
Abstract
Background
Technological advances in high throughput genome sequencing are making whole genome sequencing (WGS) available as a routine tool for bacterial typing. Standardized procedures for identification of relevant genes and of variation are needed to enable comparison between studies and over time. The core genes--the genes that are conserved in all (or most) members of a genus or species--are potentially good candidates for investigating genomic variation in phylogeny and epidemiology.
Results
We identify a set of 2,882 core genes clusters based on 73 publicly available Salmonella enterica genomes and evaluate their value as typing targets, comparing whole genome typing and traditional methods such as 16S and MLST. A consensus tree based on variation of core genes gives much better resolution than 16S and MLST; the pan-genome family tree is similar to the consensus tree, but with higher confidence. The core genes can be divided into two categories: a few highly variable genes and a larger set of conserved core genes, with low variance. For the most variable core genes, the variance in amino acid sequences is higher than for the corresponding nucleotide sequences, suggesting that there is a positive selection towards mutations leading to amino acid changes.
Conclusions
Genomic variation within the core genome is useful for investigating molecular evolution and providing candidate genes for bacterial genome typing. Identification of genes with different degrees of variation is important especially in trend analysis.
Similar content being viewed by others

Background
With the increasing number of available bacterial genome sequences, when these genomes are compared, the genetic variation within bacterial species is greater than previously predicted [1, 2]. Rapid and reliable sub-typing of bacterial pathogens is important for identification of outbreaks and monitoring of trends in order to establish population structure and to study the evolution among bacterial genomes especially within and between the outbreak strains. Today, the most widely used typing methods for bacterial genomes include multilocus sequence typing (MLST), pulsed field gel electrophoresis (PFGE), sequencing of 16S rRNA genes, and multilocus variable-number of tandem-repeat analysis (MLVA).
PFGE and MLVA have major benefits, but are time consuming and the results are difficult to standardize [3]. Other typing methods which rely on one or a few ubiquitous genes, such as the 16S rRNA gene or a set of housekeeping genes in MLST, are capable of classification at the species level and sometimes also at the subspecies level, but the biological information in a narrow selection of genes will rarely be sufficient to clearly distinguish between closely related strains such as several isolates of the same serotype [4–6]. Thus, more of the genome content should be considered rather than just one or a few genes [4].
The price and time for whole genome sequencing will soon be in the same range as the traditional typing methods mentioned above. Genome sequencing can be a powerful method in epidemiological and evolutionary investigations [7–9]. Although, to date, this has only been used in more limited epidemiological investigations where isolates suspected to be part of the same outbreak have been compared to a reference genome. In the future, it is likely that WGS will become a routine tool for identification and characterization of bacterial isolates, as hinted at in the first 'real-time' sequencing of the E. coli O104 outbreak in Germany in the summer of 2011 [10] and the Vibrio cholerae outbreak in Haiti in October 2010 [11]. This requires standard procedures for identifying variation and for analyzing similarities and differences.
Conserved genes are present across bacterial genomes of the same species (or genus). A fraction of these genes--those conserved in all (or most) of the genomes of a given bacterial taxonomic group--is called the 'core-genome' of that group. The core-genome can be identified either within a genus or species [3] and can be used to identify the variable genes in a given genome [12]. In addition, the conserved genes in general appear to evolve more slowly, and can be used for determining relationships among bacterial isolates [13].
Currently there are more than a hundred bacterial species for which sufficient genomic data are available to estimate the species core-genome (that is, there are at least three genomes sequenced from the same species) [14]. Among these, Salmonella enterica is a good candidate species for conserved gene identification because the genomes are quite similar [15]. Moreover, S. enterica is one of the most important food-borne pathogens and is responsible for global outbreaks [16] which makes international standard typing procedures of major importance in order to allow for global comparisons [17]. The Salmonella genus has only two species with sequenced genomes: Salmonella bongori and Salmonella enterica. In turn, S. enterica is divided into 6 sub-species: enterica, salamae, arizonae, diarizone, houtenae and indica. Presently, S. enterica is classified into more than 2,500 serotypes [18].
