
Abstract
Background
The degradation of plant materials by enzymes is an industry of increasing importance. For sustainable production of second generation biofuels and other products of industrial biotechnology, efficient degradation of non-edible plant polysaccharides such as hemicellulose is required. For each type of hemicellulose, a complex mixture of enzymes is required for complete conversion to fermentable monosaccharides. In plant-biomass degrading fungi, these enzymes are regulated and released by complex regulatory structures. In this study, we present a methodology for evaluating the potential of a given fungus for polysaccharide degradation.
Results
Through the compilation of information from 203 articles, we have systematized knowledge on the structure and degradation of 16 major types of plant polysaccharides to form a graphical overview. As a case example, we have combined this with a list of 188 genes coding for carbohydrate-active enzymes from Aspergillus niger, thus forming an analysis framework, which can be queried. Combination of this information network with gene expression analysis on mono- and polysaccharide substrates has allowed elucidation of concerted gene expression from this organism. One such example is the identification of a full set of extracellular polysaccharide-acting genes for the degradation of oat spelt xylan.
Conclusions
The mapping of plant polysaccharide structures along with the corresponding enzymatic activities is a powerful framework for expression analysis of carbohydrate-active enzymes. Applying this network-based approach, we provide the first genome-scale characterization of all genes coding for carbohydrate-active enzymes identified in A. niger.
Similar content being viewed by others

Background
Expression profiling of the genes encoding extracellular enzymes is of high relevance to several industries. Commercial enzyme-preparation are often targeted to a specific activity such as cellulase for the degradation of cellulose and preparation of fabrics, or amylases for the preparation of dense syrups from starch or similar compounds. The world market for industrial enzymes is a multi-billion dollar market [1]. Another emerging market is the production of second generation biofuels. Energy-efficient processes rely on enzymes produced by fungi and other degraders of dead biomass (saprobes) to produce cheap CO2-neutral fuels from non-edible plant matter [2]. For this study, we have chosen to study the saprobic fungus Aspergillus niger, a well studied, efficient, and widely used enzyme producer [3, 4].
The interest in profiling the expression patterns of genes coding for polysaccharide-active enzyme is not new, but few large-scale surveys have been done. An early study was made by one of the authors [5] using Northern blotting to create expression profiles of 26 pectinolytic genes under 16 different growth conditions. The publication of the first A. niger genome [3] provided the prediction of 171 polysaccharide-active enzymes, along with microarrays for expression profiling. This has catalyzed more research, specifically the work of Martens-Uzunova [6], where expression profiles of 21 pectinolytic genes are examined. Other studies by Yuan et al. [7–9], examined the degradation of the polysaccharide inulin and identified the regulating protein and, in one case, performed expression profiling of genes from three out of the 36 carbohydrate-acting enzyme families predicted by Pel et al. [3] to be present in A. niger. While studies of this type address interesting parts of the polysaccharide degradation potential, there is a need to evaluate the entire scope of polysaccharides and enzymes, to fully address the challenges involved in complete degradation of plant biomass for biotechnology purposes.
The degradation of plant biomass is a complex procedure requiring a cocktail of enzymes. Furthermore, the polysaccharides are rarely found independently in nature. hemicellulose, found in the cell walls of plants, are a complex three-dimensional structure of several types and structures of polysaccharides such as glucomannan, arabinan, and xylan. It has been shown in several studies [10–12], that the presence of one simple saccharide can trigger the expression of enzymes for the degradation of an entirely different structure (as has been shown to be the case with D-xylose [13]). Another possibility with industrial applications is the use of a cheap carbon-source to induce the enzymes required for degradation of a more complex and expensive one [12, 14]. One example is the work of Yuan et al. [7], where sucrose induces the genes for all of the enzymes required for degrading inulin.
With the complex regulation of expression of a large number of different enzymes and the fact that many different enzymes are needed for the utilization of certain carbon sources (see e.g. ref. [15]), it is necessary to apply a systems-wide approach for mapping the regulatory network. The enzyme expression network can be combined and cross-triggered, so being able to examine the entire system at once can shed light on systems that might not be possible to pre-empt with a hypothesis-driven approach.
In this study, we have compiled knowledge on the structure and degradation of 16 types of plant polysaccharides from ¿200 articles. This has been combined with a list of 188 genes from A. niger coding for carbohydrate-active enzymes [3] to form a systematic graphical overview.
This makes it possible to highlight directly on the polysaccharide structures e.g. which genes are actively induced on a specific carbon source. This is a network-based approach for interpretation of data, where the network is provided by the structures of the extracellular polysaccharides, in contrast to the intracellular metabolic or regulatory networks often used for data-interpretation.
We apply this reconstructed network to investigate how enzymes interact to degrade complex polysaccharides with applications within sustainable biotechnology. The correlation of gene expression analysis on three monosaccharides and three complex carbohydrates with the network allowed the detection of concerted enzymatic actions as well as cross-induction of enzymatic cocktails.
We also see the combination of the network mapping of available information on the structure of polysaccharides with the transcription analysis as a source of reference for researchers interested in the induction of specific carbohydrate-active genes on certain substrates.
Results and discussion
Polysaccharide mapping generates a graphical knowledge base on biomass degradation
In a review of the available literature on the degradation of polysaccharides by A. niger, information was compiled on the following 10 types of polysaccharides: starch, cellulose, pullulan, inulin, galactomannan, galactoglucomannan (soluble and insoluble), xyloglucan (types XXGG and XXXG), as well as the following six distinct components of pectin: smooth pectin (pectate), xylogalacturonan, arabinogalactan (type I and II, also known as protein-bound arabinogalactan), arabinan, and rhamnogalacturonan I. The last known polysaccharide component of pectin, rhamnogalacturonan II, was not included even though the structure has been elucidated [16], since no studies of its degradation by A. niger were found in the literature search. Analyses of the degradation of this polysaccharide is made more difficult by the fact that it is composed mainly of highly modified and rare sugars and thought to be the most complex polysaccharide on Earth [16].
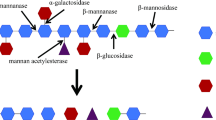
Sixteen structures have been gathered in schematic representations of each type of polysaccharide. An example of this (for soluble galactoglucomannan) is found in Figure 1. Schematic representations for all 16 structures and information on the genes are found in Additional files 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 and 16.

Schematic representation of soluble galactoglucomannan. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on the reportings of refs. [18–21].
For each of these structures, the available literature and sequence databases (Swissprot/Uniprot http://www.expasy.org/sprot/) were examined and information was gathered on the enzymes required for their degradation. This was integrated on the schematic representation of the structure as EC numbers where available, or as enzyme-names if the EC number was not available (As demonstrated in Figure 1). This was compared to the annotation of A. niger CBS 513.88 genome sequence [3] to identify putative isoenzymes for each activity. For each structure, this has resulted in a table containing the activities required for degradation, reference to literature on the characterization of the genes and enzymes, as well as information on the specificities where available. The sequence information is found as Uniprot accession numbers as well as gene IDs in the A. niger CBS 513.88 and ATCC 1015 sequence. Further information was found in an analysis of the carbohydrate degradation genes from A. nidulans[17]. An example of this is found in Table 1, with the full table and tables for all 16 structures found in Additional file 17. This annotation is a valuable reference on carbohydrate degradation by A. niger. The combination of structures and specifics on required enzymatic activities and genes makes this a significant extension of previous studies, which are dealing with a subset of the structures and generic enzymatic activities [15].
For a number of the studies found in the literature search, it was not possible to link the characterized enzymes with a sequence as only molecular mass and/or isoelectric point of the characterized enzyme(s) were given. However, as these characterizations still include valuable information on the enzymatic capabilities of A. niger, in Additional file 17: Table S17 includes an overview of the literature on characterizations of 24 enzymatic activities, where the gene could not be determined.
The mapping of these 16 structures includes information on 115 unique putative and characterized carbohydrate-active genes from the A. niger CBS 513.88 sequence [3], and 106 from the A. niger ATCC 1015 sequence (117 unique genes) [4]. Of these, the products of 57 of the genes have been previously characterized (See Additional file 17 for references). In total, the integrated information includes references to 203 articles. A full overview of the structures and the integrated genes is available in Additional file 18.
This map also includes a section with an overview of all of the 171 genes identified to code for putative or known polysaccharide-acting enzymes sorted into gene families. A plotting of expression indices directly on the structures of the map as well as in the overview section makes a systems-wide examination possible as described in the following.
Transcriptome analysis
To assess the regulatory network of genes for extracellular enzymes, A. niger was cultivated on six different carbon sources: three mono-saccharides (D-glucose, L-arabinose, and D-xylose), and three complex polysaccharides with a defined composition of sugars (starch, arabinan, and oat spelt xylan). Crude preparations of polysaccharides may include a multitude of undefined sugars and other types of compounds. The use of defined complex polysaccharides in this type of analysis allows for stronger conclusions on which saccharides induce which genes.
The batch cultivations were performed in shake flasks and samples were taken for transcriptome analysis and determination of free sugar concentrations (Table 2). Pairwise statistical comparisons of data from the sets of biological replicates were performed, and the number of statistically significantly (Benjamini-Hochberg adjusted p-values < 0.05) regulated genes in each comparison is shown in Table 3.
In an examination of the sugar-concentration measurements of Table 2, it is rather clear that the sugar-profiles at the time of sampling for transcriptome analysis are quite different, thus giving clear difference between the experimental conditions. The only exception is the profiles on the D-glucose and starch media, where the concentrations of free D-glucose are rather similar. This is most likely due to the fact that the strain employed is an amylase-producer, meaning that starch is rapidly be hydrolyzed to D-glucose, thus making the actual difference between the two carbon sources small. This is reflected in the transcriptome comparison of Table 3, where it is seen that only 27 genes show significant changes in expression between D-glucose and starch. These genes could either be false positives, genes responding to polysaccharides present in the degraded starch medium, or genes responding to the difference in D-glucose concentration between the two conditions (Table 3). While polysaccharide induction is not unlikely, A. niger is also known to have gradual responses from 0.5 – 100 mM D-glucose [38].
In a closer examination of Table 3, it is seen that a larger number of genes are responding to the polysaccharides xylan and arabinan compared to the monosaccharides and starch. This is expected since degradation of the diverse components and types of covalent bonds constituting complex carbon sources must require a larger set of genes than a simple one-sugar monosaccharide substrate. Furthermore, as can be seen in Table 2, the concentration of free sugars is roughly an order of magnitude lower in the medium containing the complex carbon sources. It has been shown that CreA mediates carbon repression of xylanolytic enzymes by D-xylose, beginning at 1 mM and increases in strength up to in the area between 30–70 mM [39, 40]. These ranges are similar to the concentrations in the comparison of D-xylose and xylan in this study. Similar effects within the same ranges of concentration are known for AraR and XlnR (L-Arabinose and D-xylose metabolism [41–44]), as well as for AmyR (Glucose-repression [38]). Cross-regulation of these carbon-repressing proteins has also been reported [40]. The free sugars are seemingly released from the polysaccharides (except starch) in a rate that effectively lessens the effects of carbon repression on these media.
The differences in induction were examined by preparing a map of the carbohydrate active enzymes for each pairwise comparison. For each map, the statistically significantly changed gene expressions are shown (Additional files 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 19, 20, 21, 22, 23 and 24). This gives maps of the enzymatic activities induced by a specific monosaccharide or the required enzymatic activities for degradation of a specific polysaccharide. A few examples of comparisons that showed results on a systemic level are discussed here:

Clustering of 161 putative and characterized genes coding for polysaccharide-active enzymes according to expression profiles. The number of genes in each cluster is shown next to the cluster number. The gene names of known genes found in a particular cluster are found in each cluster. The genes were clustered using the ClusterLustre algorithm [50].
