensity data failed to conform to the criteria. Finally, the remaining 256 samples were used for CNV detection. This study focused only on the CNVs in 26 autosomes and excluded chrX and chrUn from our analysis because the chrX sequence and annotations are still not well-known [38], furthermore, their sequences and SNPs were uncertain, including the SNP mapping.
We identified a total of 238 CNVRs in three sheep breeds using the PennCNV detection algorithm. Of these, 219 CNVRs were losses, higher than the proportion of gains (Figure 2). This result might be due to the fact that losses are easier to detect than gains, because the exponential intensity data is linearly correlated with the copy number [39]. To exclude the potential false positive signals, we used another CNV detection software, CNVpartition, which detected 52 CNVRs. Forty-seven regions were consistently identified by both CNV detection software packages. Compared to CNVpartition based on other algorithms, such as QuantiSNP [40], Birdsuite [41], and GADA [42], PennCNV combines multiple sources of information, including the total signal intensity and allelic intensity ratios at each SNP marker to generate a hidden state for copy neutral loss of heterozygosity, the distance between neighboring SNPs, and the allele frequency of SNPs. PennCNV also integrates a computational approach by fitting regression models with GC content to overcome ‘genomic waves’ [43].
We compared our CNV length dataset with another sheep CNV study that was based on a cross species cattle-sheep aCGH experiment using a tiling oligonucleotide array with approximately 385,000 probes designed using the bovine genome. Because our dataset excluded CNV calls in the chrX and chrUn, only autosomal CNVR calls (238 CNVRs) were compared to the autosomal CNV calls of that study (135 CNVRs). As in other CNV studies using aCGH experiments [15, 20, 25, 26], the number of loss events in our dataset was larger than the number of gain events. However, further validation is required as it is also possible that purifying selection occurred [5]. The CNV sizes in our study ranged from 100 to 500 kb (average, 144.6 kb), which differed from those in the work of Fontanesi et al. (average, 73.9 kb) [26]. This CNV size difference was likely due to sampling differences or to resolution and genome coverage differences between the two techniques. The SNP genotyping resolution was 50.9 kb (mean probe spacing), whereas that of the aCGH platform was 6.3 kb. A SNP array lacked non-polymorphic probes designed specifically for CNV identification. Thus, only the large CNVRs were identified with the Ovine SNP50 assay. In future experiments, high-density SNP arrays combined with improved CNV calling algorithms could remedy these differences. In addition, we investigated the distribution pattern of these CNVRs in 26 autosomes and determined whether the CNV region was common in three sheep breeds. Interestingly, 75 of the 238 CNVRs had frequency rates of > 3%, whereas 121 of the 238 CNVRs had a frequency rate of < 1% in 256 individuals. This result might have been influenced by the innate limitation of Ovine SNP 50 arrays, and therefore, many common CNVs could have been missed.
We used UCSC gene annotation (http://genome.ucsc.edu) to identify genes that were located within or partially overlapped with CNVRs. Initially, we used a database of known sheep genes to annotate the gene content of CNVRs. However, the annotation results showed only four CNVRs overlapping with five known RefSeq genes in sheep CNVRs (see Additional file 5: Table S6). These results are likely attributable to the fact that the sheep genome is not well-annotated compared to the human genome or that of other domesticated animals [44–48]. The current sheep genome version (ISGC Ovis aries 3.1) is incomplete and known related genes are rarely identified in the sheep RefSeq gene database [38]. Therefore, we performed a BLASTN search for homologous human and cattle sequences using the UCSC table browser tool, and identified 563 genes homologous to human IDs identified in DAVID. We conducted a GO analysis to determine the biological effects of the 563 copy number variant genes (homologous to human genes). Similar to other results for GO analyses, the enriched genes were related to those involved in olfactory receptor (OR) activity, sensory perception of smell, sensory perception of chemical stimulus, sensory perception, cognition, G-protein coupled receptor protein signaling pathway, neurological system processes, cell surface receptor-linked signal transduction, plasma membrane and integral membrane components. Table 4 showed that the genes involved in environmental responses were over-represented in