
Abstract
Background
Stx bacteriophages are responsible for driving the dissemination of Stx toxin genes (stx) across their bacterial host range. Lysogens carrying Stx phages can cause severe, life-threatening disease and Stx toxin is an integral virulence factor. The Stx-bacteriophage vB_EcoP-24B, commonly referred to as Ф24B, is capable of multiply infecting a single bacterial host cell at a high frequency, with secondary infection increasing the rate at which subsequent bacteriophage infections can occur. This is biologically unusual, therefore determining the genomic content and context of Ф24B compared to other lambdoid Stx phages is important to understanding the factors controlling this phenomenon and determining whether they occur in other Stx phages.
Results
The genome of the Stx2 encoding phage, Ф24B was sequenced and annotated. The genomic organisation and general features are similar to other sequenced Stx bacteriophages induced from Enterohaemorrhagic Escherichia coli (EHEC), however Ф24B possesses significant regions of heterogeneity, with implications for phage biology and behaviour. The Ф24B genome was compared to other sequenced Stx phages and the archetypal lambdoid phage, lambda, using the Circos genome comparison tool and a PCR-based multi-loci comparison system.
Conclusions
The data support the hypothesis that Stx phages are mosaic, and recombination events between the host, phages and their remnants within the same infected bacterial cell will continue to drive the evolution of Stx phage variants and the subsequent dissemination of shigatoxigenic potential.
Similar content being viewed by others

Background
Shiga toxin encoding bacteriophages (Stx phages) are responsible for converting the pathogenic profiles of their bacterial hosts. Enterohaemorrhagic Escherichia coli (EHEC), a subset of the Shigatoxigenic E. coli (STEC), differentiated by their ability to produce attachment and effacement lesions, emerged as a serious food borne threat to humans in the 1980s [1–3]. The emergence of this group of organisms was due to an Stx phage infection of a mildly pathogenic progenitor strain [4]. The severe disease (bloody diarrhoea and haemorrhagic colitis) and disease sequelae (haemolytic uraemic syndrome [HUS] and thrombotic thrombocytopenic purpura [TTP]) caused by EHEC are all linked to the activity of the Shiga toxin (Stx) [5], the expression of which is genetically coordinated by the lytic replication cycle of Stx phage [6]. Although the global incidence of EHEC infection is low, severe disease and death occurs in an unacceptably high proportion of infected individuals [7]: 10% and 3–5%, respectively [8].
Stx phages are lambdoid bacteriophages, sharing the distinct genome organisation of the archetypal bacteriophage lambda (λ) [5]. They possess two replication strategies: lysogenic, where the phage genome directs its integration into the bacterial host genome as a prophage; or lytic, where viral progeny are assembled intracellularly and released by lysis of the host cell through the action of phage encoded lysozyme, holin and pinholin proteins [1, 9, 10]. Production of Stx in the lysogen is linked to the latter, and the release of Stx from the lysogen predominantly coincides with induction of the lytic cycle and bacterial host cell lysis [6].
Bacterial genome sequencing projects have highlighted the impact that temperate phages have upon bacterial evolution, and those that impact directly on the pathogenicity of the host bacterium are known as converting phage. In addition to stx genes carried by Stx phages and expressed by E. coli, other examples of converting phage include the CTX phage encoding the cholera toxin genes expressed by Vibrio cholerae[11] and lom and bor of bacteriophage lambda, which affect E. coli adherence to human buccal epithelial cells [12] and sensitivity to serum killing [13], respectively. It can be postulated that the maintenance of converting phage in a lysogen is due to positive selection pressure for prophage carriage by the host cell in an animal host.
The bacteriophage vB_EcoP-24B[14], carrying the Shiga toxin 2 variant (Stx2) [5] (hereafter referred to as φ24B) has been well characterised [15–21] since its initial purification following induction from a clinical isolate of E. coli O157:H7 [22]. φ24B infects rough and smooth strains of E. coli[18] and can adsorb to many members of the Enterobacteriaceae, including Salmonella spp [18]. The adsorption target for this phage is an essential outer membrane protein, BamA, which is involved in the biogenesis of the Gram negative bacterial outer membrane and is not only highly conserved across members of the Enterobacteriaceae, but also conserved to some degree in all Gram negative bacteria [20]. Using a Stx phage multi loci gene typing system [21], it was demonstrated that >70% of Stx phages share a gene responsible for the short-tailed phage morphotype that enables adsorption to BamA [20]. φ24B also has the ability to multiply infect a single host cell and integrate into different sites across the E.coli chromosome [16, 17, 22], a behaviour which departs from the lambda phage immunity dogma [15]. This could act to not only increase the pathogenic profile of the host with each subsequent infection [23], but also enable recombination events between resident inducible and cryptic prophages, promoting the production and release of novel recombinant phage mosaics.