Arabinan versus L-arabinose
The comparison of these two carbon sources (Additional file 19) showed a diverse response. Regulation was found in 27 of the 36 carbohydrate-acting gene families predicted to be present in A. niger[3]. As the mapping shows, the induction on arabinan unsurprisingly includes all of the activities required to degrade arabinan (Additional file 17: Table S15), but also the entire sets of genes required to degrade the pectin components xylogalacturonan, arabinogalactan and rhamnogalacturonan. This is very likely due to the presence of small amounts of rhamnose, galactose, and galacturonic acid in the preparation of arabinan, as described in the materials section. Galacturonic acid is known to induce pectinolytic enzymes [12, 14]. The induction on arabinan also includes activities directed towards acid-residues not described in the arabinan preparation, e.g. genes putatively acting on glucuronic acid of arabinogalactan, rhamnogalacturonan acetyl esterase (rgaeA), and ferulic acid esterase B (faeB) acting on arabinan. These enzymes are reacting to compounds found in arabinan and rhamnogalacturonan I (where arabinan is a component, see Additional files 15 and 16). It is therefore likely that triggering residues are present in minute amounts in the preparation.
Arabinose versus D-glucose
Despite only 92 genes being regulated, a mapping of these genes revealed that 13 of them were coding for putative polysaccharide-active gene products (Additional file 21) and all of them induced by L-arabinose. Interestingly, all three of the induced arabinan-acting genes were exo-acting arabinofuranosidases (EC 3.2.1.55), including the characterized abfA and abfB[5, 10, 11, 39, 45–49]. However, the remaining induced genes constitute all of the activities of the xylan-acting genes (Additional file 17: Table S9), including β-galactosidase lacA, endo-xylanases, and α-galacturonase A (aguA), suggesting that L-arabinose alone can induce the entire complex of xylan-degrading enzymes.
Starch versus xylan
A comparison of these two carbon sources is especially interesting, as no bond-types are shared between the two. Mapping of the significantly regulated genes (Additional file 22) indicates just this. The regulated genes are limited to all of the activities required to degrade starch (up-regulated on starch) and the activities required for degradation of xylan (up-regulated on xylan). Those up-regulated on starch include glucoamylase A (glaA), α-glucosidase A (aglA/aglU) as well as one or more of the three genes for α-amylase (amyA, amyB, and gene ID 140567) with almost identical nucleotide-level sequences. It is not possible with an expression analysis to differentiate between them. The genes induced on xylan are the same genes as described to be induced by L-arabinose in the comparison of L-arabinose and D-glucose, with the exception that the endo-xylanase xlnB is induced on xylan instead of the putative endo-xylanase An15g04550/183088 that is induced by L-arabinose. We have thus identified a full set of extracellular polysaccharide-acting genes for the degradation of oat spelt xylan and starch and shown that with the exception of xlnB, L-arabinose induces the same array of xylan-degrading enzymes as does xylan (at the concentrations examined here).
Transcription-based clustering
To further investigate the regulation of single genes and the functions of genes that appear to be co-regulated, a clustering analysis of the expression indices of all 161 putative and characterized glucoside hydrolases, polysaccharide lyases and carbohydrate esterases on the six different carbon sources was performed, thus including also genes which could not be assigned directly to a polysaccharide structure. (Figure 2). 24 gene clusters were identified. Clustering allows the identification of genes that are only induced by the complex substrates, as well as genes that are induced equally well by a particular monosaccharide as well as the complex polysaccharide.
The transcription profile of each cluster (Figure 2) was examined and classified in terms of regulation on the different carbon sources (Additional file 17: Table S18). If a distinct regulation was evident from the clustering profile, this information was added to the table as well, making this an overview of regulation and possible polysaccharide specificity for each of the 161 genes in the survey. The same study was made for the subset of genes coding for carbohydrate-active enzymes that were significantly regulated in one or more pairwise comparisons (Additional file 23 and Additional file 17: Table S19). This subset consists of 103 of the 161 genes, and 47 of the 57 characterized genes. However, as can be seen in Additional file 23, the clustering of the subset is very similar to those of the full set of genes (Figure 2). A comparison of the clusters of the two studies are also present in Additional file 17: Table S19. Since the majority of the genes were significantly regulated, and the remainder fall mostly in the same clusters, the following detailed analysis of the clustering was made using the full set, thereby giving information on the expression patterns of as many genes as possible.
As it is evident from Figure 2, clusters 21–24 are highly similar. In making the clusters, it was attempted to use fewer clusters, and thereby combine these four clusters, however this combination required a decrease of the total numbers of clusters to 10, which increased the variation in the other clusters dramatically (Data not shown). For this reason, it was concluded that they have distinct patterns, as the small standard deviations of the clusters also suggest, and they have been kept as separate clusters. Furthermore, as clusters 21–24 are almost solely induced on arabinan, one would need to include more pectin-like substrates, e.g. polygalacturonic acid, in the analysis to be able to differentiate between the regulation of these genes. However, what one can conclude from this is that the genes of cluster 24 are specific for arabinan, and are not induced by any of the other saccharides in this study. It is interesting that axhA clusters with this group, as it was previously shown to be induced on birchwood xylan but not on D-xylose [41]. However, no induction on xylan is seen for cluster 24, suggesting that this gene is not induced by oat spelt xylan in this strain.
In examining the clusters for general trends, it was found that for most of the clusters, regulation on D-glucose and starch are very similar. This is in good agreement with the high level of free D-glucose in both cultures shown in Table 2. Exceptions are clusters 8, 9, 15, 18, and 20, which should be interesting for determining genes that are sensitive to degradation-products of starch, but not necessarily statistically significant in the comparison of starch versus D-glucose (Table 3).
For each cluster, a map marking the genes of that cluster was prepared and examined to determine an activity profile if possible (Additional file 24).
Several of the clusters are targeted to a specific type of polysaccharide. One example hereof is cluster 1. While it does not containing any characterized genes, the genes of the cluster are quite specifically predicted to code for enzymes with activity towards galactomannan and insoluble galactoglucomannan (Additional file 24). This homogeneity of functions confirms the putative annotations. As the cluster is up-regulated on the mono-sugars and the relatively easily degradable potato starch, it is likely that this cluster contains genes are induced by mono-sugar-abundance and code for a set of “scouting enzymes” with the role to liberate more substrates.
Cluster 5 has the highest level of expression on D-glucose and starch carbon sources. An examination of it with the enzyme mapping (Additional file 24), reveals that it constitutes a full set of co-regulated starch-degrading enzymes. The individual genes found in the cluster encode solely amylases and glucosidases, including glucoamylase A (glaA) and one or more of the three α-amylases with similar sequences discussed above (including amyA and amyB). The starch part of the map can be found in Figure 3. The fact that these enzymes cluster together can be seen as a validation of the transcription analysis and the clustering employed.

Map of starch-degrading enzymes in cluster 5. Genes marked with a red box are found in the cluster, gray boxes means that the gene is found in the A. niger CBS 513.88 sequence, but no bi-directional best hit is found in the A. niger ATCC 1015 sequence.
Rather interesting is the observation that the hemicellulose component arabinan seems to induce enzymes acting on cellulose and other β-glucans (Additional file 24). This is seen in clusters 17 and 19, each containing one of the characterized cellobiohydrolases (which were shown to be induced on D-xylose [51]) and several putative β-glucosidases. β-glucans are not found as components of pectins, which makes this pattern a genuine induction across carbon-sources of entirely different chemical make-up.
As discussed above, the profile of cluster 24 indicates that it mainly contains genes that can be described as pectinolytic. A map inspection (Additional file 24) confirms that these genes have a very distinct profile including all enzymatic activities for the degradation of rhamnogalacturonan I, smooth pectin, and xylogalacturonan, all components of pectin, as are arabinan.
Comparison of transcriptional regulation to previous studies
As described in the introduction, relatively few large-scale studies exist where the carbon source-based induction of extracellular enzymes have been studied. There is little overlap in the carbon sources employed in this study. Studies of polysaccharides have mainly been done with di-saccharides such as maltose or sucrose [7] or on commercial preparations of pectin for food gelling [5, 6]. However, a few have been made, which will be compared to the results of the present study here. A relatively larger number of studies including single or a few genes are published, however, these have been cited in the text above where appropriate, and in Additional file 17.
The method applied here is comparable to a study of one of the authors, where a principal component analysis (PCA) allows clustering according to expression profiles [5]. While that study focuses mainly on pectinolysis, it includes studies of growth on D-glucose, D-xylose, and L-arabinose. The study describes abfA abfB, and lacA as being co-regulated and induced by 25 mM L-arabinose, which is in good accordance with the results of this work, as all of these are found in cluster 13. Interestingly however, that work also reports abnA as being induced by L-arabinose and co-regulated with abfAB and lacA, but in the present work, only induction on arabinan is seen (cluster 24), and with a statistically significant up-regulation on arabinan in all pairwise comparisons. This may be due to our higher concentration of L-arabinose (45 mM relative to 25 mM) leading to differences in carbon-repression. The PCA also indicates a close relationship between pmeA pgaX, and abnA, which describes accurately the patterns of cluster 24.
faeA and faeB are known to be largely co-regulated, but faeA is induced on D-xylose due to an induction by the xylanolytic regulator XlnR [5, 41]. faeA and faeB are separated in clusters 10 and 14, that are very similar, but cluster 10 responds to D-xylose, whereas cluster 14 responds to the ferulic acid-containing arabinan, suggesting that faeB is more targeted to arabinan.
Another study [9] examines induction of genes on D-xylose and maltose, which has an α-1,4-glucoside bond similar to starch. In Figure two in ref. [9], eleven genes can be seen to be induced by maltose and/or D-xylose (An11g03340, An04g06930, An01g06120, An14g04190, An12g02450, An09g03070, agdA, An01g10930, glaA, An09g05880, An09g03300). All of these are regulated in a similar fashion in the present work, if maltose (Glc-α-1,4-Glc) is seen to be similar to starch ((Glc-α-1,4-Glc-α-1,4-) n ).
Conclusions
Enzyme preparations have multiple areas of application and are a billion dollar market with a low cost/profit ratio. Thus, it is of great importance and interest to gain an understanding of the processes leading to production of enzymes with a specific profile.
In this study, the first genome-scale characterization of all genes coding for carbohydrate-active enzymes identified in A. niger is provided. Based on a review of more than 200 articles and sequence database searches, information of 117 genes and enzymes has been systematized according to the polysaccharide structures they degrade, thereby providing an updated reference on extracellular enzyme-expression in A. niger. This data-integration effort has produced schematic representations (maps) of 16 types of polysaccharide-structures specifically updated with the A. niger enzymatic machinery.
An application of this knowledge was performed in a transcription study where statistical analysis as well as expression-level-based clustering and mapping were employed to provide transcription profiles of 161 genes on six defined carbon sources, thereby adding considerably to our knowledge of the transcription-level regulation of these genes. New knowledge was generated on the carbon source-based transcriptional regulation of previously characterized genes including, but not limited to, abfA, abfB, aglA, cbhA, cbhB, glaA, eglA, eglB, faeA, faeB, lacA, pelA, pelB, pelC, xlnB, and xynA as well as more than 100 uncharacterized genes predicted to code for carbohydrate-active enzymes. The mapping of transcribed genes allowed us to identify induction by mono-saccharides of complete sets of enzymes for degradation of complex substrates, as well as the induction of cellulolytic enzymes on a hemicellulose substrate, e.g. xylan.
Methods
Cultivation procedure
Strain information
The strain used was A. niger BO-1, a progenitor to high-yield amylase-producing industrial strains, obtained from Novozymes A/S. The strain is maintained as frozen spore suspensions at -80°C in 20 % glycerol.
Growth media
Complex medium: 2 g/L yeast extract, 3 g/L tryptone, 10 g/L glucose monohydrate, 20 g/L agar, 0.52 g/L KCl, 0.52 g/L MgSO4·7H2O, 1.52 g/L KH2PO4, and 1 mL/L of trace elements solution. Trace element solution: 0.4 g/L CuSO4·5H2O, 0.04 g/L Na2B4O7·10H2O, 0.8 g/L FeSO4·7H2O, 0.8 g/L MnSO4·H2O, 0.8 g/L Na2MoO4·2H2O, 8 g/L ZnSO4·7H2O.
Batch cultivation medium salt concentrations: 2.5 g/L (NH4)2SO4, 0.75 g/L KH2PO4, 1.0 g/L MgSO4·7H2O, 1 g/L NaCl, 0.1 g/L CaCl2·2H2O, 0.05 mL/L antifoam 204 (Sigma), and 1 mL/L trace element solution. Trace element solution composition: 7.2 g/L ZnSO4·7H2O, 0.3 g/L NiCl2·6H2O, 6.9 g/L FeSO4·7H2O, 3.5 g/L MnCl2·4H2O, and 1.3 g/L CuSO4·5H2O. The carbon sources used for the cultivations were D-glucose H2O (20 g/L), D-xylose (20 g/L), L-arabinose (19.5 g/L + 0.5 g/L D-glucose), sugar beet arabinan (Megazyme, P-ARAB) (16.3 g/L), oat spelt xylan (Sigma, no 95590) (16.2 g/L), and potato starch (Sigma, no 85650) (16.5 g/L). These concentrations all equal 0.61 Cmole/L. Cultivations with L-arabinose were added D-glucose in order to make the spores germinate.