the CNVRs. In this study, functional analysis results were similar to those previously reported in CNV studies in human and other mammals [11, 15, 19, 26, 49]. This significant (p < 3.51 × 10-158) enrichment for OR activity might represent the frequent occurrence of CNVs in gene clusters for OR [50], as previously observed in cattle [19]. In addition, we observed 563 human orthologous genes in sheep CNVRs, of which 341 genes were previously reported in the DGV database (Additional file 6: Table S7). Moreover, we identified CNV regions that may overlap with Online Mendelian Inheritance in Man (OMIM) genes influencing disease susceptibility (Table 4). Notwithstanding, in the great majority of cases, these CNVs encompass genes and thereby directly affect gene dosage by loss or gain in the level of gene expression [2, 3, 51]. However, some other reports have suggested that CNVs are located preferably in gene-poor regions [52, 53] or in noncoding regions, and some studies provide evidence that the CNVs are considered nonpathogenic. Recent studies have described genome-wide distribution of CNVs in regions that encompass noncoding sequences, thereby affecting the regulation of distant target genes [54–57]. This suggested impact of CNVs in noncoding regions requires further elucidation in future studies. As discussed above, functional analysis studies indicated that the CNV genes possess a wide spectrum of molecular functions. However, since the sheep genome is less well-defined, Sheep CNVRs warrant further investigation for their roles in complex traits.
To verify CNV obtained by PennCNV, we conducted quantitative PCR (qPCR) on ten selected CNVRs and compared them with a sample control region known to have no CNVs (the DGAT1 gene fragment). We found that 26.7% of our qPCR assays (16 confirmed/60 total) agree with our CNVRs predictions in these regions. In total, six regions of these CNVRs were validated in at least one qPCR assay. However, it should be noted that another four CNVRs were not confirmed by qPCR (Additional file 4: Table S5). Reasons for this discrepancy between the CNVR analysis of Ovine SNP50 BeadChip data and the qPCR experiment may be due to the fact that the low probe density of the Ovine SNP50 BeadChip made it difficult to identify the true boundaries of CNVRs, resulting in an overestimation of their actual sizes. In addition, the primer pairs might have been designed outside the copy number polymorphic region.
In this research, only a small proportion of CNVRs identified overlapped with other studies. A similar situation was also encountered in human and other mammal CNV studies [10, 16, 58]. This indicated that a vast amount of CNVs existing had not been detected in the Ovine genome. Furthermore, a comparative analysis of CNVRs identified in sheep, goat and cattle could reveal the evolutionary mechanisms determining CNV formation during the mammalian evolution.
Conclusions
We firstly constructed a sheep CNV map based on the Ovine SNP50 array. 238 CNVRs were totally identified in the sheep autosomal genome, these CNVRs were non-randomly distributed on chromosomes, and 70% of which (166/238 CNVRs) were shared in > two animals. We also confirmed 6 CNVRs in total of 10 selected CNVRs by qPCR method. Compared with the results of CNV studies based on aCGH, the currently available genome wide SNP assays can infer Ovine CNV efficiently. However, it should be noticed that smaller size CNVs (< 10 kb) were not identified using this SNP panel and only 16 out of the 60 qPCR assays confirmed in this study was likely to be a overestimation of the true number and true boundary of CNVRs in the sheep genome. On the other hand, in this study, we using two detection programs: PennCNV and CNVpartition, to identify the sheep genome CNVs, results indicated that 47 CNVRs identified by the PennCNV (19.7%) overlapped with the CNVpartition data (90.4%), which highlighted the differences and commonalities of the two detection methods. Follow-up studies for high-resolution CNV mapping require improved SNP assay and next-generation sequencing to improve the accuracy of CNV calling. In addition, integration of different algorithms can enhance the detection of genomic structure variations. Furthermore, our findings would be of help for understanding the ovine genome and provide preliminary foundation for carrying out the CNVs association studies with economically important phenotypes of sheep in the future.
Methods
The experiments were performed on trait records and DNA samples that had been collected previously from animals born in 2010. Because no new animals were handled in these experiments, there are no needs of Animal Care and Use Committee approval for this study.