The objectives of this study were to sequence the genome of φ24B and apply comparative genomic analyses to highlight important genetic similarities and differences across the Stx phages sequenced to date. The ultimate aim is to identify potential effectors controlling the biology of these phages and the expression of genes that provide a selective advantage to either the bacterial lysogen or to the phages themselves.
Results and discussion
Genome annotation
Phage genes are usually small in size (< 1 kb), and very few of them have been subjected to detailed biochemical/functional characterisation, which makes the definitive annotation of phage genomes challenging. Notwithstanding the difficulties inherent in the production of informative phage genome annotation, the sequencing and subsequent annotation of the φ24B genome is reported here [HM208303]. Its genomic organisation confirms that φ24B is a lambdoid phage sharing similar overall genetic context with bacteriophage lambda (Figure 1). Annotation of the 57,677 bp genome revealed 88 putative coding regions (CDS, including the stx 2 AB genes of which the B gene is not annotated due to allelic replacement with the chloramphenicol resistance gene from pLysS [Novagen]), comprised of 26 CDS (30%) that shared a high level of sequence similarity with those of known function in other lambdoid phages (with or without stx genes); three CDS (vb_24B 2c, 4c and 25c), which have never been identified previously; eleven CDS sharing some, but not complete, homology to those genes with poorly defined roles in lambdoid phage biology; and 48 CDS encoding proteins of unknown function (55%), but are found in association with other lambdoid phages ( Additional file 1). A comparison of the number of genes encoding proteins of undetermined function in Stx phages and the number of hypothetical proteins encoded by sequenced E. coli isolates (Figure 2), demonstrates that Stx phages carry a greater percentage of hypothetical genes than their E. coli hosts, 55% verses 24%, respectively (Figure 2), especially remarkable considering the size differential between the bacteriophage and bacterial genomes, but not an uncommon occurrence in bacteriophage genomes [24]. An analysis of the annotated Ф24B genome with CGView [25, 26] (Figure 1) shows that hypothetical genes are particularly common in the late gene region of the phage; downstream of the antiterminator Q, 44 φ24B genes were annotated (both strands) of which 32 (73%) are designated as hypothetical. Because of their location in the late gene region, their expression is likely to be linked to prophage induction/phage replication unless they are morons (horizontally acquired genes with no function for the phage, but usually beneficial to the bacterial host), uncoupled from the standard regulatory networks [27]. Expression analyses of these 32 genes is necessary to determine if they have been carried along via in situ recombination events without impacting the bacterial host or phage replication machinery or if they have been retained in the genome under their own expression control (or linked to other regulatory networks) because they benefit the bacterial host or phage replication.

CGView-derived schematic of the Ф24 B genome; the concentric rings include the annotation, location and direction of expression. Genes that are detailed in the centre of the genome and suffixed with a ‘c’ are expressed from the complimentary strand. The internal concentric rings indicate +/- GC skew and GC content.

Percentage levels of unknown/hypothetical genes with no inferred function from all sequenced and annotated E. coli strains and Stx phages available on Genbank. The open box encompasses the current levels of annotated genes with no inferred function in Stx phage genomes.