In order to investigate an outbreak caused by Salmonella, characterization of Salmonella isolates from genome data is a crucial step. Salmonella genomes are highly similar, particularly within subspecies enterica, where little variance exists in the genomes [15]. This high similarity presents a challenge for typing and classification.
In their pioneering work Tettelin et al.[1] defined the core genes of a species by being those genes found present in (nearly) all known members of the species. Since then others have studied core and pan genomes at the genus level or even at the kingdom level [19], but for our purposes the original definition at the species level is suitable. In this work we identify the core genes within S. enterica genomes and determine variation between the different available genomes, both in terms of sequence and presence/absence of non-core genes; in the latter case using a method originally published by Snipen & Ussery [20]. We evaluate the value of different approaches for classification of isolates in epidemiological settings and compare our findings to currently used sequencing methods, both in long term trend analysis and outbreak investigations.
Results and discussion
The 73 Salmonella genomes used in this study are summarized in Additional file 1: Table S1. The set comprises 21 completed genomes and 52 nearly completed genomes. Of these, 35 genomes are closely-related S. Montevideo strains pertaining to an outbreak of salmonellosis from Italian-style spiced meat [21]. All genomes were retrived from GenBank [22] except S. Typhimurium str. DT104, which was received from the Sanger Institute's bacterial genome database. All Salmonella genomes are from subspecies enterica with the exception of the single S. enterica subsp. Arizonae.
Evaluation of traditional bacterial sequence-based typing
The ribosomal genes are essential for the survival of all cells, and their structure cannot change much because of their involvement in protein synthesis [23]. Thus, 16S rRNA genes are highly conserved among isolates belonging to the same bacterial species [4]. Exceptions may be N. meningitidis[24] and Mycoplasma[25]. However, due to limited variation within a given species, the 16S sequencing is often not useful for epidemiological studies, where the classification of highly similar strains is needed. Jacobsen et al. shows a phylogenetic tree based on 16S rRNA genes, extracted from 26 Salmonella enterica genomes, using RNAmmer [15, 26]. As expected, there is not sufficient resolution to distinguish among the Salmonella subspecies enterica.
Genes such as rpoB or sodA have been suggested as substitutes for 16S rRNA and have shown improved efficacy in species identification [27], although it remains unlikely that a single gene can always reflect the subtle differences between genomes of the same species.
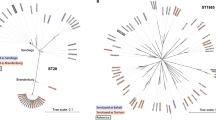
The limitations of using a single gene may be improved by the simultaneous analysis of multiple genes. Multi Locus Sequence Typing (MLST) has found wide applications, especially in phylogenetic studies and is most commonly based on seven housekeeping genes - each bacterial species having its own set. For Salmonella these are: aroC, dnaN, hemD, hisD, purE, sucA and thrAhttp://www.mlst.net. A MLST tree, based on an in silico analysis of the 73 available Salmonella enterica genomes in Genbank, is shown in Figure 1. Strains of the same serovar generally cluster into distinct groups, although exceptions exist; for example the S. Weltevreden str. HI_N05-537 is mixed with S. Montivideo. Futhermore, recent work on 61 sequenced E. coli genomes [4], found that the 16S rRNA tree cannot resolve well within the genus level and also that MLST cannot differentiate pathogenic strains from non- pathogenic strains. Still, MLST has proven useful for long-term analysis of population structures, but often fails to detect differences between closely related strains [28]. Indeed, improved MLST schemes that include more than 7 genes have been suggested [4].

In silico MLST tree. Seven housekeeping genes were extracted from Salmonella genomes. Concatenated sequences were aligned by MUSCLE. The phylogenetic trees were generated by MEGA5 using bootstrap maximum likelihood method. Each color represents a different serogroup (O antigen). The confidence value is the bootstrap value calculated by sampling with replacement from the multiple sequence alignment.
For Salmonella, sequencing specific short repeats and virulence genes have recently been suggested as an alternative and improved method for typing of S. Enteritidis[29]. The usefulness of this approach in epidemiological studies and typing is currently unknown, although the choice of repeats must be tailored for the specific bacterial species studies.