Samples and genotyping
Blood samples were collected from 329 six-month-old lambs from three breeds including 161 German Mutton sheep (71 males, 90 females), 99 Dorper (49 males, 50 females), and 69 Sunite sheep (57 males, 12 females) using the TIANamp Blood DNA Kit (Tiangen Biotech Co. Ltd. Beijing, China). Whole Genomic DNAs were genotyped using the Illumina Ovine SNP50 BeadChip that contained 54,241 single nucleotide polymorphisms (SNPs) and the average spacing was 50.9 kb. Two individuals were excluded because of call rate less than 98%. The DNA from the remaining 327 individuals that passed the sample quality control criteria were entered into the subsequent CNV detection and analysis procedures.
CNV identification and filter quality control
All signal intensity (log R ratio: LRR) and allelic intensity (B allele frequency: BAF) ratios of samples were reported using Illumina GenomeStudio1.0 software for each SNP. The PFB file was calculated based on the BAF of each marker in these populations. The sheep GC model file was generated by calculating the GC content of the 1 Mb genomic region surrounding each marker (500 Kb each side). CNVs were inferred using a Hidden Markov Model (PennCNV, http://www.openbioinformatics.org/penncnv/) [39]. PennCNV quality filters were applied after CNV detection. We used high quality samples with a standard deviation (SD) of LRR < 0.30 and with the default set: BAF drift as 0.01 and waviness factor value between -0.05 and 0.05, respectively. Appropriate LRR adjustments based on the GC model were incorporated in PennCNV. In addition, we used the program argument: the “lastchr 26” in the “detect” argument for specific CNVRs. CNV CNVRs were determined by aggregating overlapping CNVs identified in different animals, as reported previously [2, 4]. After filtering the samples, the CNVRs identified by PennCNV were shown (Additional file 2: Table S4). For construction of the CNVR map, we classified the status of these CNVR into three categories, ‘Loss’ (CNVR containing deletion), ‘Gain’ (CNVR containing duplication) and ‘Both’ (CNVR containing both deletion and duplications). In addition,we employed the LiftOver tool to map sheep CNVRs coordinates on the Btau_4.0 version so as to compare the CNVRs detected in sheep genome by SNP array with CNVRs in ruminants.
CNV calling using CNVpartition
In order to verify the CNVs detected by PennCNV, we employed the CNVpartition software [59] to analyze the same data set as well, with the confidence score threshold of 35. The CNVRs detected were cross-validated by CNVpartition and PennCNV. Furthermore, we applied a ‘reciprocal any overlapping’ method to the CNVRs detected by the two software analyses, and determined the sharing regions by both software programs.
Gene content and functional analysis
We used the UCSC table browser tool (ISGC ovis aries version 1.0) to identify the gene content located within or partially overlapping with the CNVRs. However, because the sheep genome is not well-annotated compared to the human genome, we used the human orthologs for gene ontology analyses, and DAVID Bioinformatics Resources (http://david.abcc.ncifcrf.gov) was used for further GO functional analysis, including Gene Ontology [13], KEGG pathway [14] and OMIM.
To investigate the gene functions of orthologous human genes in the CNVRs by BLAST search using the UCSC genome browser, we compared these genes in the CNVRs identified by PennCNV with genes that are disease-trait related in the Database of Genomic Variations (DGV) [60]. For this analysis, we used the latest data from the DGV (varation.hg19.v10.nov.2010.txt) downloaded from the DGV website (http://projects.tcag.ca/varation/).
Validation of CNVR by qPCR
Quantitative PCR experiments (qPCR) were performed using the SYBR green chemistry on the CFX96 Detection System (Bio-Rad, Hercules, CA, USA). The DGAT1 gene was chosen as a reference location for all qPCR experiments [26]. Primer version 5.0 was used to design PCR primers for the selected target CNVs and reference gene. Primer pairs for the control gene fragments and analyzed CNVRs are listed in the Additional file 4: Table S5. qPCR conditions were as follows: The following reagents were used for amplification in 20 μL: 2 μL of DNA, 10 μL of UltraSYBR Mixture (2×), 0.4 μL forward primer (10 μM), 0.4 μL reverse primer (10 μM); thermal cycling was initiated with a 2 min incubation at 50°C followed by a denaturation step from 10 min at 95°C, to 40 cycles of 15 sec at 95°C, and lastly to 1 min at 60°C. All reactions were run in triplicate and included controls without template. The 2-△△Cq method was used to calculate the copy number [61–63], where Cq is the threshold cycle. The CN of the target gene in test sample against reference samples is given by 2 × 2-ΔΔCq, where ΔΔCq = [(Cq of target gene in each test sample - Cq of DGAT1 in each test sample) - (average Cq of target gene in reference samples - average Cq of DGAT1 in reference samples)] [23].