Two unexpectedly large genes were identified in the Ф24B genome sequence. The first of these large genes, vb_24B 48, is predicted to encode a protein of 2,808 aa and is located close to the right end of the genome (Figure 1). This gene is also carried by other Stx phages including 933W, VT2-Sa, Stx2 II, Stx2 converting bacteriophage 86 and Min27. Gene vb_24B 48 homologues have also been identified within bacterial genomes carrying non-Stx prophages, e.g. Salmonella enterica subsp. enterica serovar Kentucky isolate (ZP_0258689) encodes a gene sharing 1128 of the 1611 amino acid residues. The predicted protein of vb_24B 48 has no easily assignable function, but does possess a partial COG1483 domain (associated with the AAA + superfamily of ATPases by general function prediction) between residues 345 and 1176; SignalP analysis [28] indicates that the first 15 nucleotides might function as a leader peptide. The protein encoded by vb_24B 48 has no homology with any protein subjected to conventional functional analysis, but TMPred [29, 30] predicts that the protein possesses membrane-spanning domains. This protein has many of the characteristics of the giant genes that typically encode surface proteins involved in bacterial fitness [31], and this could be relevant to its conservation among Stx phages. The second large gene, P (2906 bp), encodes the polymerase for Ф24B replication. PФ24B possesses a number of well characterised and conserved domains, including an intact TOPRIM_primase domain (cd01029) at the amino terminus and an intact P loop NTPase superfamily domain (cl09099) at the carboxyl terminus, specifically harbouring the GP4d_helicase domain (cd01122). An orthologue of PФ24B has been found in association with a Shigella flexneri prophage (YP_690085.1, sharing 955 of the 968 amino acid residues), and in Stx phage Min27 (YP_001648921.1) with an amino acid identity of 87%. PФ24B carries an intein [32, 33], interrupting amino acid residues 372–702, and includes an intact HintN domain (cl12032), specifically of the smart00306 superfamily. Comparison of the CDS sequence excluding the intein shows that PФ24B shares significant identity with a number of prophage or bacteriophage encoded proteins including ZP_0795005.1 and ZP_04535347.1 associated with an unclassified member of the Enterobacteriaceae (NZ_ADCU00000000), and an unidentified Escherichia isolate (NZ_DS999462.1), respectively. It is very likely that the activation of the intein will play a role in the post-translational regulation of the replication protein, but this, as well as the basic function of the intein, has yet to be experimentally determined.
φ24B also harbours the two accessory genes, lom and bor[5], that in bacteriophage lambda are not involved in phage replication, but do affect the fitness of lambda lysogens in mammalian hosts [13, 34] and are expected to play similar fitness roles in this Stx phage and other Stx phages that carry these genes.
Genome comparisons
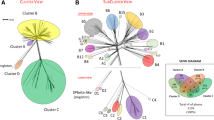
Ф24B was compared to eleven previously sequenced Stx phages [35–43] (though Stx2 bacteriophage 86 ([AB255436] is unpublished) and bacteriophage lambda [39]. The analysis presented in Figure 3 highlights the mosaic nature of these lambdoid phages. The most similar Stx phages to Ф24B are Min27 [36], 933W [41], VT2-Sakai [42], and the Stx2 converting phages 1 and 2 [35], which like Ф24B all possess a Podoviridae-like morphology. These phages represent a global collection of Stx phages associated with incidents of human STEC infection from around the world e.g. 933W (US), Sakai and all Stx2 converting phages I, II and 86 (Japan), Stx 2 converting phage 1717 (Canada), Ф24B (UK) and Min27 (China). These phages all share regions of homology with one another, but the degree of shared identity differs between phages, and no two phage are identical.

Multi-genome comparison of all sequenced Stx phages, the archetypal lambdoid phage, Lambda and Ф24 B . A. Circos map depicting the MuMer alignment results with respect to Stx2 phage Ф24B. Each coloured segment represents a phage genome with the numbers on the external surface indicating genome size in kb. Inside the genome ring are hatch marks indicating gene locations and their respective coding strands. The inner circle is composed of coloured blocks that are indicative of gene conservation with Ф24B. The coloured swept arcs indicate sequence conservation and orientation of those sequences with respect to Ф24B. B. A multi-loci comparison [21]. Loci corresponding to the genome annotation that have been marked are loci that have been used in previous multi-loci typing of Stx bacteriophages or are defined in Additional file 1: Table S 1.
There is evidence that most circulating Stx phages are short-tailed Podoviridae[5, 20, 21, 44, 45], which have evolved an almost perfect infection strategy utilising an essential, highly conserved, outer membrane protein BamA (previously YaeT) for host cell recognition and adsorption [20]. This essential adsorption target, the fact that many outbreak strains carry more than one Stx phage [46, 47], and the capacity of at least some Stx phages to multiply infect a single host cell [15–17, 22, 48] is likely to foster many opportunities to drive phage evolution through in situ recombination events. Thus the similarities in genome content across the short-tailed phages depicted in Figure 3, excluding lambda and Phi 27 that lie outside this group, may be a consequence of such recombination [49].