Identification of core genes
Determining gene conservation across multiple genomes is not overly difficult, but certain choices must be made which will affect the final outcome. Using a previously published method [20, 30, 31] which employs single-linkage clustering on top of BLASTp alignments, sets of pan- and core-genomes were estimated, based on all 73 Salmonella genomes. The progression of the pan- and core-genomes is shown in Figure 2A. The number of novel gene clusters in the pan-genome gradually increases when more genomes are considered, while the number of conserved gene clusters constituting the core genome decreases slightly. When all Salmonella genomes have been considered, there are 10,581 pan gene clusters and 2,882 core gene clusters (Additional file 2) in species enterica. In the step going from S. Typhimurium to S. Typhi, the number of core genes drops suddenly, most likely because the S. Typhi genome has undergone considerable pseudogene formation resulting in gene loss [32]. The number of core genes drops again when adding a genome of the subspecies arizonae which is associated with cold-blooded animals. This technique has previously been applied successfully in finding core genomes for Proteobacteria genera Burkholderia[33], Escherichia coli[4], Vibrionaceae[34] and Campylobacter jenuni[30], as well as Bacteroides [35] and Lactic acid bacteria [36].

Pan- core-genome plot and variation plot. (A) Pan- and core-genome plot of 73 Salmonella enterica. The plot shows an increase of the pan-genome (blue line) and a decrease of the core-genome (red line) as more genomes are added. The last points show the total number of gene clusters in the pan-genome and the core-genome. (B) Variation plot. This plot shows the variation within core gene clusters in amino acid levels (green dots) and nucleotide levels (red dots). Black dots show the distribution of housekeeping genes in the core genes. The Y- and X-axes represent average percent identity and numerical core gene cluster name respectively.
Genomic variation within the core genes
The core genes as calculated above were used for constructing a gene variation plot by performing all-against-all BLAST alignments between 2,882 core gene clusters and all 73 Salmonella enterica genomes. The resulting average identities within each core gene cluster is displayed in Figure 2B. From this figure, the average percent identity was very high (> 98%) in most of the core genes, but dropped sharply for around 5% of the core genes. From this plot, the identified core genes can be divided into two categories: a small group of highly variable genes and the majority of genes which show little variation.
For the highly variable core genes, the variation in amino acid sequences (Figure 2B, green dots) was higher than for the nucleotide sequences (Figure 2B, red dots), whereas the opposite was the case for the more conserved core genes. This indicates that for core genes with low variation there is a selection against mutations leading to amino acid changes, whereas for the highly variable genes, positive selection for amino acid changes seems to be the case. In order to confirm these hypothesis, the approximation of dN/dS has been performed by dividing the number of non-synonymous changes per non-synonymous sites with the number of synnonymous changes per synonymous sites [37] using S. Typhimurium str. LT2 as a reference genome. The median dN/dS ratio for conserved and highly variable core genes are 1.0 and 1.25 respectively. Therefore, the amino acid changes in highly variable core genes might be due to an increase in positive selection at some sites. Nonetheless, the importance of this needs to be confirmed by additional analysis, although one could imagine, for example, a selective pressure to vary the surface proteins to avoid immune response.
The seven genes used for MLST are marked in the Figure 2B, and are scattered throughout the highly conserved part of the core genes (Figure 2B, black dots) and, as expected, little variation exists in these genes. Including core genes from both the highly conserved and variable regions might be beneficial in evolution studies. On the one hand, the more slowly evolving genes are useful in distinguishing between divergent and convergent evolution, while faster evolving genes can help in strain identification.
Functional analysis of conserved genes
In order to determine the functional profile of core genes, the core gene clusters were aligned against UniProt [30]. Functional profiles were determined based on Gene Ontology (GO) terms and visualized in Figure 3. Though the difference is generally small, some terms common in conserved core genes tend to be less frequent in highly variable core genes; for example, electron carrier activity, structural molecule activity and metallochaperone activity. These functions are essential for living cells and are therefore enriched in conserved core genes. On the other hand, highly variable core genes encode many proteins that are associated with the extracellular region. In general, genes located outside the cell are known to be more variable [38].

Gene Ontology term summary of core genes. Gene Ontology terms for conserved core genes (blue bars) and highly variable core genes (red bars) are shown in 3 categories (from top to bottom): biological processes (green labels), cellular component (pink labels) and molecular function (black labels). GO are assigned from blast all-against-all between core genes and protein sequences from Uniprot based on 50/50 rule. All conclusions drawn about the variable set are relative to the fraction of like sequences in the conserved set, and not in any way absolute.