Web resources
UCSC: http://genome.ucsc.edu
PennCNV: http://www.openbioinformatics.org/penncnv/
DAVID Bioinformatics Resources: http://david.abcc.ncifcrf.gov
The Database of Genomic Variations website (DGV): http://projects.tcag.ca/varation/
R program: http://www.R-project.org/
References
Manolio TA, Collins FS: The HapMap and genome-wide association studies in diagnosis and therapy. Annu Rev Med. 2009, 60: 443-456.
Feuk L, Carson AR, Scherer SW: Structural variation in the human genome. Nat Rev Genet. 2006, 7 (2): 85-97.
Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P: Origins and functional impact of copy number variation in the human genome. Nature. 2010, 464 (7289): 704-712.
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W: Global variation in copy number in the human genome. Nature. 2006, 444 (7118): 444-454.
Zhang F, Gu W, Hurles ME, Lupski JR: Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009, 10: 451-481.
Stankiewicz P, Lupski JR: Structural Variation in the Human Genome and its Role in Disease. Annu Rev Med. 2010, Palo Alto: Annual Reviews, 437-455. Volume 61
Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK: Mapping copy number variation by population-scale genome sequencing. Nature. 2011, 470 (7332): 59-65.
Graubert TA, Cahan P, Edwin D, Selzer RR, Richmond TA, Eis PS, Shannon WD, Li X, McLeod HL, Cheverud JM: A high-resolution map of segmental DNA copy number variation in the mouse genome. PLoS Genet. 2007, 3 (1): e3-
She X, Cheng Z, Zollner S, Church DM, Eichler EE: Mouse segmental duplication and copy number variation. Nat Genet. 2008, 40 (7): 909-914.
Perry GH, Tchinda J, McGrath SD, Zhang J, Picker SR, Caceres AM, Iafrate AJ, Tyler-Smith C, Scherer SW, Eichler EE: Hotspots for copy number variation in chimpanzees and humans. Proc Natl Acad Sci USA. 2006, 103 (21): 8006-8011.
Lee AS, Gutierrez-Arcelus M, Perry GH, Vallender EJ, Johnson WE, Miller GM, Korbel JO, Lee C: Analysis of copy number variation in the rhesus macaque genome identifies candidate loci for evolutionary and human disease studies. Hum Mol Genet. 2008, 17 (8): 1127-1136.
Emerson JJ, Cardoso-Moreira M, Borevitz JO, Long M: Natural selection shapes genome-wide patterns of copy-number polymorphism in Drosophila melanogaster. Science. 2008, 320 (5883): 1629-1631.
Maydan JS, Lorch A, Edgley ML, Flibotte S, Moerman DG: Copy number variation in the genomes of twelve natural isolates of Caenorhabditis elegans. BMC Genomics. 2010, 11: 62-
Bae JS, Cheong HS, Kim LH, NamGung S, Park TJ, Chun JY, Kim JY, Pasaje CF, Lee JS, Shin HD: Identification of copy number variations and common deletion polymorphisms in cattle. BMC Genomics. 2010, 11: 232-
Fadista J, Thomsen B, Holm LE, Bendixen C: Copy number variation in the bovine genome. BMC Genomics. 2010, 11: 284-
Hou Y, Liu GE, Bickhart DM, Cardone MF, Wang K, Kim ES, Matukumalli LK, Ventura M, Song J, VanRaden PM: Genomic characteristics of cattle copy number variations. BMC Genomics. 2011, 12: 127-
Kijas JW, Barendse W, Barris W, Harrison B, McCulloch R, McWilliam S, Whan V: Analysis of copy number variants in the cattle genome. Gene. 2011, 482 (1–2): 73-77.