Sugar beet arabinan (Megazyme) is 95% pure 1,5-α-L-arabinan containing arabinose:galactose:rhamnose:galacturonic acid in the ratios 88:4:2:6. The documentation further describes the arabinan as being “a polymer of 1,5-alpha-L-linked arabinofuranose units which is highly substituted by 1,3- and 1,2-linked single α-L-arabinofuranose residues. About 50% of 1,5-linked arabinosyl residues in the main chain are substituted by 1,3 or 1,2 linked arabinofuranosyl branches.”
Oat spelt xylan (Sigma) contains ≤10% L-arabinose, ≤15% D-glucose, and ≥70% D-xylose.
Shake flask cultivations
Shake flasks were initiated by spore inoculation to a final concentration of 2×109 spores/L. Spores were propagated on complex medium plates and incubated for 7–8 days at 30°C before being harvested with 10 mL of 0.01% Tween 80. All cultivations were carried out in 500 mL baffled shake flasks with a total volume of 200 mL liquid medium at 30°C and 150 rpm. The initial pH was set to pH 2.5 to avoid pellet formation. The entire content of the shake flask was harvested when the biomass concentration was approximately half the maximum biomass concentration on the given medium (mid-growth phase). All cultivations were performed in biological triplicates except starch, where a duplicate culture was performed.
Sampling
Cell dry weight was determined using nitrocellulose filters (pore size 0.45 μ m, Gelman Sciences). The filters were pre-dried in a microwave oven at 150 W for 15 minutes, cooled in a desiccator and subsequently weighed. A known volume of cell culture was filtered and the residue was washed with 0.9% NaCl and dried on the filter for 15 minutes in a microwave oven at 150 W and cooled in a desiccator. The filtrate was saved for quantification of sugars and extracellular metabolites and stored at -80°C. The filter was weighed again and the cell mass concentration was calculated. These values were used to calculate maximum specific growth rates. For gene expression analysis, mycelium was harvested at the mid-late exponential phase (at approximately half the maximum concentration of biomass) by filtration through sterile Mira-Cloth (Calbiochem) and washed with a PBS buffer (8 g/L NaCl, 0.20 g/L KCl, 1.44 g/L Na2HPO4, and 0.24 g/L KH2PO4 in distilled water). The mycelium was quickly dried by squeezing, and subsequently frozen in liquid nitrogen. Samples were stored at -80°C until RNA extraction.
Quantification of sugars
The concentrations of sugar in the filtrates were determined using HPLC on an Aminex HPX-87H ion-exclusion column (BioRad, Hercules, CA). The column was eluted at 60°C with 5 mM H2SO4 at a flow rate of 0.6 mL/min. Sugars were detected with a refractive index detector and a UV detector.
Transcriptome analysis
Extraction of total RNA
40–50 mg of frozen mycelium was placed in a 2 mL Eppendorf tube, pre-cooled in liquid nitrogen, containing three steel balls (two balls with a diameter of 2 mm and one ball with a diameter of 5 mm). The tubes were then shaken in a Mixer Mill, at 5°C for 10 minutes, until the mycelium was ground to powder. Total RNA was isolated from the powder using the Qiagen RNeasy Mini Kit, according to the protocol for isolation of total RNA from plant and fungi. The quality of the extracted total RNA was assessed using a BioAnalyzer 2100 (Agilent Technologies Inc., Santa Clara, CA, USA) and the quantity determined using a spectrophotometer (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). The total RNA was stored at -80°C until further processing.
Preparation of biotin-labeled cRNA and microarray processing
15 μ g of fragmented biotin-labeled cRNA was prepared from 5 μ g of total RNA and hybridized to the 3AspergDTU GeneChip [13] according to the Affymetrix GeneChip Expression Analysis Technical Manual [52].
cRNA was quantified in a spectrophotometer (same as above). cRNA quality was assessed using a BioAnalyzer. A GeneChip Fluidics Station FS-400 (fluidics protocol FS450_001) and a GeneChip Scanner 3000 were used for hybridization and scanning.
The scanned probe array images (.DAT files) were converted into .CEL files using the GeneChip Operating Software (Affymetrix).
Analysis of transcription data
Affymetrix CEL-data files were preprocessed using the statistical language and environment R [53] version 2.6.1. The probe intensities were normalized for background using the RMA method [54] using only perfect match (PM) probes. Normalization was performed subsequently using the quantiles algorithm [55]. Gene expression values were calculated from the PM probes with the medianpolish summary method [54]. All statistical preprocessing methods were used by invoking them through the affy package [56].
Statistical analysis was applied to determine genes subject to differential transcriptional regulation. The limma package [57] was used to perform moderated t-tests between two sets of triplicates from each pH level. Empirical Bayesian statistics were used to moderate the standard errors within each gene and Benjamini-Hochberg’s method [58] to adjust for multi-testing. A cut-off value of adjusted p<0.05 was set to assess statistical significance.
Normalized and raw data-values are deposited with GEO as series GSE11930.
Clustering
Genes were clustered using the clustering algorithm ClustreLustre [50], using k-means clustering, a pearson-based distance measure and accounting for biological replicates.
References
Novozymes: The Novozymes Report 2007. 2008, [www.novozymes.com].
Galbe M, Zacchi G: Pretreatment of lignocellulosic materials for efficient bioethanol production. Adv Biochem Eng Biotechnol. 2007, 108: 41-65.
Pel H, de Winde J, Archer D, Dyer P, Hofmann G, Schaap P, Turner G, de Vries R, Albang R, Albermann K, Andersen M, Bendtsen J, Benen J, van den Berg M, Breestraat S, Caddick M, Contreras R, Cornell M, Coutinho P, Danchin E, Debets A, Dekker P, van Dijck P, van Dijk A, Dijkhuizen L, Driessen A, d’Enfert C, Geysens S, Goosen C, Groot G, de Groot P, Guillemette T, Henrissat B, Herweijer M, van den Hombergh J, van den Hondel C, van der Heijden R, van der Kaaij R, Klis F, Kools H, Kubicek C, van Kuyk P, Lauber J, Lu X, van der Maarel M, Meulenberg R, Menke H, Mortimer M, Nielsen J, Oliver S, Olsthoorn M, Pal K, van Peij N, Ram A, Rinas U, Roubos J, Sagt C, Schmoll M, Sun J, Ussery D, Varga J, Vervecken W, van de Vondervoort P, Wedler H, Wosten HA, Zeng A, van Ooyen A, Visser J, Stam H: Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol. 2007, 25 (2): 221-231. 10.1038/nbt1282.
Andersen MR, Salazar MP, Schaap PJ, van de Vondervoort PJI, Culley D, Thykaer J, Frisvad JC, Nielsen KF, Albang R, Albermann K, Berka RM, Braus GH, Braus-Stromeyer SA, Corrochano LM, Dai Z, van Dijck PWM, Hofmann G, Lasure LL, Magnuson JK, Menke H, Meijer M, Meijer SL, Nielsen JB, Nielsen ML, van Ooyen AJJ, Pel HJ, Poulsen L, Samson RA, Stam H, Tsang A, van den Brink JM, Atkins A, Aerts A, Shapiro H, Pangilinan J, Salamov A, Lou Y, Lindquist E, Lucas S, Grimwood J, Grigoriev IV, Kubicek CP, Martinez D, van Peij NNME, Roubos JA, Nielsen J, Baker SE: Comparative genomics of citric-acid-producing Aspergillus niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Res. 2011, 21 (6): 885-897. 10.1101/gr.112169.110.
de Vries RP, Jansen J, Aguilar G, Parenicova L, Joosten V, Wulfert F, Benen JA, Visser J: Expression profiling of pectinolytic genes from Aspergillus niger. FEBS Lett. 2002, 530 (1–3): 41-47.
Martens-Uzunova E, Zandleven J, Benen J, Awad H, Kools H, Beldman G, Voragen A, van den Berg J, Schaap P: A new group of exo-acting family 28 glycoside hydrolases of Aspergillus niger that are involved in pectin degradation. Biochem J. 2006, 400: 43-52. 10.1042/BJ20060703.
Yuan X, Goosen C, Kools H, van der Maarel M, van den Hondel C, Dijkhuizen L, Ram A: Database mining and transcriptional analysis of genes encoding inulin-modifying enzymes of Aspergillus niger. Microbiology. 2006, 152 (Pt 10): 3061-3073.
Yuan X, Roubos J, van den Hondel C, Ram A: Identification of InuR, a new Zn(II)2Cys6 transcriptional activator involved in the regulation of inulinolytic genes in Aspergillus niger. Mol Genet Genomics. 2008, 279: 11-26. 10.1007/s00438-007-0290-5.
Yuan X, van der Kaaij R, van den Hondel C, Punt P, van der Maarel M, Dijkhuizen L, Ram A: Aspergillus niger genome-wide analysis reveals a large number of novel α-glucan acting enzymes with unexpected expression profiles. Mol Genet Genomics. 2008, 279 (6): 545-561. 10.1007/s00438-008-0332-7.
vd Veen P, Flipphi MJ, Voragen AG, Visser J: Induction of extracellular arabinases on monomeric substrates in Aspergillus niger. Arch Microbiol. 1993, 159: 66-71. 10.1007/BF00244266.
Flipphi M, Visser J, van der Veen P, de Graaff L: Arabinase gene expression in Aspergillus niger: indications for coordinated regulation. Microbiology. 1994, 140 (10): 2673-2682. 10.1099/00221287-140-10-2673.
de Vries RP, Parenicova L, Hinz SW, Kester HC, Beldman G, Benen JA, Visser J: The β-1,4-endogalactanase A gene from Aspergillus niger is specifically induced on arabinose and galacturonic acid and plays an important role in the degradation of pectic hairy regions. Eur J Biochem. 2002, 269 (20): 4985-4993. 10.1046/j.1432-1033.2002.03199.x.
Andersen M, Vongsangnak W, Panagiotou G, Salazar M, Lehmann L, Nielsen J: A tri-species Aspergillus micro array: comparative transcriptomics of three Aspergillus species. Proc Natl Acad Sci USA. 2008, 105 (11): 4387-4392. 10.1073/pnas.0709964105.
Ruttkowski E, Khanh NQ, Wientjes FJ, Gottschalk M: Characterization of a polygalacturonase gene of Aspergillus niger RH5344. Mol Microbiol. 1991, 5 (6): 1353-1361. 10.1111/j.1365-2958.1991.tb00782.x.
van den Brink J, de Vries R: Fungal enzyme sets for plant polysaccharide degradation. Appl Microbiol Biotechnol. 2011, 91 (6): 1477-1492. 10.1007/s00253-011-3473-2.
Perez S, Rodriguez-Carvajal M, Doco T: A complex plant cell wall polysaccharide: rhamnogalacturonan II. A structure in quest of a function. Biochimie. 2003, 85 (1–2): 109-121.
Coutinho PM, Andersen MR, Kolenova K, vanKuyk PA, Benoit I, Gruben BS, Trejo-Aguilar B, Visser H, van Solingen P, Pakula T, Seiboth B, Battaglia E, Aguilar-Osorio G, de Jong JF, Ohm RA, Aguilar M, Henrissat B, Nielsen J, Stalbrand H, de Vries RP: Post-genomic insights into the plant polysaccharide degradation potential of Aspergillus nidulans and comparison to Aspergillus niger and Aspergillus oryzae. Fungal Genet Biol. 2009, 46 (Suppl 1): S161-S169.
Sims I, Craik D, Bacic A: Structural characterisation of galactoglucomannan secreted by suspension-cultured cells of Nicotiana plumbaginifolia. Carbohydr Res. 1997, 303: 79-92. 10.1016/S0008-6215(97)00144-4.