Consensus tree based on core gene clusters
Figure 4 shows a phylogenetic tree generated from the sequence of all 2,882 Salmonella core gene clusters. The tree generally divides the serotypes up well, but the bootstrap value in several branches is very low. This uncertainty could be due to the large number of core gene trees being analyzed individually; the low bootstrap values near the root reflect a lack of consensus at the higher levels. In contrast, the low bootstrap values found in S. Montevideo strains likely reflect uncertainty due to the high similarity of gene sequence of the clonal outbreak. All S. Montevideo strains sequenced were from a single outbreak [21] and as expected this analysis confirmed the almost complete identity of these isolates.

Consensus tree based on 2,882 core gene clusters. Phylogenetic trees were constructed from all core genes using PAUP. All trees were combined and the consensus trees were generated using the Phylip software package. The percentage of branches present in all trees is shown. The colors represent different serogroups, as in Figure 1.
A previous study described that there are 69 genes unique to Salmonella[39]. Instead of using all core genes, we generated a consensus tree based on these 69 Salmonella- specific genes (Additional file 3: Figure S1). We also constructed an additional four consensus trees based on sets of 69 core genes randomly picked from different areas in the variation plot (Figure 2B): from a mixture of high, medium and low variable core genes (Additional file 4: Figure S2), from medium variable core genes (Additional file 5: Figure S3), from highly variable core genes (Additional file 6: Figure S4) and from the area where the curve decreases in the variation plot (Additional file 7: Figure S5). The appearance of these 5 consensus trees was similar to the tree from Figure 4, with two exceptions: the trees based on the 69 specific genes (Additional file 3: Figure S1) and the highly variable core genes (Additional file 6: Figure S4). In the former, S. arizonae, which is not part of the subspecies enterica, was still mixed in with other enterica, while for the latter, S. Agona str. SL483 clustered away from the other subspecies enterica. Thus, based on these results, it appears that using only Salmonella unique genes or highly variable genes does not provide phylogenetically useful information and should probably not be used for future WGS studies. Comparisons using more genomes in more species can further test this.
Pan-genome tree
In principle, genome similarity is not only measurable by shared genes, but also by the absence of genes. Figure 5 is another tree, based on gene presence/absence across all the Salmonella genomes [20]. This tree bears a striking resemblance to the consensus tree based on core genes (Figure 4), although the bootstrap values are higher in many of the branches, especially near the root. Of all methods investigated in this study, the pan-genome tree presents itself as the best solution for a tree that can resolve strain differences in a biologically meaningful way, even if it would be expected to correlate more with phenotype than phylogeny. It is, however, important to note that creating pan-genome trees requires higher quality sequencing data and assemblies than what are typically obtained using short reads from second-generation sequencing methodologies. Even so, we have found that pan-genome trees with good correspondence to known bacterial types can be constructed from Solexa data (100 bp reads), if care is taken to ensure good assembly and gene finding (data not shown).

Pan-genome tree. This tree does not produce a sequence-based alignment tree but it is generated from the presence or absence of gene clusters across all Salmonella genomes [31]. The bootstrap values are shown in red.
The power to discriminate between variants differs between the methods used. The phylogenetic analysis for the MLST tree is based on the identified informative sites among the seven housekeeping genes, for the pan-genome tree on presence and absence of genes and for the consensus tree based on the informative sites of core gene clusters from alignments of all core genes trees. The number of infomative sites for in silico MLST tree, pan-genome tree and consensus tree based on core gene clusters were 877 bp (10,008 total base-pairs in the seven genes), 7,699 genes (10,581 total genes) and 880,832 bp (2,868,821 bp in all core genes), respectively. The pan genome and core gene analysis were based on much more variation than the MLST analysis and have a much stronger power to discriminate closely related strains.
Conclusions
Bacterial typing should provide meaningful information for both epidemiological and evolutionary studies. For epidemiology, the ability to differentiate unrelated isolates (discriminatory power) and the ability to cluster related isolates are crucial. 16S rRNA and the MLST genes rarely provide separation between closely related strains. The performance of the pan-genome tree, however, is valid for epidemiological investigation in both discriminatory and clustering abilities. One caveat is that this method depends on good quality genomic data.