Genomic comparison has also shown that although many of the genes carried by Stx phages encode hypothetical proteins, there are recognisable accessory genes with activities that have been characterised in other systems, e.g. exo gam bet lar lom bor and stk. The genes exo, gam and bet are the three components of the lambda-encoded Red recombinase system [50]. The products of these genes increase DNA recombination rates, which is likely to drive the creation of novel phages and extend bacterial host ranges through in situ recombination events between resident inducible and cryptic prophages, as well as infecting phages in the bacterial lysogen [5]. The gene lar encodes a protein involved in the alleviation of restriction systems [51], which are often used by bacteria as a primary defense against phage infection [52]. The genes lom and bor encode products that enhance the lysogen’s ability to colonise its host [13, 34], and stk encodes a kinase with an as yet unidentified impact on the lysogen or the lysogen’s host [53], but it is clear that stk expression is controlled by the pRM promoter, and its expression occurs only under conditions of stable lysogeny [54].
The genes associated with the genetic switch, controlling the behaviour of these phages and their decision to enter the lysogenic or lytic replicative cycles (e.g. cI, Q and N), are present across all lambdoid phages, though distinct sequence variants are known (Figure 3). A PCR-based multilocus characterisation system developed for Stx phages [21] was applied to the 11 sequenced Stx phages and lambda (Figure 3B). The integrase gene of Ф24B[16, 17] is also carried by the Stx2 converting phages 86 and 1717. All three phages possess the int genes in a genomic orientation opposite to the lambda phage integrase gene. The Ф24B-like integrase gene is under the control of its own promoter region [55] in all three phages from where it is likely to drive high frequency superinfection events [17]. The Ф24BcIII gene is not present in P27, but in the other phages it is well conserved sharing at least 99% aa identity. The antiterminator, N, involved in early gene expression, is present in one of three forms in all but phage P27. N1 [21] is present in Ф24B, 933W, Stx2 converting phage I, Min27 and BP 4795, all sharing at least 98% identity, and N2 [21] is carried by VT2Sa, Stx2 phage 1717 and YYZ-2008, whilst Lambda possesses a third variant ( Additional file 1: Table S1).; the three variants can share as little as 22% sequence identity. The cI gene product, the regulator controlling maintenance of lysogeny through repression of the lytic life cycle, was identified in five variant forms. The repressor of Stx phages 933W, Min27, Stx2 converting I and Stx2 converting phage 86 all possess cI 1b, while BP-4795 possesses cI 1a , which shares 69% overall identify with the cI1a protein and 100% identity at the carboxy terminal half. Sequence and structure/function predictions mean that the altered amino terminus is likely to have different DNA binding properties, whilst retaining similar dimerization properties that are key to its function [56]. The cI 2c genes from Vt2-Sa, Stx2 phage II, YYZ-2008 and Stx2 phage 1717 all share sequence identity across the entire coding region of the cI gene, though they are currently annotated with different amino termini. The VT2-Sa cI gene amplifies with the cI 2c primers, but a single nucleotide polymorphism has introduced a stop codon and thus ablates 60 amino acids from the amino terminus, probably destroying the ability of this repressor protein to bind DNA; this may, at least partly, explain the non-inducible nature of this prophage [57]. The archetypal Lambda repressor (CI2a) shares 100% identity at its carboxy terminus with the CI2c variants, but its amino terminal end is unique, and again implies that it binds DNA differently from the CI2c variants. The Stx2 converting phage I possesses the cI 7 variant ( Additional file 1: Table S1) not previously included in the Stx phage multilocus PCR typing system [21]. Orthologues of the cro gene product (Cro3) are carried by Stx phages 933W, Stx2 converting phage 86, Ф24B and Min27 and are all identical at the aa level. The cro gene variant (cro 4 ) is carried by Stx2 converting phages 1717 and II as well as VT2-Sa, again sharing 100% amino acid identity. Lambda phage encodes Cro1; BP4795, Cro9; YYZ2008, Cro10; Stx2 converting phage I, Cro11 and P27, Cro12. All the diversity seen across the cI variants and the lack of association of specific cI genes with specific cro genes (Figure 3B) has been predicted [58], providing evidence of repressor/operator coevolution. This coevolution has been predicted to drive superinfection immunity groups and thus effect the production of new and novel Stx phage mosaics [5]. Only the CII from Min27 is completely identical to that of Ф24B; all the other phages in the Circos comparison, apart from P27 and Lambda, have CII proteins that are approximately 86% identical at the protein level. Lambda CII has the lowest sequence identity at 36% and no orthologue was identified in P27.