Schroder R, Nicolas P, Vincent S, Fischer M, Reymond S, Redgwell R: Purification and characterisation of a galactoglucomannan from kiwifruit (Actinidia deliciosa). Carbohydr Res. 2001, 331 (3): 291-10.1016/S0008-6215(01)00046-5.
de Vries R, Visser J: Aspergillus enzymes involved in degradation of plant cell wall polysaccharides. Microbiol Mol Biol Rev. 2001, 65 (4): 497-522. 10.1128/MMBR.65.4.497-522.2001.
Capek P, Alfoldi J, Liskova D: An acetylated galactoglucomannan from Picea abies L. Karst. Carbohydr Res. 2002, 337 (11): 1033-1037. 10.1016/S0008-6215(02)00090-3.
Ademark P, Varga A, Medve J, Harjunpaa V, Drakenberg T, Tjerneld F, Stalbrand H: Softwood hemicellulose-degrading enzymes from Aspergillus niger: purification and properties of a β-mannanase. J Biotechnol. 1998, 63 (3): 199-210. 10.1016/S0168-1656(98)00086-8.
Ademark P, Lundqvist J, Hagglund P, Tenkanen M, Torto N, Tjerneld F, Stalbrand H: Hydrolytic properties of a β-mannosidase purified from Aspergillus niger. J Biotechnol. 1999, 75 (2–3): 281-289.
Ademark P, de Vries RP, Hagglund P, Stalbrand H, Visser J: Cloning and characterization of Aspergillus niger genes encoding an α-galactosidase and a β-mannosidase involved in galactomannan degradation. Eur J Biochem. 2001, 268 (10): 2982-2990. 10.1046/j.1432-1327.2001.02188.x.
Bouquelet S, Spik G, Montreuil J: Properties of a β-D-mannosidase from Aspergillus niger. Biochim Biophys Acta. 1978, 522 (2): 521-530. 10.1016/0005-2744(78)90084-0.
de Vries RP, van den Broeck HC, Dekkers E, Manzanares P, de Graaff LH, Visser J: Differential expression of three α-galactosidase genes and a single β-galactosidase gene from Aspergillus niger. Appl Environ Microbiol. 1999, 65 (6): 2453-2460.
Manzanares P, de Graaff LH, Visser J: Characterization of galactosidases from Aspergillus niger: purification of a novel α-galactosidase activity. Enzyme Microb Technol. 1998, 22 (5): 383-390. 10.1016/S0141-0229(97)00207-X.
den Herder I, Rosell AM, van Zuilen CM, Punt PJ, van den Hondel CA: Cloning and expression of a member of the Aspergillus niger gene family encoding α-galactosidase. Mol Gen Genet. 1992, 233 (3): 404-410.
Wallis GL, Easton RL, Jolly K, Hemming FW, Peberdy JF: Galactofuranoic-oligomannose N-linked glycans of α-galactosidase A from Aspergillus niger. Eur J Biochem. 2001, 268 (15): 4134-4143. 10.1046/j.1432-1327.2001.02322.x.
Witte K, Wartenberg A: Purification and properties of two β-glucosidases isolated from Aspergillus niger. Acta Biotechnol. 1989, 2: 179-190.
Dan S, Marton I, Dekel M, Bravdo B, He S, Withers S, Shoseyov O: Cloning, expression, characterization, and nucleophile identification of family 3, Aspergillus niger β-glucosidase. J Biol Chem. 2000, 275 (7): 4973-4980. 10.1074/jbc.275.7.4973.
Ramirez-Coronel MA, Viniegra-Gonzalez G, Darvill A, Augur C: A novel tannase from Aspergillus niger with β-glucosidase activity. Microbiology. 2003, 149 (Pt 10): 2941-2946.
Seidle HF, Marten I, Shoseyov O, Huber RE: Physical and kinetic properties of the family 3 β-glucosidase from Aspergillus niger which is important for cellulose breakdown. Protein J. 2004, 23: 11-23.
Seidle H, Huber R: Transglucosidic reactions of the Aspergillus niger family 3 β-glucosidase: qualitative and quantitative analyses and evidence that the transglucosidic rate is independent of pH. Arch Biochem Biophys. 2005, 436 (2): 254-264. 10.1016/j.abb.2005.02.017.
Seidle H, McKenzie K, Marten I, Shoseyov O, Huber R: Trp-262 is a key residue for the hydrolytic and transglucosidic reactivity of the Aspergillus niger family 3 β-glucosidase: substitution results in enzymes with mainly transglucosidic activity. Arch Biochem Biophys. 2005, 444: 66-75. 10.1016/j.abb.2005.09.013.
Seidle H, Allison S, George E, Huber R: Trp-49 of the family 3 β-glucosidase from Aspergillus niger is important for its transglucosidic activity: creation of novel β-glucosidases with low transglucosidic efficiencies. Arch Biochem Biophys. 2006, 455 (2): 110-108. 10.1016/j.abb.2006.09.016.
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B: The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37 (Database issue): D233-D238.
Vankuyk PA, Benen JAE, Wosten HAB, Visser J, de Vries RP: A broader role for AmyR in Aspergillus niger: regulation of the utilisation of D-glucose or D-galactose containing oligo- and polysaccharides. Appl Microbiol Biotechnol. 2012, 93: 285-293. 10.1007/s00253-011-3550-6.
Ruijter GJG, Vanhanen SA, Gielkens MMC, van de Vondervoort PJI, Visser J: Isolation of Aspergillus niger creA mutants and effects of the mutations on expression of arabinases and L-arabinose catabolic enzymes. Microbiology. 1997, 143: 2991-2998. 10.1099/00221287-143-9-2991.
de Vries RP, Visser J, de Graaff LH: CreA modulates the XlnR-induced expression on xylose of Aspergillus niger genes involved in xylan degradation. Res Microbiol. 1999, 150: 281-285. 10.1016/S0923-2508(99)80053-9.
van Peij N, Gielkens M, de Vries R, Visser J, de Graaff L: The transcriptional activator XlnR regulates both xylanolytic and endoglucanase gene expression in Aspergillus niger. Appl Environ Microbiol. 1998, 64 (10): 3615-3619.
Hasper A, Visser J, de Graaf L: The Aspergillus niger transcriptional activator XlnR, which is involved in the degradation of the polysaccharides xylan and cellulose, also regulates D-xylose reductase gene expression. Mol Microbiol. 2000, 36: 193-200. 10.1046/j.1365-2958.2000.01843.x.
Battaglia E, Visser L, Nijssen A, van Veluw GJ, Wosten HAB, de Vries RP: Analysis of regulation of pentose utilisation in Aspergillus niger reveals evolutionary adaptations in Eurotiales. Stud Mycol. 2011, 69: 31-38. 10.3114/sim.2011.69.03.
Battaglia E, Hansen SF, Leendertse A, Madrid S, Mulder H, Nikolaev I, de Vries RP: Regulation of pentose utilisation by AraR, but not XlnR, differs in Aspergillus nidulans and Aspergillus niger. Appl Microbiol Biotechnol. 2011, 91 (2): 387-397. 10.1007/s00253-011-3242-2.
vd Veen P, Flipphi MJ, Voragen AG, Visser J: Induction, purification and characterisation of arabinases produced by Aspergillus niger. Arch Microbiol. 1991, 157: 23-28.
Flipphi MJ, van HM, der van V, Visser J, de Graaff LH: Cloning and characterization of the abfB gene coding for the major α-L-arabinofuranosidase (AbfB) of Aspergillus niger. Curr Genet. 1993, 24 (6): 525-532. 10.1007/BF00351717.
Flipphi M, Visser J, van der Veen P, de Graaff L: Cloning of the Aspergillus niger gene encoding α-L-arabinofuranosidase A. Appl Microbiol Biotechnol. 1993, 39 (3): 335-340.
Crous JM, Pretorius I, van Zyl WH: Cloning and expression of the α-L-arabinofuranosidase gene (ABF2) of Aspergillus niger in Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 1996, 46: 256-260. 10.1007/s002530050813.
de Groot MJL, van de Vondervoort PJI, de Vries RP, P A vanKuyk GJR, Visser J: Isolation and characterization of two specific regulatory Aspergillus niger mutants shows antagonistic regulation of arabinan and xylan metabolism. Microbiology. 2003, 149: 1183-1191. 10.1099/mic.0.25993-0.
Grotkjaer T, Winther O, Regenberg B, Nielsen J, Hansen L: Robust multi-scale clustering of large DNA microarray datasets with the consensus algorithm. Bioinformatics. 2006, 22: 58-67. 10.1093/bioinformatics/bti746.
Gielkens MM, Dekkers E, Visser J, de Graaff LH: Two cellobiohydrolase-encoding genes from Aspergillus niger require D-xylose and the xylanolytic transcriptional activator XlnR for their expression. Appl Environ Microbiol. 1999, 65 (10): 4340-4345.
Affymetrix: GeneChip Expression Analysis Technical Manual - With Specific Protocols for Using the GeneChip Hybridization, Wash, and Stain Kit. 2007, Santa Clara, California, USA: Affymetrix, P/N 702232 Rev. 2
R Development Core Team: R: A Language and Environment for Statistical Computing. 2007, Vienna, Austria: R Foundation for Statistical Computing
Irizarry R, Hobbs B, Collin F, Beazer-Barclay Y, Antonellis K, Scherf U, Speed T: Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003, 4 (2): 249-264. 10.1093/biostatistics/4.2.249.
Bolstad B, Irizarry R, Astrand M, Speed T: A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003, 19 (2): 185-193. 10.1093/bioinformatics/19.2.185.
Gautier L, Cope L, Bolstad B, Irizarry R: affy — analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004, 20 (3): 307-315. 10.1093/bioinformatics/btg405.
Smyth G: Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004, 3: Article 3-
Benjamini Y, Hochberg Y: Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B. 1995, 57: 289-300.
Polysaccharides – Structural diversity and functional versatility. Edited by: Dumitriu S. 2005, New York: Marcel Dekker
Persson I, Tjerneld F, Hahn-Hagerdal B: Fungal cellulolytic enzyme production: A review. Process Biochem. 1991, 26: 65-74. 10.1016/0032-9592(91)80019-L.
Doman-Pytka M, Bardowski J: Pullulan degrading enzymes of bacterial origin. Crit Rev Microbiol. 2004, 30 (2): 107-121. 10.1080/10408410490435115.
Niness KR: Inulin and oligofructose: what are they?. J Nutr. 1999, 129 (7): 1402S-1406S.
Daas PJ, Schols HA, de Jongh HH: On the galactosyl distribution of commercial galactomannans. Carbohydr Res. 2000, 329 (3): 609-619. 10.1016/S0008-6215(00)00209-3.
le Goff A, Renard C, Bonnin E, Thibault JF: Extraction, purification and chemical characterization of xylogalacturonans from pea hulls. Carbohydr Polym. 2001, 45 (4): 325-334. 10.1016/S0144-8617(00)00271-X.
Nakamura A, Furuta H, Maeda H, Takao T, Nagamatsu Y: Analysis of the molecular construction of xylogalacturonan isolated from soluble soybean polysaccharides. Biosci Biotechnol Biochem. 2002, 66 (5): 1155-1158. 10.1271/bbb.66.1155.
Zandleven J, Beldman G, Bosveld M, Benen J, Voragen A: Mode of action of xylogalacturonan hydrolase towards xylogalacturonan and xylogalacturonan oligosaccharides. Biochem J. 2005, 387 (Pt 3): 719-725.
Zandleven J, Sorensen S, Harholt J, Beldman G, Schols H, Scheller H, Voragen A: Xylogalacturonan exists in cell walls from various tissues of Arabidopsis thaliana. Phytochemistry. 2007, 68 (8): 1219-1226. 10.1016/j.phytochem.2007.01.016.
Biely P: Handbook of Food Enzymology. 2003:, Inc, New York: Marcel Dekker, 879–915. chap. 71: Xylanolytic enzymes
Eriksson K, Blanchette RA, Ander P: Microbial and Enzymatic Degradation of Wood and Wood Components. 1990:, Heidelberg: Springer-Verlag, 181–224. chap. 3: Biodegradation of Hemicelluloses
York W, Harvey L, Guillen R, Albersheim P, Darvill A: Structural analysis of tamarind seed xyloglucan oligosaccharides using β-galactosidase digestion and spectroscopic methods. Carbohydr Res. 1993, 248: 285-301.