Comparative genomics can determine the conserved genes (core-genome) among bacterial genomes at either genus or species level. Genomic variation within the core-genome can then be used to reveal highly variable genes (fast evolving genes) and conserved genes (slow evolving genes). These core genes are useful for investigating molecular evolution and remain useful as candidate genes for bacterial genome typing--even if they cannot be expected to differentiate highly similar isolates from e.g. outbreak cases, such is not always desirable. Even in cases where a deeper distinction of isolates is of interest, e.g. in mapping outbreaks, core genes might still be useful as a reference fragment for SNPs calling instead of using whole genome analysis. However, in term of computational costs, the consensus tree based on core genes requires more computational time than the other methods.
In the near future, global real-time surveillance of Salmonella and other pathogens giving simultaneous information on population structure and evolution, as well as outbreak detection, may well be possible.
Methods
Salmonella genome data and gene annotation
From public genome databases (NCBI and Sanger Institute's bacterial genome databases), 83 Salmonella enterica genomes available at the time (April, 2011) were downloaded. These genomes consisted of 21 completed genomes and 62 draft genomes. Due to the large number of contigs in some genomes, only 73 genomes were selected for this study (Additional file 1: Table 1). The gene finder Prodigal was used on DNA sequences of all genomes to eliminate biases in annotation quality and to standardize the genes found in all genomes [15]. Gene clusters were then inferred according to [15, 20, 30]
In silico MLST trees
The in silico MLST tree was constructed from seven housekeeping genes: aroC, dnaN, hemD, hisD, purE, sucA and thrAhttp://www.mlst.net. These genes were extracted from Salmonella genomes and concatenated. The concatenated sequences were aligned using MUSCLE [40]. Phylogenetic trees were generated by MEGA5 using the maximum likelihood method [41]. The confidence value is, in this case, the same as the bootstrap value, calculated by sampling with replacement from the multiple sequence alignments [42]. Thus, the in silico MLST differs from traditional MLST in that complete genes are used and not just the MLST alleles. However, since the alleles typically cover the majority of the genes, the difference is small.
Consensus trees
All core gene clusters from 73 Salmonella genomes were used for generating a consensus tree. Multiple alignments for each core gene cluster from all strains were performed using MUSCLE [40]. A phylogenetic tree for each core gene was generated using PAUP [43]. The Phylip package was used to construct the consensus tree from all the trees [44]. The bootstrap values are shown in the consensus tree.
GO annotation
The core gene clusters were compared in an all-against-all BLAST with protein sequences from UniProt based on the '50/50 rule' [30]. Functional profiles were summarized from BLAST results by mapping UniProt IDs to Gene Ontology (GO) terms. Mapping GO parental terms were performed using publicly available GO-PERL modules for searching through a graph structure of ontology data [45, 46]
Pan-genome trees
The Pan-genome matrix consists of gene clusters (rows) and genomes (columns). The absence and presence of genes across genomes are represented by 0's and 1's respectively. The relative Manhattan distance between genomes was calculated and used for hierarchical clustering. The bootstrap values are calculated in order to represent the confidence of branches [20].
References
Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, Margarit Y, Ros I, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N, Khouri H, Radune D, Dimitrov G, Watkins K, O'Connor KJ, Smith S, Utterback TR, White O, Rubens CE, Grandi G, Madoff LC, Kasper DL, Telford JL, Wessels MR, Rappuoli R, Fraser CM: Genome analysis of multiple pathogenic isolates of Streptococcus agalactia: Implications for the microbial "pan-genome". Proc Natl Acad Sci USA. 2005, 102 (39): 13950-13955. 10.1073/pnas.0506758102.
Binnewies TT, Motro Y, Hallin PF, Lund O, Dunn D, La T, Hampson DJ, Bellgard M, Wassenaar TM, Ussery DW: Ten years of bacterial genome sequencing: comparative-genomics- based discoveries. Funct Integr Genomics. 2006, 6: 165-185. 10.1007/s10142-006-0027-2.