Only Stx phage Min27 carries O and P genes (O2 P2; Additional file 1: Table S1) like those carried by Ф24B (99 and 98% identity, respectively). Across all of the phages, there were five distinct DNA replication systems encoded, with little homology shared between each system. O1/P1 is carried by Lambda phage, 933W, Stx2 converting phage I and BP-4795; O3/P3 is carried by Stx2 converting phage II, VT2-Sa and Stx2 converting phage 86, O4/P4 is carried by P27 and O5/P5 is carried by Stx2 converting phage 1717 (Figure 3B). These two-protein systems would therefore be a suitable additional diversity marker for phage characterisation (Additional file 1: Table S1). The lytic induction enhancer, Ant, [55] can also be identified in genomic context within the genomes of Min27 (97%), VT2Sa and Stx2 converting phage II (78%) and Stx2 converting phage 1717 (73%) (Figure 3A). Downstream of Ant is a gene encoding a protein of similar predicted conformation, Roi, which shares its 125 amino-terminal amino acid sequence (242 a.a. in total) with Roi from bacteriophage HK022 [59]. In bacteriophage HK022, Roi has been implicated in phage lytic growth [59]. RoiФ24B is identical at the protein level to RoiMin27, and possesses 99% sequence identity to the Roi genes of five of the other Stx phages. Roi encoded by genes carried by Stx2 converting phage II and VT2Sa, and Stx converting bacteriophage 86 are still distinctly similar but share lower identity to RoiФ24B (89 and 83%, respectively); in all cases the genomic context of Roi in these Stx phages is preserved. The protein product of the antiterminator gene Q is widely conserved (≥98% identity) throughout the Stx phages, as it is in all lambdoid phages [60]. The well conserved short tail of Ф24B is widespread across Stx phages [21] due to its outer membrane protein adsorption target that is itself highly conserved and an essential gene in the bacterial host [20]. Examination of the distribution and similarity of the gene encoding this short tail structure across the sequenced Stx phages, 933W, VT2Sa, Min27, Stx2 converting phage 2 and Ф24B reveals 99% sequence identity at the protein level. This 1% difference is simply due to different start codons. Stx2 converting phage 1 possesses a tail gene with 95% identity to Ф24B.
A Jaccard dissimilarity dendrogram (Figure 3B) was created from data on the presence or absence of the gene variants associated with each sequenced genome. The dendrogram illustrates the high level of genetic diversity that exists amongst these 11 Stx phages, with no two phage possessing an identical genetic profile. This further demonstrates the genetic heterogeneity of Stx phages previously revealed by PCR multilocus typing of phage pools induced from STEC strains (55).
The most challenging question in phage genomics is: What is the function of the uncharacterised genes that dominate bacteriophage genomes? Phage genomes are normally small and compact, and it is likely that many of the genes of unknown function have been maintained in this dynamic pool by positive selection pressure. Most Stx phages have larger genomes than bacteriophage lambda, so carry more genes that are not required for core lambdoid phage replication and life cycle control. The suggestion that these accessory genes have roles in the fitness of either the Stx phages themselves or their bacterial hosts can be made with some confidence.
Conclusions
Over the last 10 years, the phage research community has begun to use genomic analyses to compare double stranded DNA phages, most extensively with respect to the comparative genomics of mycobacteriophages or their lysogens [61–69]. Bacteriophages are significant drivers of bacterial evolution because of their ability to disseminate DNA across their host range, either as converting phages [70] or through both generalised (59) and specialised (25) transduction. By identifying genetic variation in groups of phage which impact upon the phenotypic profiles of their hosts, it may be possible to infer biological roles for the numerous hypothetical proteins identified in translated bacteriophage genome sequences.