Vincken JP, York WS, Beldman G, Voragen AG: Two general branching patterns of xyloglucan, XXXG and XXGG. Plant Physiol. 1997, 114: 9-13. 10.1104/pp.114.1.9.
Hinz SW, Verhoef R, Schols HA, Vincken JP, Voragen AG: Type I arabinogalactan contains β-D-Galp-(1-3)-β-D-Galp structural elements. Carbohydr Res. 2005, 340 (13): 2135-2143. 10.1016/j.carres.2005.07.003.
Tischer C, Iacomini M, Gorin P: Structure of the arabinogalactan from gum tragacanth (Astralagus gummifer). Carbohydr Res. 2002, 337: 1647-1655. 10.1016/S0008-6215(02)00023-X.
Redgwell R, Curti D, Fischer M, Nicolas P, Fay L: Coffee bean arabinogalactans: acidic polymers covalently linked to protein. Carbohydr Res. 2002, 337 (3): 239-253. 10.1016/S0008-6215(01)00316-0.
Haque M, Kotake T, Tsumuraya Y: Mode of action of β-glucuronidase from Aspergillus niger on the sugar chains of arabinogalactan-protein. Biosci Biotechnol Biochem. 2005, 69 (11): 2170-2177. 10.1271/bbb.69.2170.
Martinelli SD, Kinghorn JR: Aspergillus: 50 years on. 1994, Amsterdam: Elsevier Science
Rombouts F, Voragen A, Searle-van Leeuwen M, Geraeds C, Scholds H, Pilnik W: The arabinases of Aspergillus niger — Purification and characterization of two α-L-arabinofuranosidases and an endo-1,5-α-L-arabinase. Carbohydr Polym. 1988, 9: 25-47. 10.1016/0144-8617(88)90075-6.
Beldman G, Searle-van Leeuwen M, Ruiter GD, Siliha H, Voragen A: Degradation of arabinans by arabinases from Aspergillus aculeatus and Aspergillus niger. Carbohydr Polym. 1993, 20: 159-168. 10.1016/0144-8617(93)90146-U.
An J, Zhang L, O’Neill M, Albersheim P, Darvill A: Isolation and structural characterization of endo-rhamnogalacturonase-generated fragments of the backbone of rhamnogalacturonan I. Carbohydr Res. 1994, 264: 83-96. 10.1016/0008-6215(94)00186-3.
Renard CM, Lahaye M, Mutter M, Voragen FG, Thibault JF: Isolation and structural characterisation of rhamnogalacturonan oligomers generated by controlled acid hydrolysis of sugar-beet pulp. Carbohydr Res. 1997, 305 (2): 271-280. 10.1016/S0008-6215(97)10028-3.
Abdel-Naby MA, Osman MY, Abdel-Fattah AF: Purification and properties of three cellobiases from Aspergillus niger A20. Appl Biochem Biotechnol. 1999, 76 (1): 33-44. 10.1385/ABAB:76:1:33.
Acuna-Argüelles ME, Gutiérrez-Rojas M, Viniegra-Gonzalez G, Favela-Torres E: Production and properties of three pectinolytic activities produced by Aspergillus niger in submerged and solid-state fermentation. Appl Microbiol Biotechnol. 1995, 43 (5): 808-814. 10.1007/BF02431912.
Ademark P, Larsson M, Tjerneld F, Stalbrand H: Multiple α-galactosidases from Aspergillus niger: purification, characterization and substrate specificities. Enzyme Microb Technol. 2001, 29 (6–7): 441-448.
Adikane HV, Patil MB: Isolation and properties of β-glucosidase from Aspergillus niger. Indian J Biochem Biophys. 1985, 22 (2): 97-101.
Adya S, Elbein AD: Glucoprotein enzymes secreted by Aspergillus niger: Purification and properties of α-galactosidase. J Bacteriol. 1977, 129 (2): 850-856.
Akeboshi H, Tonozuka T, Furukawa T, Ichikawa K, Aoki H, Shimonishi A, Nishikawa A, Sakano Y: Insights into the reaction mechanism of glycosyl hydrolase family 49. Site-directed mutagenesis and substrate preference of isopullulanase. Eur J Biochem. 2004, 271 (22): 4420-4427. 10.1111/j.1432-1033.2004.04378.x.
Akiba S, Kimura Y, Yamamoto K, Kumagai H: Purification of a proteaseresistant cellulase from Aspergillus niger. J Ferment Bioeng. 1995, 79 (2): 125-130. 10.1016/0922-338X(95)94078-6.
Akimoto H, Kushima T, Nakamura T, Ohta K: Transcriptional analysis of two endoinulinase genes inuA and inuB in Aspergillus niger and nucleotide sequences of their promoter regions. J Biosci Bioeng. 1999, 88 (6): 599-604. 10.1016/S1389-1723(00)87086-3.
Alfani F, Cantarella L, Gallifuoco A, Pezzullo L, Scardi V, Cantarella M: Characterization of the β-glucosidase activity associated with immobilized cellulase of Aspergillus niger. Ann N Y Acad Sci. 1987, 501: 503-507. 10.1111/j.1749-6632.1987.tb45762.x.
Aliwan FO, Kroon PA, Faulds CB, Pickersgill R, Williamson G: Ferulic acid esterase-III from Aspergillus niger does not exhibit lipase activity. J Sci Food Agric. 1999, 79: 457-459. 10.1002/(SICI)1097-0010(19990301)79:3<457::AID-JSFA283>3.0.CO;2-G.
Altaner C, Saake B, Puls J: Specificity of an Aspergillus niger esterase deacetylating cellulose acetate. Cellulose. 2003, 10: 85-93. 10.1023/A:1023010330865.
Amirul AA, Khoo SL, Nazalan MN, Razip MS, Azizan MN: Purification and properties of two forms of glucoamylase from Aspergillus niger. Folia Microbiol (Praha). 1996, 41 (2): 165-174. 10.1007/BF02814694.
Aoki H, Yopi, Padmajanti A, Sakano Y, Yopi: Two components of cellbound isopullulanase from Aspergillus niger ATCC 9642-their purification and enzymatic properties. Biosci Biotechnol Biochem. 1996, 60 (11): 1795-1798. 10.1271/bbb.60.1795.
Aoki H, Yopi, Sakano Y, Yopi: Molecular cloning and heterologous expression of the isopullulanase gene from Aspergillus niger A.T.C.C. 9642. Biochem J. 1997, 323 (Pt 3): 757-764.
Bahl OP: Glucosidases of Aspergillus niger: II. Purification and general properties of 1,2-α-L-fucosidase. J Biol Chem. 1970, 245 (2): 199-304.
Barbe C, Dubourdieu D: Characterisation and purification of a cinnamate esterase from Aspergillus niger industrial pectinase preparation. J Sci Food Agric. 1998, 78: 471-478. 10.1002/(SICI)1097-0010(199812)78:4<471::AID-JSFA141>3.0.CO;2-0.
Benen JA, Kester HC, Parenicova L, Visser J: Characterization of Aspergillus niger pectate lyase A. Biochem. 2000, 39 (50): 15563-15569. 10.1021/bi000693w.
Benen JA, Kester HC, Visser J: Kinetic characterization of Aspergillus niger N400 endopolygalacturonases I , II and C. Eur J Biochem. 1999, 259: 577-585.
Biely P, Vrsanska M, Gorbacheva IV: The active site of an acidic endo-1,4-β- xylanase of Aspergillus niger. Biochim Biophys Acta. 1983, 743 (1): 155-161. 10.1016/0167-4838(83)90429-6.
Boel E, Hansen MT, Hjort I, Hoegh I, Fiil NP: Two different types of intervening sequences in the glucoamylase gene from Aspergillus niger. EMBO J. 1984, 3: 1581-1585.
Boel E, Hjort I, Svensson B, Norris F, Norris KE, Fiil NP: Glucoamylases G1 and G2 from Aspergillus niger are synthesized from two different but closely related mRNAs. EMBO J. 1984, 3 (5): 1097-1102.
Bonnin E, Lahaye M, Vigouroux J, Thibault JF: Preliminary characterization of a new exo-β-(1,4)-galactanase with transferase activity. Int J Biol Macromol. 1995, 17 (6): 345-351. 10.1016/0141-8130(96)81844-7.
Brillouet JM, Williams P, Moutounet M: Purification and some properties of a novel endo-β-(1$\rightarrow $→6)-Dgalactanase from Aspergillus niger. Agric Biol Chem. 1991, 55 (6): 1565-1571. 10.1271/bbb1961.55.1565.
Bussink HJ, Brouwer KB, de Graaff LH, Kester HC, Visser J: Identification and characterization of a second polygalacturonase gene of Aspergillus niger. Curr Genet. 1991, 20 (4): 301-307. 10.1007/BF00318519.
Bussink HJ, Buxton FP, Visser J: Expression and sequence comparison of the Aspergillus niger and Aspergillus tubigensis genes encoding polygalacturonase II. Curr Genet. 1991, 19 (6): 467-474. 10.1007/BF00312738.
Bussink HJD, Buxton FP, Fraaye BA, de Graaff LH, Visser J: The polygalacturonases of Aspergillus niger are encoded by a family of diverged genes. Eur J Biochem. 1992, 208: 83-90. 10.1111/j.1432-1033.1992.tb17161.x.
Bussink HJD, Kester HCM, Visser J: Molecular cloning, nucleotide sequence and expression of the gene encoding prepro-polygalacturonase II of Aspergillus niger. FEBS Lett. 1990, 273: 127-130. 10.1016/0014-5793(90)81066-W.
Claeyssens M, Loontiens FG, Kersters-Hilderson H, De Bruyne CK: Partial purification and properties of an Aspergillus niger β-Dxylosidase. Enzymologia. 1970, 40 (3): 177-198.
Claeyssens M, Loontiens FG, Kersters-Hilderson H, de Bruyne CK: Partial purification and properties of an Aspergillus niger β-Dxylosidase. Enzymologia. 1971, 40 (3): 177-198.
Cooke RD, Ferber CEM, Kanagasabapathy L: Purification and characterization of polygalacturonases from a commercial Aspergillus niger preparation. Biochim Biophys Acta. 1976, 452: 440-451. 10.1016/0005-2744(76)90194-7.
Costa-Ferreira M, Dias A, Maximo C, Morgado MJ, Sena-Martins G, Duarte JC: Xylanolytic enzyme production by an Aspergillus niger isolate. Appl Biochem Biotechnol. 1994, 44 (3): 231-242. 10.1007/BF02779659.
de Vries RP, Michelsen B, Poulsen CH, Kroon PA, van den Heuvel RHH, Faulds CB, Williamson G, van den Hombergh JPTW, Visser J: The faeA genes from Aspergillus niger and Aspergillus tubingensis encode ferulic acid esterases involved in degradation of complex cell wall polysaccharides. Appl Environ Microbiol. 1997, 63 (12): 4638-4644.
de Vries RP, van de Vondervoort P, Hendriks L, van de Belt M, Visser J: Regulation of the α- glucuronidase-encoding gene (aguA) from Aspergillus niger. Mol Genet Genomics. 2002, 268 (1): 96-102. 10.1007/s00438-002-0729-7.
de Vries RP, Kester HC, Poulsen CH, Benen JA, Visser J: Synergy between accessory enzymes of Aspergillus involved in the degradation of sugar beet pectin. Carbohydr Res. 2000, 327: 401-410. 10.1016/S0008-6215(00)00066-5.
de Vries RP, vanKuyk PA, Kester HC, Visser J: The Aspergillus niger faeB gene encodes a second feruloyl esterase involved in pectin and xylan degradation and is specifically induced in the presence of aromatic compounds. Biochem J. 2002, 363: 377-386. 10.1042/0264-6021:3630377.
de Vries RP, Visser J: Regulation of the feruloyl esterase (faeA) gene from Aspergillus niger. Appl Environ Microbiol. 1999, 65 (12): 5500-5503.
Elbein AD, Adya S, Lee YC: Purification and properties of a β-mannosidase from Aspergillus niger. J Biol Chem. 1977, 252 (6): 2026-2031.
Faulds CB, Williamson G: Ferulic acid esterase from Aspergillus niger: purification and partial characterization of two forms from a commercial source of pectinase. Biotechnol Appl Biochem. 1993, 17 (Pt 3): 349-359.