Malorny B: New Approaches in Subspecies-level Salmonella Classification. Salmonella From Genome to Function. Edited by: Porwollik S. 2011, Norwich United Kingdom: Caister Academic Press, 1-23.
Lukjancenko O, Wassenaar TM, Ussery DW: Comparison of 61 Sequenced Escherichia coli Genomes. Microb Ecol. 2010, 60 (4): 708-720. 10.1007/s00248-010-9717-3.
Harris SR, Feil EJ, Holden MT, Quail MA, Nickerson EK, Chantratita N, Gardete S, Tavares A, Day N, Lindsay JA, Edgeworth JD, de Lencastre H, Parkhill J, Peacock SJ, Bentley SD: Evolution of MRSA During Hospital Transmission and Intercontinental Spread. Science. 2010, 327 (5964): 469-474. 10.1126/science.1182395.
Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R: The microbial pan-genome. Curr Opin Genet Dev. 2005, 15 (6): L589-L594. 10.1016/j.gde.2005.09.006.
Gardy JL, Johnston JC, Ho Sui SJ, Cook VJ, Shah L, Brodkin E, Rempel S, Moore R, Zhao Y, Holt R, Varhol R, Birol I, Lem M, Sharma MK, Elwood K, Jones SJ, Brinkman FS, Brunham RC, Tang P: Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med. 2011, 364 (8): 730-739. 10.1056/NEJMoa1003176.
Rasko DA, Worsham PL, Abshire TG, Stanley ST, Bannan JD, Wilson MR, Langham RJ, Decker RS, Jiang L, Read TD, Phillippy AM, Salzberg SL, Pop M, Van Ert MN, Kenefic LJ, Keim PS, Fraser-Liggett CM, Ravel J: Bacillus anthracis comparative genome analysis in support of the Amerithrax investigation. Proc Natl Acad Sci. 2011, 108 (12): 5027-5030. 10.1073/pnas.1016657108.
Pallen MJ, Loman NJ, Penn CW: High-throughput sequencing and clinical microbiology: progress, opportunities and challenges. Curr Opin Microbiol. 2010, 13 (5): 625-631. 10.1016/j.mib.2010.08.003.
Mellmann A, Harmsen D, Cummings CA, Zentz EB, Leopold SR, Rico A, Prior K, Szczepanowski R, Ji Y, Zhang W, McLaughlin SF, Henkhaus JK, Leopold B, Bielaszewska M, Prager R, Brzoska PM, Moore RL, Guenther S, Rothberg JM, Karch H: Prospective Genomic Characterization of the German Enterohemorrhagic Escherichia col O104:H4 Outbreak by Rapid Next Generation Sequencing Technology. PLoS One. 2011, 6 (7): e22751-10.1371/journal.pone.0022751.
Chin CS, Sorenson J, Harris JB, Robins WP, Charles RC, Jean-Charles RR, Bullard J, Webster DR, Kasarskis A, Peluso P, Paxinos EE, Yamaichi Y, Calderwood SB, Mekalanos JJ, Schadt EE, Waldor MK: The origin of the Haitian cholera outbreak strain. N Engl J Med. 2011, 364 (1): 33-42. 10.1056/NEJMoa1012928.
Adékambi T, Butler RW, Hanrahan F, Delcher AL, Drancourt M, Shinnick TM: Core gene set as the basis of multilocus sequence analysis of the subclass Actinobacteridae. PLoS One. 2011, 6 (3): e14792-10.1371/journal.pone.0014792.
Urwin R, Maiden MC: Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol. 2003, 11 (10): 479-487. 10.1016/j.tim.2003.08.006.
Kyrpides NC: Fifteen years of microbial genomics: meeting the challenges and fulfilling the dream. Nat Biotechnol. 2009, 27 (7): 627-632. 10.1038/nbt.1552.
Jacobsen A, Hendriksen RS, Aaresturp FM, Ussery DW, Friis C: The Salmonella enteric Pan-genome. Microb Ecol. 2011, 62 (3): 487-504. 10.1007/s00248-011-9880-1.
Foley SL, Zhao S, Walker RD: Comparison of molecular typing methods for the differentiation of salmonella foodborne pathogens. Foodborne Pathog Dis. 2007, 4 (3): 253-276. 10.1089/fpd.2007.0085.