In this full genomic comparison of eleven Stx phages we have demonstrated that no two sequenced Stx phage are identical. All of the lambdoid phages are mosaics, sharing genomic loci and genomic synteny, but to varying degrees. The short-tailed Stx phages possess more genomic relatedness, which may be driven by their shared host range (due to the adsorption target, BamA) enabling appreciable levels of genomic recombination, facilitating efficient recombination of and selection for genetic material carried by these phages. The phage backbone of P27 is very different from the other Stx phages and may be the result of a productive recombination even between a non-lambdoid and a lambdoid phage, as many key regulatory lambdoid phage elements cannot be identified within the P27 genome. However, the Shiga toxin genes remain linked to the Q gene. It has been reported before that lambdoid phages appear to possess most genetic morons within the late gene region [27], and the Stx phages hold true to this observation. The conserved nature of many of these morons, which are likely to confer some as yet unidentified property to their host cell, indicate that Stx phages are likely to contribute more to their pathogenic bacterial host than toxin production. Understanding these factors is likely to be important to understanding the evolution of EHEC and other Shiga toxin producing enteric pathogens.
Genomic approaches to phage biology provide the means to examine the growing number of novel bacteriophages isolated directly from different environments, induced from their bacterial hosts or identified as prophages in sequenced bacterial genomes. Deep pyrosequencing technologies, enabling metaviral analyses of environmental samples, are further driving our understanding and appreciation of bacteriophage genomics and the bacteriophage pan-genome [71, 72]. Assigning definitive or putative functions to the hypothetical proteins that are the expressed products of the majority of bacteriophage genes remains the main barrier to significant progress in unravelling bacteriophage biology.
Methods
Bacterial strains and bacteriophages
The E. coli C derivative strain WG5rif+ and the E. coli K12 strain DM1187 have been used to isolate and propagate a number of Stx phages previously [15, 16, 18, 21, 22, 73]. Unless stated otherwise, these bacterial strains were grown in Luria-Bertani broth (VWR) or on plates prepared by addition of 1.5% (w/v) agar (Difco). The engineered variant of Ф24B sequenced in this study, Ф24B::Cat [22], possesses a stx operon that has been replaced with the cat gene, which confers chloramphenicol resistance upon its lysogen.
VB_ECOP-24B::Cat (Ф24B::Cat) DNA extraction for genome sequencing
Agar plates with semi-confluent plaques of Ф24B::Cat were flooded with 3 ml of SM buffer (50 mM Tris Cl [pH 7.5], 0.1 M NaCl, 10 mM MgSO4,) [74] and gently agitated overnight at 4°C. The SM buffer was harvested and the plate flooded again with SM buffer. The top agar containing the plaques and the second volume of SM buffer were then scraped from the agar plates and added to the former sample. This mixture was vortexed, and the top agar and bacterial debris pelleted by centrifugation (10,000 g, 10 min). Chloroform (30 μl 10 mL-1) was added to the recovered supernatant to inactivate any remaining bacterial cells. Contaminating bacterial DNA and RNA were removed by the addition of DNAse (Ambion; 5 μg mL-1) and RNAse (1 μg mL-1), and the mixtures were incubated at 37°C for 1 hr. The phages present were precipitated in the presence of 33% PEG 8000 (Sigma) on ice for 30 min and recovered by centrifugation at 10,000 g for 10 min. The resulting phage pellet was suspended in 500 μl of SM per 30 ml original vol followed by a further DNAse and RNAse digestion. The viral nucleic acid was purified following two extractions with an equal vol 25:24:1 phenol:chloroform:isoamyl alcohol and centrifugation (14,500 g, 30 min). The DNA present was precipitated by the addition of 0.6 vol isopropanol. The DNA was harvested by centrifugation (14,500 g for 30 min), washed with 70% ethanol and allowed to air dry. It was then suspended in 100 μl of distilled H2O [60].
Ф24B::Cat Sequencing and annotation
The Ф24B::Cat phage genome was sequenced at the Welcome Trust Sanger Institute. The phage DNA was randomly sheared by sonication and a library produced by cloning fragments into the plasmid pUC19 (New England Biolabs). The phage genome was sequenced to provide 10x coverage using the ABI3730 sequencer (Applied Biosystems). Assembly of the sequence was accomplished using Phrap, and contiguous sequence was assembled using GAP4. The phage DNA predicted coding genes were identified using ORPHEUS28 and GLIMMER29 and these predictions were combined and annotated in Artemis [75] by comparison against the non redundant database using BLASTN and TBLASTX [76]. Putative coding sequences were added to the annotation if they contained both start and stop codons and a probable ribosome binding site.