Zhan BJ, Fadista J, Thomsen B, Hedegaard J, Panitz F, Bendixen C: Global assessment of genomic variation in cattle by genome resequencing and high-throughput genotyping. BMC Genomics. 2011, 12:
Seroussi E, Glick G, Shirak A, Yakobson E, Weller JI, Ezra E, Zeron Y: Analysis of copy loss and gain variations in Holstein cattle autosomes using BeadChip SNPs. BMC Genomics. 2010, 11: 673-
Liu GE, Hou Y, Zhu B, Cardone MF, Jiang L, Cellamare A, Mitra A, Alexander LJ, Coutinho LL, Dell’Aquila ME: Analysis of copy number variations among diverse cattle breeds. Genome Res. 2010, 20 (5): 693-703.
Alvarez CE, Akey JM: Copy number variation in the domestic dog. Mamm Genome. 2012, 23 (1–2): 144-163.
Wang X, Nahashon S, Feaster T, Bohannon-Stewart A, Adefope N: An initial map of chromosomal segmental copy number variations in the chicken. BMC Genomics. 2010, 11 (1): 351-
Fadista J, Nygaard M, Holm LE, Thomsen B, Bendixen C: A snapshot of CNVs in the pig genome. PLoS One. 2008, 3 (12): e3916-
Ramayo-Caldas Y, Castello A, Pena RN, Alves E, Mercade A, Souza CA, Fernandez AI, Perez-Enciso M, Folch JM: Copy number variation in the porcine genome inferred from a 60 k SNP BeadChip. BMC Genomics. 2010, 11: 593-
Fontanesi L, Martelli PL, Beretti F, Riggio V, Dall’Olio S, Colombo M, Casadio R, Russo V, Portolano B: An initial comparative map of copy number variations in the goat (Capra hircus) genome. BMC Genomics. 2010, 11: 639-
Fontanesi L, Beretti F, Martelli PL, Colombo M, Dall’olio S, Occidente M, Portolano B, Casadio R, Matassino D, Russo V: A first comparative map of copy number variations in the sheep genome. Genomics. 2011, 97 (3): 158-165.
Fontanesi L, Martelli PL, Scotti E, Russo V, Rogel-Gaillard C, Casadio R, Vernesi C: Exploring copy number variation in the rabbit (Oryctolagus cuniculus) genome by array comparative genome hybridization. Genomics. 2012, 100 (4): 245-251.
Peiffer DA, Le JM, Steemers FJ, Chang W, Jenniges T, Garcia F, Haden K, Li J, Shaw CA, Belmont J: High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006, 16 (9): 1136-1148.
Rincon G, Weber K, Van Eenennaam A, Golden B, Medrano J: Hot topic: Performance of bovine high-density genotyping platforms in Holsteins and Jerseys. J Dairy Sci. 2011, 94 (12): 6116-6121.
McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PI, Maller JB, Kirby A: Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008, 40 (10): 1166-1174.
Rhead B, Karolchik D, Kuhn RM, Hinrichs AS, Zweig AS, Fujita PA, Diekhans M, Smith KE, Rosenbloom KR, Raney BJ: The UCSC Genome Browser database: update 2010. Nucleic Acids Res. 2010, 38: D613-619.
Wain LV, Armour JA, Tobin MD: Genomic copy number variation, human health, and disease. Lancet. 2009, 374 (9686): 340-350.
Schoumans J, Ruivenkamp C, Holmberg E, Kyllerman M, Anderlid BM, Nordenskjold M: Detection of chromosomal imbalances in children with idiopathic mental retardation by array based comparative genomic hybridisation (array-CGH). J Med Genet. 2005, 42 (9): 699-705.
He YJ, Hoskins JM, McLeod HL: Copy number variants in pharmacogenetic genes. Trends Mol Med. 2011, 17 (5): 244-251.
Lou H, Li S, Yang Y, Kang L, Zhang X, Jin W, Wu B, Jin L, Xu S: A map of copy number variations in Chinese populations. PLoS One. 2011, 6 (11): e27341-
McCarroll SA: Copy number variation and human genome maps. Nat Genet. 2010, 42 (5): 365-366.