Boxrud D, Monson T, Stiles T, Besser J: The role, challenges, and support of pulsenet laboratories in detecting foodborne disease outbreaks. Public Health Rep. 2010, 125 (Suppl 2): 57-62.
Popoff MY, Le Minor L: Taxonomy of the genus Salmonella. Changes in serovars nomenclature. Antigenic formulas of the Salmonella serovars, 7th revision. Edited by: Popoff MY, Le Minor L. 1997, Paris, France: WHO Collaborating Centre for Reference and Research on Salmonella. Institut Pasteur, 5-
Lapierre P, Gogarten JP: Estimating the size of the bacterial pan-genomes. Trends Genet. 2009, 25 (3): 107-110. 10.1016/j.tig.2008.12.004.
Snipen L, Ussery DW: Standard operation procedure for computing pangenome trees. Stand Genomics Sci. 2009, 2: 135-141.
Lienau EK, Strain E, Wang C, Zheng J, Ottesen AR, Keys CE, Hammack TS, Musser SM, Brown EW, Allard MW, Cao G, Meng J, Stones R: Identification of a Salmonellosis outbreak by means of molecular sequencing. N Engl J Med. 2011, 364 (10): 981-982. 10.1056/NEJMc1100443.
Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW: GenBank. Nucleic Acids Res. 2011, 39: D32-D37. 10.1093/nar/gkq1079.
Woose CR: Bacterial evolution. Microbiol Rev. 1987, 51 (2): 221-271.
Sacchi CT, Whitney AM, Reeves MW, Mayer LW, Popovic T: Sequence diversity of Neisseria meningitidi 16S rRNA genes and use of 16S rRNA gene sequencing as a molecular subtyping tool. J Clin Microbiol. 2002, 40 (12): 4520-4527. 10.1128/JCM.40.12.4520-4527.2002.
Königsson MH, Bölske G, Johansson KE: Intraspecific variation in the 16S rRNA gene sequences of Mycoplasma agalactia and Mycoplasma bovi strains. Vet Microbiol. 2002, 85 (3): 209-220. 10.1016/S0378-1135(01)00517-X.
Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW: RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35 (9): 3100-3108. 10.1093/nar/gkm160.
De Clerck E, De Vos P: Genotypic diversity among Bacillus licheniformi strains from various sources. FEMS Microbiol Lett. 2004, 231 (1): 91-98. 10.1016/S0378-1097(03)00935-2.
Li W, Raoult D, Fournier PE: Bacterial strain typing in the genomic era. FEMS Microbiol Rev. 2009, 33 (5): 892-916. 10.1111/j.1574-6976.2009.00182.x.
Liu F, Kariyawasam S, Jayarao BM, Barrangou R, Gerner-Smidt P, Ribot EM, Knabel SJ, Dudley EG: Subtyping Salmonella enterica Serovar Enteritidis Isolates from Different Sources by Using Sequence Typing Based on Virulence Genes and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs). Appl Environ Microbiol. 2011, 77 (13): 4520-4526. 10.1128/AEM.00468-11.
Friis C, Wassenaar TM, Javed MA, Snipen L, Lagesen K, Hallin PF, Newell DG, Toszeghy M, Ridley A, Manning G, Ussery DW: Genomic characterization of Camphylobacter jejun M1. PLoS One. 2010, 5 (8): e12253-10.1371/journal.pone.0012253.
Ussery DW, Wassenaar TM, Borini S: Computing for Comparative Genomics: Bioinformatics for Microbiologists (Computational Series). 2008, London: Springer Verlag
Holt KE, Thomson NR, Wain J, Langridge GC, Hasan R, Bhutta ZA, Quail MA, Norbertczak H, Walker D, Simmonds M, White B, Bason N, Mungall K, Dougan G, Parkhill J: Pseudogene accumulation in the evolutionary histories of Salmonella enterica serovars Paratyphi A and Typhi. BMC Genomics. 2009, 10: 36-10.1186/1471-2164-10-36.