Flipphi MJ, Panneman H, van der Veen P, Visser J, de Graaff LH: Molecular cloning, expression and structure of the endo-1,5-α-L-arabinase gene of Aspergillus niger. Appl Microbiol Biotechnol. 1993, 40 (2–3): 318-326.
Frederick MM, Frederick JR, Fratzke AR, Reilly PJ: Purification and characterization of a xylobiose- and xylose-producing endo-xylanase from Aspergillus niger. Carbohydr Res. 1981, 97: 87-103. 10.1016/S0008-6215(00)80527-3.
Frederick MM, Kiang C-H, Frederick JR, Reilly PJ: Purification and characterization of endo-xylanases from Aspergillus niger. I. Two isoenzymes active on xylan backbones near branch points. Biotechnol Bioeng. 1985, 27: 525-532. 10.1002/bit.260270420.
Freedberg IM, Levin Y, Kay CM, McCubbin WD, Katchalski-Katzir E: Purification and characterization of Aspergillus niger exo-1,4-glucosidase. Biochim Biophys Acta. 1975, 391 (2): 361-381. 10.1016/0005-2744(75)90260-0.
Galas E, Romanowska I: Purification and some properties of β-glucosidase from Aspergillus niger IBT-90. Acta Microbiol Pol. 1997, 46 (3): 241-252.
Gielkens MM, Visser J, de Graaff LH: Arabinoxylan degradation by fungi: characterization of the arabinoxylan-arabinofuranohydrolase encoding genes from Aspergillus niger and Aspergillus tubingensis. Curr Genet. 1997, 31 (1): 22-29. 10.1007/s002940050172.
Goosen C, Yuan XL, van Munster JM, Ram AF, van der Maarel MJ, Dijkhuizen L: Molecular and biochemical characterization of a novel intracellular invertase from Aspergillus niger with transfructosylating activity. Eukaryot Cell. 2007, 6 (4): 674-681. 10.1128/EC.00361-06.
Gorbacheva IV, Rodionova NA: Studies on xylan degrading enzymes. I. Purification and characterization of endo- 1,4-β-xylanase from Aspergillus niger str. 14. Biochim Biophys Acta. 1977, 484 (1): 79-93. 10.1016/0005-2744(77)90114-0.
Gorbacheva IV, Rodionova NA: Studies on xylan-degrading enzymes. II. Action pattern of endo-1,4-β-xylanase from Aspergillus niger str. 14 on xylan and xylooligosaccharides. Biochim Biophys Acta. 1977, 484 (1): 94-102. 10.1016/0005-2744(77)90115-2.
Gottschalk TE, Nielsen JE, Rasmussen P: Detection of endogenous β-glucuronidase activity in Aspergillus niger. Appl Microbiol Biotechnol. 1996, 45 (1–2): 240-244.
Greenberg NA, Mahoney RR: Rapid purification of β-galactosidase (Aspergillus niger) from a commercial preparation. J Food Sci. 1981, 46: 684-687. 10.1111/j.1365-2621.1981.tb15324.x.
Gunata Z, Brillouet J-M, Voirin S, Baumes R, Cordonnier R: Purification and some properties of an α- L-Arabinofuranosidase from Aspergillus niger. Action on grape monoterpenyl arabinofuranosylglucosides. J Agric Food Chem. 1990, 38: 772-776. 10.1021/jf00093a040.
Gysler C, Harmsen JAM, Kester HCM, Visser J, Heim J: Isolation and structure of the pectin lyase D-encoding gene from Aspergillus niger. Gene. 1990, 89: 101-108. 10.1016/0378-1119(90)90211-9.
Hagglund P, Sabini E, Boisset C, Wilson K, Chanzy H, Stålbrand H: Degradation of mannan I and II crystals by fungal endo-β-1,4-mannanases and a β-1,4- mannosidase studied with transmission electron microscopy. Biomacromolecules. 2001, 2 (3): 694-699. 10.1021/bm010006w.
Harmsen JAM, Kurster-Van Sommeren MA, Visser J: Cloning and expression of a second Aspergillus niger pectin lyase gene (pelA): indications of a pectin lyase gene family in A. niger. Curr Genet. 1990, 18: 161-166. 10.1007/BF00312604.
Hasper AA, Dekkers E, van Mil M, van de Vondervoort PJI, de Graaf LH: EglC, a new endoglucanase from Aspergillus niger with major activity towards xyloglucan. Appl Environ Microbiol. 2002, 68 (4): 1556-1560. 10.1128/AEM.68.4.1556-1560.2002.
Heinrichova K, Rexova-Benkova L: Purification and characterization of an extracellular exo-D-galacturonanase of Aspergillus niger. Biochim Biophys Acta. 1976, 422 (2): 349-356. 10.1016/0005-2744(76)90146-7.
Hessing JG, van Rotterdam C, Verbakel JM, Roza M, Maat J, van Gorcom RF, van den Hondel CA: Isolation and characterization of a 1,4-β- endoxylanase gene of A. awamori. Curr Genet. 1994, 26 (3): 228-232. 10.1007/BF00309552.
Himmel ME, Adney WS, Fox JW, Mitchell DJ, Baker JO: Isolation and characterization of two forms of β-D-glucosidase from Aspergillus niger. Appl Biochem Biotechnol. 1993, 39–40: 213-225.
Hong J, Tamaki H, Akiba S, Yamamoto K, Kumagai H: Cloning of a gene encoding a highly stable endo-β- 1,4-glucanase from Aspergillus niger and its expression in yeast. J Biosci Bioeng. 2001, 92 (5): 434-441.
Hurst PL, Nielsen J, Sullivan PA, Shepherd MG: Purification and properties of a cellulase from Aspergillus niger. Biochem J. 1977, 165 (1): 33-41.
Hurst PL, Sullivan PA, Shepherd MG: Substrate specificity and mode of action of a cellulase from Aspergillus niger. Biochem J. 1978, 169 (2): 389-395.
Jenkins J, Scott M, Mayans O, Pickersgill R, Harris G, Connerton I, Gravesen T: Crystallization and preliminary X-ray analysis of pectin lyase A from Aspergillus niger. Acta Crystallogr D Biol Crystallogr. 1996, 52 (Pt 2): 402-404.
John M, Schmidt B, Schmidt J: Purification and some properties of five endo-1,4-β-D-xylanases and a β- D-xylosidase produced by a strain of Aspergillus niger. Can J Biochem. 1979, 57 (2): 125-134. 10.1139/o79-016.
Kaji A, Tagawa K: Purification, crystallization and amino acid composition of α-L-arabinofuranosidase from Aspergillus niger. Biochim Biophys Acta. 1970, 207 (3): 456-464.
Kaji A, Talawa K, Ichimi T: Properties of purified α-L-arabinofuranosidase from Aspergillus niger. Biochim Biophys Acta. 1969, 171 (1): 186-188. 10.1016/0005-2744(69)90118-1.
Kaneko R, Kusakabe I, Ida E, Murakami K: Substrate specificity of α- galactosidase from Aspergillus niger 5-16. Agric Biol Chem. 1991, 55 (1): 109-115. 10.1271/bbb1961.55.109.
Kaneko S, Shimasaki T, Kusakabe I: Purification and some properties of intracellular α-L-arabinofuranosidase from Aspergillus niger 5-16. Biosci Biotechnol Biochem. 1993, 57 (7): 1161-1165. 10.1271/bbb.57.1161.
Keon JPR, Waksman G: Common amino acid domain among endopolygalacturonases of ascomycete fungi. Appl Environ Microbiol. 1990, 56 (8): 2522-2528.
Kester HC, Benen JA, Visser J, Warren ME, Orlando R, Bergmann C, Magaud D, Anker D, Doutheau A: Tandem mass spectrometric analysis of Aspergillus niger pectin methylesterase: mode of action on fully methyl-esterified oligogalacturonates. Biochem J. 2000, 346 (Pt 2): 469-474.
Kester HCM, Visser J: Purification and characterization of polygalacturonases produced by the hyphal fungus Aspergillus niger. Biotechnol Appl Biochem. 1990, 12: 150-160.
Khademi S, Zhang D, Swanson SM, Wartenberg A, Witte K, Meyer EF: Determination of the structure of an endoglucanase from Aspergillus niger and its mode of inhibition by palladium chloride. Acta Crystallogr D Biol Crystallogr. 2002, 58: 660-667. 10.1107/S0907444902003360.
Khanh NQ, Ruttkowski E, Leidinger K, Albrecht H, Gottschalk M: Characterization and expression of a genomic pectin methyl esterase-encoding gene in Aspergillus niger. Gene. 1991, 106 (1): 71-77. 10.1016/0378-1119(91)90567-U.
Kinoshita K, Takano M, Koseki T, Ito K, Iwano K: Cloning of the xynNB gene encoding xylanase B from Aspergillus niger and its expression in Aspergillus kawachii. J Ferment Bioeng. 1995, 79 (5): 422-428. 10.1016/0922-338X(95)91255-4.
Kiryu T, Nakano H, Kiso T, Murakami H: Purification and characterization of a novel α-glucuronidase from Aspergillus niger specific for O-α-Dglucosyluronic acid 2005-D-glucosiduronic acid. Biosci Biotechnol Biochem. 2005, 69 (3): 522-529. 10.1271/bbb.69.522.
Kobayashi T, Sakano Y: Pullanase from Aspergillus. 1973, Patent: JP 48008989.
Korman DR, Bayliss FT, Barnett CC, Carmona CL, Kodama KH, Royer TJ, Thompson SA, Ward M, Wilson LJ, Berka RM: Cloning, characterization, and expression of two α-amylase genes from Aspergillus niger var. awamori. Curr Genet. 1990, 17 (3): 203-212. 10.1007/BF00312611.
Kormelink FJM, Gruppen H, Voragen AGJ: Mode of action of (1- >4)-β-arabinoxylan arabinofuranohydrolase (AXH) and α-L-arabinofuranosidases on alkali-extractable wheat-flour arabinoxylan. Carbohydr Res. 1993, 249 (2): 345-353. 10.1016/0008-6215(93)84099-R.
Kormelink FJM, Lefebve B, Strozyk F, Voragen AGJ: Purification and characterization of an acetyl xylan esterase from Aspergillus niger. J Biotechnol. 1993, 27: 267-282. 10.1016/0168-1656(93)90090-A.
Krengel U, Dijkstra BW: Threedimensional structure of Endo-1,4-β- xylanase I from Aspergillus niger: molecular basis for its low pH optimum. J Mol Biol. 1996, 263 (1): 70-78. 10.1006/jmbi.1996.0556.
Krisana A, Rutchadaporn S, Jarupan G, Lily E, Sutipa T, Kanyawim K: Endo-1,4-β-xylanase B from Aspergillus cf. niger BCC14405 isolated in Thailand: purification, characterization and gene isolation. J Biochem Mol Biol. 2005, 38 (1): 17-23. 10.5483/BMBRep.2005.38.1.017.
Kroon PA, Williamson G: Release of ferulic acid from sugar-beet pulp by using arabinanase, arabinofuranosidase and an esterase from Aspergillus niger. Biotechnol Appl Biochem. 1996, 23 (Pt 3): 263-267.
Kumar V, Ramakrishnan S, Teeri TT, Knowles JKC, Hartley BS: Saccharomyces cerevisiae cells secreting an Aspergillus niger β-galactosidase grow on whey permeate. Biotechnology (N Y). 1992, 10: 82-85. 10.1038/nbt0192-82.
Kurosawa Y, Ikeda K, Egami F: α-L-Rhamnosidases of the liver of Turbo cornutus and Aspergillus niger. J Biochem. 1973, 73: 31-37.
Kusters-Van Someren M, Flipphi M, de Graaff L, van den Broeck H, Kester H, Hinnen A, Visser J: Characterization of the Aspergillus niger pelB gene: structure and regulation of expression. Mol Gen Genet. 1992, 234: 113-120.
Kusters-Van Someren MA, Harmsen JAM, Kester HCM, Visser J: Structure of the Aspergillus niger pelA gene and its expression in Aspergillus niger and Aspergillus nidulans. Curr Genet. 1991, 20: 293-299. 10.1007/BF00318518.
Limberg G, Korner R, Buchholt HC, Christensen TM, Roepstorff P, Mikkelsen JD: Analysis of different de-esterification mechanisms for pectin by enzymatic fingerprinting using endopectin lyase and endopolygalacturonase II from A. niger. Carbohydr Res. 2000, 327 (3): 293-307. 10.1016/S0008-6215(00)00067-7.