Jakobsson M, Scholz SW, Scheet P, Gibbs JR, VanLiere JM, Fung HC, Szpiech ZA, Degnan JH, Wang K, Guerreiro R: Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008, 451 (7181): 998-1003.
Archibald AL, Cockett NE, Dalrymple BP, Faraut T, Kijas JW, Maddox JF, McEwan JC, Hutton Oddy V, Raadsma HW, Wade C: The sheep genome reference sequence: a work in progress. Anim Genet. 2010, 41 (5): 449-453.
Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M: PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 17 (11): 1665-1674.
Colella S, Yau C, Taylor JM, Mirza G, Butler H, Clouston P, Bassett AS, Seller A, Holmes CC, Ragoussis J: QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007, 35 (6): 2013-2025.
Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins PJ, Darvishi K: Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet. 2008, 40 (10): 1253-1260.
Pique-Regi R, Monso-Varona J, Ortega A, Seeger RC, Triche TJ, Asgharzadeh S: Sparse representation and Bayesian detection of genome copy number alterations from microarray data. Bioinformatics. 2008, 24 (3): 309-318.
Diskin SJ, Li M, Hou C, Yang S, Glessner J, Hakonarson H, Bucan M, Maris JM, Wang K: Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. Nucleic Acids Res. 2008, 36 (19): e126-
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W: Initial sequencing and analysis of the human genome. Nature. 2001, 409 (6822): 860-921.
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA: The sequence of the human genome. Science. 2001, 291 (5507): 1304-1351.
Hillier LW, Miller W, Birney E, Warren W, Hardison RC, Ponting CP, Bork P, Burt DW, Groenen MAM, Delany ME: Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004, 432 (7018): 695-716.
Elsik CG, Tellam RL, Worley KC, Gibbs RA, Muzny DM, Weinstock GM, Adelson DL, Eichler EE, Elnitski L, Guigo R: The genome sequence of taurine cattle: a window to ruminant biology and evolution. Science. 2009, 324 (5926): 522-528.
Humphray SJ, Scott CE, Clark R, Marron B, Bender C, Camm N, Davis J, Jenks A, Noon A, Patel M: A high utility integrated map of the pig genome. Genome Biol. 2007, 8 (7): R139-
Moon S, Kim YJ, Hong CB, Kim DJ, Lee JY, Kim BJ: Data-driven approach to detect common copy-number variations and frequency profiles in a population-based Korean cohort. Eur J Hum Genet. 2011, 19 (11): 1167-1172.
Nei M, Niimura Y, Nozawa M: The evolution of animal chemosensory receptor gene repertoires: roles of chance and necessity. Nat Rev Genet. 2008, 9 (12): 951-963.
McCarroll SA, Bradner JE, Turpeinen H, Volin L, Martin PJ, Chilewski SD, Antin JH, Lee SJ, Ruutu T, Storer B: Donor-recipient mismatch for common gene deletion polymorphisms in graft-versus-host disease. Nat Genet. 2009, 41 (12): 1341-1344.
Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK: A high-resolution survey of deletion polymorphism in the human genome. Nat Genet. 2006, 38 (1): 75-81.
Freeman JL, Perry GH, Feuk L, Redon R, McCarroll SA, Altshuler DM, Aburatani H, Jones KW, Tyler-Smith C, Hurles ME: Copy number variation: new insights in genome diversity. Genome Res. 2006, 16 (8): 949-961.
Kantaputra PN, Klopocki E, Hennig BP, Praphanphoj V, Le Caignec C, Isidor B, Kwee ML, Shears DJ, Mundlos S: Mesomelic dysplasia Kantaputra type is associated with duplications of the HOXD locus on chromosome 2q. Eur J Hum Genet. 2010, 18 (12): 1310-1314.
Kurth I, Klopocki E, Stricker S, van Oosterwijk J, Vanek S, Altmann J, Santos HG, van Harssel JJ, de Ravel T, Wilkie AO: Duplications of noncoding elements 5’ of SOX9 are associated with brachydactyly-anonychia. Nat Genet. 2009, 41 (8): 862-863.