Ussery DW, Kill K, Lagesen K, Sicheritz-Ponten T, Bohlin J, Wassenaar TM: The Genus Burkholderia: Analysis of 56 Genomic Sequences. Microbial Pathogenomics. Microbial Pathogenomics. Edited by: Reuse Hd, Bereswill S. 2009, Basel, Karger, 140-157.
Vesth T, Wassenaar TM, Hallin PF, Snipen L, Lagesen K, Ussery DW: On the Origins of a Vibrio Species. Microb Ecol. 2010, 59 (1): 1-13. 10.1007/s00248-009-9596-7.
Karlsson FH, Ussery DW, Nielsen J, Nookaew I: A closer look at bacteroides: phylogenetic relationship and genomic implications of a life in the human gut. Microb Ecol. 2011, 61 (3): 473-485. 10.1007/s00248-010-9796-1.
Lukjancenko O, Ussery DW, Wassenaar TM: Comparative genomics of Bifidobacterium, Lactobacillus and related probiotic genera. Microb Ecol. 2011,
Yi S: Synonymous and Nonsynonymous Rates. eLS. 2007, doi: 10.1002/9780470015902.a0005110.pub2
Julenius K, Pedersen AG: Protein evolution is faster outside the cell. Mol Biol Evol. 2006, 23 (11): 2039-2048. 10.1093/molbev/msl081.
Lukjancenko O, Ussery DW: Design of an Enterobacteriaceae Pan-Genome Microarray Chip. Proceeding of CSBio 2010: Thailand. 2010, 115: 174-189.
Edgar RC: MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004, 5: 113-10.1186/1471-2105-5-113.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28 (10): 2731-2739. 10.1093/molbev/msr121.
Wróbel B: Statistical measures of uncertainty for branches in phylogenetic trees inferred from molecular sequences by using model-based methods. J Appl Genet. 2008, 49 (1): 49-67. 10.1007/BF03195249.
Swofford DL: PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. 2004, Sunderland: Sinauer Associates
Felsenstein J: PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. Department of Genome Sciences. 2005, Seattle: University of Washington
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000, 25: 25-29. 10.1038/75556.
Leekitcharoenphon P, Taweemuang U, Palittapongarnpim P, Kotewong R, Supasiri T, Sonthayanon B: Predicted sub-populations in a marine shrimp proteome as revealed by combined EST and cDNA data from multiple Penaeu species. BMC Res Notes. 2010, 3: 295-10.1186/1756-0500-3-295.
Acknowledgements
This study was supported by the Center for Genomic Epidemiology (09-067103/DSF) http://www.genomicepidemiology.org and by grant 3304-FVFP-08- from the Danish Food Industry Agency. PL and OL would like to acknowledge funding from the Technical University of Denmark. The authors would like to thank Colleen Ussery for editorial assistance in preparing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PL planned the study, carried out all bioinformatics analysis and drafted the manuscript. OL participated in consensus tree based on core genes. CF participated in the planning of the study, the core genes identification and drafted the manuscript. FMA supervised and planned the study and drafted the manuscript. DWU supported the supervision, participated in the design of the study and drafted the manuscript. All authors have read and approved the final manuscript.
Electronic supplementary material
12864_2011_3990_MOESM4_ESM.PDF
Additional file 4: Figure S2 Consensus tree based on 69 Salmonella core genes randomly picked up from high, medium and low variable core genes. (PDF 5 KB)
12864_2011_3990_MOESM5_ESM.PDF
Additional file 5: Figure S3 Consensus tree based on 69 Salmonella core genes randomly picked up from medium variable core genes. (PDF 5 KB)
12864_2011_3990_MOESM6_ESM.PDF
Additional file 6: Figure S4 Consensus tree based on 69 Salmonella core genes randomly picked up from highly variable core genes. (PDF 5 KB)
12864_2011_3990_MOESM7_ESM.PDF
Additional file 7: Figure S5 Consensus tree based on 69 Salmonella core genes randomly picked up from decreasing curve in the variation plot. (PDF 5 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Leekitcharoenphon, P., Lukjancenko, O., Friis, C. et al. Genomic variation in Salmonella enterica core genes for epidemiological typing. BMC Genomics 13, 88 (2012). https://doi.org/10.1186/1471-2164-13-88
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-13-88