Limberg G, Korner R, Buchholt HC, Christensen TM, Roepstorff P, Mikkelsen JD: Quantification of the amount of galacturonic acid residues in blocksequences in pectin homogalacturonan by enzymatic fingerprinting with exo- and endo-polygalacturonase II from Aspergillus niger. Carbohydr Res. 2000, 327 (3): 321-332. 10.1016/S0008-6215(00)00068-9.
Linden J, Samara M, Decker S, Johnson E, Boyer M, Pecs M, Adney W, Himmel M: Purification and characterization of an acetyl esterase from Aspergillus niger. Appl Biochem Biotechnol. 1994, 45–46: 383-393.
Markovic O, Joernvall H: Tomato and Aspergillus niger pectinesterases. Correlation of differences in existing reports: large species variations. Protein Seq Data Anal. 1990, 3: 513-515.
Master ER, Zheng Y, Storms R, Tsang A, Powlowski J: A xyloglucanspecific family 12 glycosyl hydrolase from Aspergillus niger: recombinant expression, purification and characterization. Biochem J. 2008, 411 (1): 161-170. 10.1042/BJ20070819.
Matsushita J, Kato Y, Matsuda K: Purification and properties of an α-Dxylosidase from Aspergillus niger. J Biochem (Tokyo). 1985, 98 (3): 825-832.
Matsushita J, Kato Y, Matsuda K: Characterization of α-D-xylosidase II from A. niger. Agric Biol Chem. 1987, 51 (7): 2015-2016. 10.1271/bbb1961.51.2015.
Mayans O, Scott M, Connerton I, Gravesen T, Benen J, Visser J, Pickersgill R, Jenkins J: Two crystal structures of pectin lyase A from Aspergillus reveal a pH driven conformational change and striking divergence in the substratebinding clefts of pectin and pectate lyases. Structure. 1997, 5: 677-689. 10.1016/S0969-2126(97)00222-0.
McCleary BV: Modes of action of β- mannanase enzymes of diverse origin on legume seed galactomannans. Phytochemistry. 1979, 18: 757-763. 10.1016/0031-9422(79)80009-6.
Meagher MM, Tao BY, Chow JM, Reilly PJ: Kinetics and subsite mapping of a D-xylobiose- and D-xyloseproducing Aspergillus niger endo-(1-4)-β- D-xylanase. Carbohydr Res. 1988, 173 (2): 273-283. 10.1016/S0008-6215(00)90823-1.
Mizuno M, Koide A, Yamamura A, Akeboshi H, Yoshida H, Kamitori S, Sakano Y, Nishikawa A, Tonozuka T: Crystal structure of Aspergillus niger isopullulanase, a member of glycoside hydrolase family 49. J Mol Biol. 2008, 376 (1): 210-220. 10.1016/j.jmb.2007.11.098.
Moriyama S, Tanaka H, Uwataki M, Muguruma M, Ohta K: Molecular cloning and characterization of an exoinulinase gene from Aspergillus niger strain 12 and its expression in Pichia pastoris. J Biosci Bioeng. 2003, 96 (4): 324-331.
Nakamura A, Nishimura I, Yokoyama A, Lee DG, Hidaka M, Masaki H, Kimura A, Chiba S, Uozumi T: Cloning and sequencing of an α- glucosidase gene from Aspergillus niger and its expression in A. nidulans. J Biotechnol. 1997, 53 (1): 75-84. 10.1016/S0168-1656(97)01664-7.
Ohta K, Akimoto H, Matsuda S, Toshimitsu D, Nakamura T: Molecular cloning and sequence analysis of two endoinulinase genes from Aspergillus niger. Biosci Biotechnol Biochem. 1998, 62 (9): 1731-1738. 10.1271/bbb.62.1731.
Pages S, Kester HC, Visser J, Benen JA: Changing a single amino acid residue switches processive and non-processive behavior of Aspergillus niger endopolygalacturonase I and II. J Biol Chem. 2001, 276 (36): 33652-33656. 10.1074/jbc.M105770200.
Pagès S, Heijne WH, Kester HC, Visser J, Benen JA: Subsite mapping of Aspergillus niger endopolygalacturonase II by site-directed mutagenesis. J Biol Chem. 2000, 275 (38): 29348-29353. 10.1074/jbc.M910112199.
Parenicova L, Benen JA, Kester HC, Visser J: pgaA and pgaB encode two constitutively expressed endopolygalacturonases of Aspergillus niger. Biochem J. 2000, 345: 637-644. 10.1042/0264-6021:3450637.
Parenicova L, Benen JAE, Kester HCM, Visser J: pgaE encodes a fourth member of the endopolygalacturonase gene family from Aspergillus niger. Eur J Biochem. 1998, 251: 72-80. 10.1046/j.1432-1327.1998.2510072.x.
Parenicova L, Kester HC, Benen JA, Visser J: Characterization of a novel endopolygalacturonase from Aspergillus niger with unique kinetic properties. FEBS Lett. 2000, 467 (2–3): 333-336.
Pazur JH, Simpson DL, Knull HR: Biosynthesis of glucohydrolase I , a glycoenzyme from Aspergillus niger. Biochem Biophys Res Commun. 1969, 36 (3): 394-400. 10.1016/0006-291X(69)90577-4.
Pellerin P, Brillouet JM: Purification and properties of an exo-(1->3)-β-Dgalactanase from Aspergillus niger. Carbohydr Res. 1994, 264 (2): 281-291. 10.1016/S0008-6215(05)80012-6.
Pitson SM, Voragen AG, Vincken JP, Beldman G: Action patterns and mapping of the substrate-binding regions of endo-(1->5)-α-L-arabinanases from Aspergillus niger and Aspergillus aculeatus. Carbohydr Res. 1997, 303 (2): 207-218. 10.1016/S0008-6215(97)00159-6.
Raab B: Characterization of endopolygalacturonase (EC 3.2.1.15) from Aspergillus niger as glycoprotein by electrophoretic methods and lectin affinoblotting. Electrophoresis. 1992, 13 (9–10): 807-808.
Regalado C, Garcia-Almendarez BE, Venegas-Barrera LM, Téllez-JuradoA A, Rodriguez-Serrano G, Huerta-Ochoa S, Whitaker JR: Production, partial purification and properties of β-mannanases obtained by solid substrate fermentation of spent soluble coffee wastes and copra paste using Aspergillus oryzae and Aspergillus niger. J Sci Food Agric. 2000, 80: 1343-1350. 10.1002/1097-0010(200007)80:9<1343::AID-JSFA651>3.0.CO;2-#.
Rodionova NA, Tavobilov IM, Bezbodorow AM: β-Xylosidase from Aspergillus niger 15: Purification and Properties. J Appl Biochem. 1983, 5: 300-312.
Sakamoto T, Bonnin E, Quemener B, Thibault J-F: Purification and characterisation of two exo-polygalacturonases from Aspergillus niger able to degrade xylogalacturonan and acetylated homogalacturonan. Biochim Biophys Acta. 2002, 1572: 10-18. 10.1016/S0304-4165(02)00277-5.
Sakano Y, Higuchi M, Kobayashi T: Pullulan 4-glucanohydrolase from Aspergillus niger. Arch Biochem Biophys. 1972, 153 (1): 180-187. 10.1016/0003-9861(72)90434-1.
Sanchez-Torres P, Visser J, Benen JA: Identification of amino acid residues critical for catalysis and stability in Aspergillus niger family 1 pectin lyase A. Biochem J. 2003, 370 (Pt 1): 331-337.
Schroder M, Soe JB, Zargahi MR, Rouau X: Production of substrate for galactose oxidase by depolymerization of an arabinogalactan-peptide from wheat flour. J Agric Food Chem. 1999, 47 (4): 1483-1488. 10.1021/jf980813v.
Schroter KH, Arkema A, Kester HC, Visser J, Dijkstra BW: Crystallization and preliminary crystallographic characterization of endo-polygalacturonase II from Aspergillus niger. J Mol Biol. 1994, 243 (2): 351-352. 10.1006/jmbi.1994.1660.
Shei JC, Fratzke AR, Frederick MM, Frederick JR, Reilly PJ: Purification and characterization of endo-xylanases from Aspergillus niger. II. An enzyme of pI 4.5. Biotechnol Bioeng. 2985, 27: 533-538.
Fournier Shei R, Frederick MM, Frederick JR, Reilly PJ: Purification and characterization of endo-xylanases from Aspergillus niger. III. An enzyme of pI 3.65. Biotechnol Bioeng. 1985, 27: 539-546. 10.1002/bit.260270422.
Singh A, Agrawal AK, Abidi AB, Darmwal NS: Properties of exoglucanase from A. niger. J Gen Appl Microbiol. 1990, 36: 245-253. 10.2323/jgam.36.245.
Singh A, Agrawal AK, Abidi AB, Darmwal NS: General and kinetic properties of endoglucanase from Aspergillus niger. FEMS Microbiol Lett. 1990, 59 (1–2): 221-224.
Singh SA, Appu Rao AG: A simple fractionation protocol for, and a comprehensive study of the molecular properties of two major endopolygalacturonases from Aspergillus niger. Biotechnol Appl Biochem. 2002, 35 (Pt 2): 115-123.
Somiari RI, Balogh E: Properties of an extracellular glycosidase of Aspergillus niger suitable for removal of oligosaccharides from cowpea meal. Enzyme Microb Technol. 1995, 17 (4): 311-316. 10.1016/0141-0229(94)00006-9.
Suykerbuyk MEG, Kester HCM, Schaap PJ, Stam H, Musters W, Visser J: Cloning and characterization of two rhamnogalacturonan hydrolase genes from Aspergillus niger. Appl Environ Microbiol. 1997, 63 (7): 2507-2515.
Svensson B, Larsen K, Gunnarsson A: Characterization of a glucoamylase G2 from Aspergillus niger. Eur J Biochem. 1986, 154 (3): 497-502. 10.1111/j.1432-1033.1986.tb09425.x.
Tahir TA, Berrin J-G, Flatman R, Roussel A, Roepstorff P, Williamson G, Juge N: Specific characterization of substrate and inhibitor binding sites of a glycosyl hydrolase family 11 xylanase from Aspergillus niger. J Biol Chem. 2002, 277 (46): 44035-44043. 10.1074/jbc.M205657200.
Uchida H, Nanri T, Kawabata Y, Kusakabe I, Murakami K: Purification and characterization of intracellular α-glucuronidase from A. niger. Biosci Biotechnol Biochem. 1992, 56 (10): 1608-1615. 10.1271/bbb.56.1608.
Unno T, Ide K, Yazaki T, Tanaka Y, Nakakuki T, Okada G: High recovery purification and some properties of a β-glucosidase from A. niger. Biosci Biotechnol Biochem. 1993, 57 (12): 2172-2173. 10.1271/bbb.57.2172.
van Alebeek GJ, van Scherpenzeel K, Beldman G, Schols HA, Voragen AG: Partially esterified oligogalacturonides are the preferred substrates for pectin methylesterase of Aspergillus niger. Biochem J. 2003, 372 (Pt 1): 211-218.
van Alebeek G-JWM, Christensen TMIE, Schols HA, Mikkelsen JD, Voragen AGJ: Mode of action of pectin lyase A of Aspergillus niger on differently C(6)-substituted oligogalacturonides. J Biol Chem. 2002, 277 (29): 25929-25936. 10.1074/jbc.M202250200.
van de Vis JW, Searle van Leeuwen MJF, Siliha HA, Kormelink FJM, Voragen AGJ: Purification and characterization of endo-1,4-β- galactanases from Aspergillus niger and Aspergillus aculeatus: Use in combination with arabinanases from Aspergillus niger in enzymic conversion of potato arabinogalactan. Carbohydr Polym. 1991, 16 (2): 167-187. 10.1016/0144-8617(91)90101-H.
van den Broeck HC, de Graaff LH, Hille JDR, van Ooyen AJ, Visser J, Harder A: Cloning and expression of xylanase genes from fungal origin. European Patent Application. 1992, Patent EP463706-A1: 55 pages-
van Ooyen AJJ, de Graaff LH, van den Broeck HC, Visser J: Cloning and expression of xylanase B. International Patent Application. 1994, Patent WO9414965-A1: 49 pages-
van Peij NN, Brinkmann J, Vrsanska M, Visser J, de Graaff LH: betaxylosidase activity, encoded by xlnD, is essential for complete hydrolysis of xylan by Aspergillus niger but not for induction of the xylanolytic enzyme spectrum. Eur J Biochem. 1997, 245 (1): 164-173. 10.1111/j.1432-1033.1997.00164.x.
van Pouderoyen, Snijder HJ, Benen JAE, Dijkstra BW: Structural insights into the processivity of endopolygalacturonase I from Aspergillus niger. FEBS Lett. 2003, 554: 462-466. 10.1016/S0014-5793(03)01221-3.
van Santen Y, Benen JAE, Schroeter KH, Kalk KH, Armand S, Visser J, Dijkstra BW: 1.68 Å crystal structure of endopolygalacturonase II from Aspergillus niger and identification of active site residues by site-directed mutagenesis. J Biol Chem. 1999, 274: 30474-30480. 10.1074/jbc.274.43.30474.