Dathe K, Kjaer KW, Brehm A, Meinecke P, Nurnberg P, Neto JC, Brunoni D, Tommerup N, Ott CE, Klopocki E: Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2. Am J Hum Genet. 2009, 84 (4): 483-492.
Wieczorek D, Pawlik B, Li Y, Akarsu NA, Caliebe A, May KJ, Schweiger B, Vargas FR, Balci S, Gillessen-Kaesbach G: A specific mutation in the distant sonic hedgehog (SHH) cis-regulator (ZRS) causes Werner mesomelic syndrome (WMS) while complete ZRS duplications underlie Haas type polysyndactyly and preaxial polydactyly (PPD) with or without triphalangeal thumb. Hum Mutat. 2010, 31 (1): 81-89.
Perry GH, Yang F, Marques-Bonet T, Murphy C, Fitzgerald T, Lee AS, Hyland C, Stone AC, Hurles ME, Tyler-Smith C: Copy number variation and evolution in humans and chimpanzees. Genome Res. 2008, 18 (11): 1698-1710.
Dellinger AE, Saw SM, Goh LK, Seielstad M, Young TL, Li YJ: Comparative analyses of seven algorithms for copy number variant identification from single nucleotide polymorphism arrays. Nucleic Acids Res. 2010, 38 (9): e105-
Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C: Detection of large-scale variation in the human genome. Nat Genet. 2004, 36 (9): 949-951.
Bodin L, Beaune PH, Loriot MA: Determination of cytochrome P450 2D6 (CYP2D6) gene copy number by real-time quantitative PCR. J Biomed Biotechnol. 2005, 2005 (3): 248-253.
D’haene B, Vandesompele J, Hellemans J: Accurate and objective copy number profiling using real-time quantitative PCR. Methods. 2010, 50 (4): 262-270.
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods. 2001, 25 (4): 402-408.
Acknowledgements
We thank Hongguang Qian, Yunhua Li (Institute of Animal Science, Inner Mongolia Academy of Agricultural & Animal Husbandry Sciences), Monke-Bater, A-Rong, Tubuxin (Sunite County, Inner Mongolia Autonomous Region) and Hua Chen (Aoqun Animal Husbandry, Co. Ltd) for helping collect samples. We especially thank Xiaojing Miao (Shanghai South Gene Technology Co. Ltd.) and Yanjun Xiao (Shanghai Biotechnology Co. Ltd) for technical assistance. We also greatly appreciate Fontanesi (University of Bologna) and anonymous reviewers for insightful comments on the early version of the manuscript. This work was supported by the National Modern Agricultural Industry Technology Fund for Scientists in Sheep Industry System (CARS-39-04B), the National Natural Science Foundation of China (No. 30972094), the National Nonprofit Institute Research Grant (2012jc-2) and“Twelfth Five-Year-Plan” of the Chinese 863 Program (No. 2011AA100307-02).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare no competing financial interests.
Authors’ contributions
JL performed all statistical and computational analyses and wrote the manuscript. LZ provided SNP genotype data and coordinated the SNP analysis. LX and JL contributed by giving advice on analyses or by giving advice on the content of the paper. LD and FZ conceived and designed the experiments. XZ, SZ, CW, XZ, participated in phenotype data collection. All authors read and approved the final manuscript.
Jiasen Liu, Li Zhang contributed equally to this work.
Electronic supplementary material
12864_2012_4905_MOESM1_ESM.doc
Additional file 1: Table S1: Size distribution of CNVs identified by PennCNV. Table S2. Size distribution of CNVR identified by PennCNV. Table S3. Distribution pattern of CNVR across sheep 26 autosome (a The size of chromosome was obtained from NCBI website). (DOC 86 KB)
Additional file 2: Table S4: Summary of CNV regions and their frequency in sheep genome. (XLS 80 KB)
12864_2012_4905_MOESM7_ESM.xls
Additional file 7: Table S8: Comparisons between identified 238CNVRs in this study and the existing other ruminants CNVRs dataset. (XLS 75 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Liu, J., Zhang, L., Xu, L. et al. Analysis of copy number variations in the sheep genome using 50K SNP BeadChip array. BMC Genomics 14, 229 (2013). https://doi.org/10.1186/1471-2164-14-229
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-14-229