Vidmar S, Turk V, Kregar I: Cellulytic complex of Aspergillus niger under conditions for citric acid production. Isolation and characterization of two β-(1-4)-glucan hydrolases. Appl Microbiol Biotechnol. 1984, 20: 326-330. 10.1007/BF00270594.
Vitali J, Schiek B, Kester HCM, Visser J, Jurnak F: The three dimensional structure of A. niger pectin lyase B at 1.7-Å resolution. Plant Physiol. 116: 69-80.
Vrsanska M, Gorbacheva IV, Kratký Z, Biely P: Reaction pathways of substrate degradation by an acidic endo-1,4-β-xylanase of Aspergillus niger. Biochim Biophys Acta. 1982, 704 (1): 114-122. 10.1016/0167-4838(82)90138-8.
Waibel R, Amadò R, Neukom H: Purification of an α-L-arabinofuranosidase from Aspergillus niger by substrate affinity chromatography. J Chromatogr. 1980, 197 (1): 86-91. 10.1016/S0021-9673(00)80539-7.
Wallis GL, Hemming FW, Peberdy JF: Secretion of two β- fructofuranosidases by Aspergillus niger growing in sucrose. Arch Biochem Biophys. 1997, 345 (2): 214-222. 10.1006/abbi.1997.0228.
Watanabe T, Sato T, Yoshioka S, Koshijima T, Kuwahara M: Purification and properties of Aspergillus niger β- glucosidase. Eur J Biochem. 1992, 209 (2): 651-659. 10.1111/j.1432-1033.1992.tb17332.x.
Xie Y, Gao Y, Chen Z: Purification and characterization of an extracellular β-glucosidase with high transglucosylation activity and stability from Aspergillus niger No. 5.1. Appl Biochem Biotechnol. 2004, 119 (3): 229-240. 10.1007/s12010-004-0004-y.
Yamaguchi F, Inoue S, Hatanaka C: Purification and properties of endo-β- 1,4-D-galactanase from Aspergillus niger. Biosci Biotechnol Biochem. 1995, 59 (9): 1742-1744. 10.1271/bbb.59.1742.
Yan TR, Lin CL: Purification and characterization of a glucose-tolerant β- glucosidase from Aspergillus niger CCRC 31494. Biosci Biotechnol Biochem. 1997, 61 (6): 965-970. 10.1271/bbb.61.965.
Yan TR, Lin YH, Lin CL: Purification and characterization of an extracellular β-glucosidase II with high hydrolysis and transglucosylation activities from Aspergillus niger. J Agric Food Chem. 1998, 46 (2): 431-437. 10.1021/jf9702499.
Yang Y, Bergmann C, Benen J, Orlando R: Identification of the glycosylation site and glycan structures of recombinant endopolygalacturonase II by mass spectrometry. Rapid Commun Mass Spectrom. 1997, 11: 1257-1262. 10.1002/(SICI)1097-0231(199708)11:12<1257::AID-RCM19>3.0.CO;2-X.
Yeoh HH, Tan TK, Chua SL, Lim G: Properties of β-glucosidase purified from Aspergillus niger. MIRCEN J Appl Microbiol Biotechnol. 1988, 4: 425-430. 10.1007/BF00940168.
Zhang L, Zhao C, Zhu D, Ohta Y, Wang Y: Purification and characterization of inulinase from Aspergillus niger AF10 expressed in Pichia pastoris. Protein Expr Purif. 2004, 35 (2): 272-275. 10.1016/j.pep.2004.02.015.
Acknowledgements
Lene Holberg Blicher is acknowledged for assistance with array preparations.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests
Authors’ contributions
MRA conceived the study, designed the experiments, analyzed the data and wrote the manuscript. MGJ conducted the experiments, designed the experiments and analyzed data. RPdV analyzed data and wrote the manuscript. JN conceived the study, designed the experiments, and wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2012_4195_MOESM1_ESM.pdf
Additional file 1: Schematic representation of starch. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on ref. [59]. (PDF 34 KB)
12864_2012_4195_MOESM2_ESM.pdf
Additional file 2: Schematic representation of cellulose. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on refs. [20, 60]. (PDF 28 KB)
12864_2012_4195_MOESM3_ESM.pdf
Additional file 3: Schematic representation of pullulan. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on refs. [59, 61]. (PDF 33 KB)
12864_2012_4195_MOESM4_ESM.pdf
Additional file 4: Schematic representation of inulin. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on ref. [62]. (PDF 34 KB)
12864_2012_4195_MOESM5_ESM.pdf
Additional file 5: Schematic representation of galactomannan. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on refs. [20, 63]. (PDF 34 KB)
12864_2012_4195_MOESM6_ESM.pdf
Additional file 6: Schematic representation of insoluble galactoglucomannan. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on ref. [20]. (PDF 37 KB)
12864_2012_4195_MOESM7_ESM.pdf
Additional file 7: Schematic representation of soluble galactoglucomannan. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on ref. [18–21]. (PDF 42 KB)
12864_2012_4195_MOESM8_ESM.pdf
Additional file 8: Schematic representation of smooth pectin. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on refs. [20, 60]. (PDF 39 KB)
12864_2012_4195_MOESM9_ESM.pdf
Additional file 9: Schematic representation of xylogalactouronan. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on refs. [20, 64–67]. (PDF 41 KB)
12864_2012_4195_MOESM10_ESM.pdf
Additional file 10: Schematic representation of xylan. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on refs. [20, 60, 68, 69]. (PDF 42 KB)
12864_2012_4195_MOESM11_ESM.pdf
Additional file 11: Schematic representation of xyloglucan type XXGG. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on refs. [20, 70, 71]. (PDF 34 KB)
12864_2012_4195_MOESM12_ESM.pdf
Additional file 12: Schematic representation of xyloglucan type XXXG. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on refs. [20, 70, 71]. (PDF 43 KB)
12864_2012_4195_MOESM13_ESM.pdf
Additional file 13: Schematic representation of arabinogalactan type I. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on refs. [12, 72, 73]. (PDF 32 KB)
12864_2012_4195_MOESM14_ESM.pdf
Additional file 14: Schematic representation of arabinogalactan type II (protein bound arabinogalactan). The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on [12, 72, 74, 75]. (PDF 43 KB)
12864_2012_4195_MOESM15_ESM.pdf
Additional file 15: Schematic representation of arabinan. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on refs. [76–78]. (PDF 38 KB)
12864_2012_4195_MOESM16_ESM.pdf
Additional file 16: Schematic representation of rhamnogalacturonan type I. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. The structure is based on refs. [20, 76, 77, 79, 80]. (PDF 43 KB)
12864_2012_4195_MOESM17_ESM.pdf
Additional file 17: Tables of enzymatic activities required for the degradation of 16 different plant-derived polysaccharides. For each isoenzyme, putative or characterized, is noted literature references and the gene ID in the whole genome sequencings of A. niger CBS 513.88 and ATCC 1015 [3–7, 10–12, 14, 18–36, 39–41, 45–49, 51, 59–68, 70–226]. (PDF 96 KB)
12864_2012_4195_MOESM18_ESM.pdf
Additional file 18: Full map of 16 polysaccharide structures with the addition of genes from A. niger ATCC 1015 specific for each type of bond. The nature of the bonds between the sugar units are noted where they are known. The number of sides on the sugar polygons reflect the number of carbons of the sugar. The first carbon is indicated on all sugars with a 1 to clarify the bond configurations. Carbons are numbered clockwise from the first carbon with the two last carbons of the sugar (hexose or pentose) on the same corner of the polygon. The oligos are hypothetical hydrolysis products and sugars that appear due to the action of exo-acting enzymes. Type and approximate 2D orientation is shown for each of the chemical bonds connecting sugars and/or acids. Next to each bond, one can find verified and hypothetical enzymes in A. niger ATCC 1015 capable of catalyzing the hydrolysis of the bond. The bottom panel denotes CAZy-families for each of the genes. See Additional Files 1-16 for details on literature references for structures. (PDF 161 KB)
12864_2012_4195_MOESM19_ESM.pdf
Additional file 19: Map of statistically significantly regulated genes coding for carbohydrate-active enzymes in a pairwise comparison of growth on arabinan versus L-arabinose. Genes marked with a red box are significantly up-regulated on arabinan relative to L-arabinose, a green box denotes the opposite, gray boxes means that the gene is found in the A. niger CBS 513.88 sequence, but no bi-directional best hit is found in the A. niger ATCC 1015 sequence. The absence of a box mean that this gene is not statistically significantly regulated in this comparison. (PDF 201 KB)
12864_2012_4195_MOESM20_ESM.pdf
Additional file 20: Map of statistically significantly regulated genes coding for carbohydrate-active enzymes in a pairwise comparison of growth on starch versus D-glucose. Genes marked with a red box are significantly up-regulated on starch relative to D-glucose, a green box denotes the opposite, gray boxes means that the gene is found in the A. niger CBS 513.88 sequence, but no bi-directional best hit is found in the A. niger ATCC 1015 sequence. The absence of a box mean that this gene is not statistically significantly regulated in this comparison. (PDF 201 KB)
12864_2012_4195_MOESM21_ESM.pdf
Additional file 21: Map of statistically significantly regulated genes coding for carbohydrate-active enzymes in a pairwise comparison of growth on L-arabinose versus D-glucose. Genes marked with a red box are significantly up-regulated on L-arabinose relative to D-glucose, a green box denotes the opposite, gray boxes means that the gene is found in the A. niger CBS 513.88 sequence, but no bi-directional best hit is found in the A. niger ATCC 1015 sequence. The absence of a box mean that this gene is not statistically significantly regulated in this comparison. (PDF 201 KB)
12864_2012_4195_MOESM22_ESM.pdf
Additional file 22: Map of statistically significantly regulated genes coding for carbohydrate-active enzymes in a pairwise comparison of growth on starch versus xylan. Genes marked with a red box are significantly up-regulated on starch relative to xylan, a green box denotes the opposite, gray boxes means that the gene is found in the A. niger CBS 513.88 sequence, but no bi-directional best hit is found in the A. niger ATCC 1015 sequence. The absence of a box mean that this gene is not statistically significantly regulated in this comparison. (PDF 147 KB)
12864_2012_4195_MOESM23_ESM.pdf
Additional file 23: Clustering of 103 significantly regulated genes coding for enzymes with putative and characterized polysaccharide-activities. All genes were found to be significantly regulated in at least one pairwise-comparison of two carbon sources. Cluster numbers in parentheses is the number of the corresponding cluster(s) in Figure 2. The number of genes in each cluster is shown next to the cluster number. The gene names of known genes found in a particular cluster are found at the bottom of the transcription profile graph of each cluster. The genes were clustered using the ClusterLustre algorithm [50]. (PDF 36 KB)
12864_2012_4195_MOESM24_ESM.zip
Additional file 24: 24 maps of enzymatic activities present in the expression-based clusters 1-24 of Figure2. Genes marked with a red box are found in the cluster, gray boxes means that the gene is found in the A. niger CBS 513.88 sequence, but no bi-directional best hit is found in the A. niger ATCC 1015 sequence. (ZIP 3 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Andersen, M.R., Giese, M., de Vries, R.P. et al. Mapping the polysaccharide degradation potential of Aspergillus niger. BMC Genomics 13, 313 (2012). https://doi.org/10.1186/1471-2164-13-313
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-13-313