Genome comparison
The accession numbers for the Stx phages used for the genome comparison were: Ф24B::Cat (HM208303), 933W (AF125520), P27 (AJ298298), Min27 (EU311208), Stx2 Converting phage I (AP004402), Stx2 Converting phage II (AP005154), Stx2 Converting phage 86 (AB255436), Stx2 Converting phage 1717 (FJ188381), VT2-Sakai (AP000363) YYZ 2008 (FJ184280), BP-4795 (AJ556162) and non-Stx encoding bacteriophage Lambda (J02459). Comparative genome analysis was performed using MUMmer version 3 [77] and visualized using CIRCOs [78]. Coordinates were generated using NUCmer [77] with the parameters breaklen, maxgap, mincluster, and minmatch set to 200, 90, 65 and 20, respectively.
R-based loci comparisons
The presence of bands from each individual amplification reaction, using primer pairs specific for variant loci [21], was used as the data for construction of a binary similarity matrix. Computation script was written using R version 2.11.1, to enable visualisation of the variant of each genetic locus present.
References
Pang T, Savva C, Fleming K, Struck D, Young R: Structure of the lethal phage pinhole. Proc Natl Acad Sci U S A. 2009, 106: 18966-18971. 10.1073/pnas.0907941106.
Riley LW, Remis RS, Helgerson SD, McGee HB, Wells JG, Davis BR, Hebert RJ, Olcott ES, Johnson LM, Hargrett NT, et al: Hemorrhagic colitis associated with a rare Escherichia coli serotype. N Engl J Med. 1983, 308: 681-685. 10.1056/NEJM198303243081203.
Strockbine NA, Marques LR, Newland JW, Smith HW, Holmes RK, O'Brien AD, et al: Two toxin-converting phages from Escherichia coli O157:H7 strain 933 encode antigenically distinct toxins with similar biologic activities. Infect Immun. 1986, 53: 135-140.
Whittam TS, Wachsmuth IK, Wilson RA: Genetic evidence of clonal descent ofEscherichia coliO157:H7 associated with hemorrhagic colitis and hemolytic uremic syndrome. J Infect Dis. 1988, 157: 1124-1133. 10.1093/infdis/157.6.1124.
Allison H: Stx-phages: drivers and mediators of the evolution of STEC and STEC-like pathogens. Future Microbiol. 2007, 2: 165-174. 10.2217/17460913.2.2.165.
Wagner PL, Acheson DWK, Waldor MK: Isogenic lysogens of diverse Shiga toxin 2-encoding bacteriophages produce markedly different amounts of Shiga toxin. Infect Immun. 1999, 67: 6710-6714.
Watanabe Y, Ozasa K, Mermin JH, Griffin PM, Masuda K, Imashuku S, Sawada T: Factory outbreak ofEscherichia coliO157:H7 infection in Japan. Emerg Infect Dis. 1999, 5: 424-428. 10.3201/eid0503.990313.
Enterohaemorrhagic E. coli (EHEC) WHO Media Centre: www.who.int/mediacentre/factsheets/fs125/en
Berry J, Summer E, Struck D, Young R: The final step in the phage infection cycle: the Rz and Rz1 lysis proteins link the inner and outer membranes. Mol Microbiol. 2008, 70: 341-351. 10.1111/j.1365-2958.2008.06408.x.
Savva C, Dewey J, Deaton J, White R, Struck D, Holzenburg A, Young R: The holin of bacteriophage lambda forms rings with large diameter. Mol Microbiol. 2008, 69: 784-793. 10.1111/j.1365-2958.2008.06298.x.
Davis B, Moyer K, Boyd E, Waldor M: CTX prophages in classical biotype Vibrio cholerae: functional phage genes but dysfunctional phage genomes. J Bacteriol. 2000, 182: 6992-6998. 10.1128/JB.182.24.6992-6998.2000.
Vica Pacheco S, Garcia Gonzalez O, Paniagua Contreras GL: Thelomgene of bacteriophage lambda is involved inEscherichia coliK12 adhesion to human buccal epithelial cells. FEMS Microbiol Lett. 1997, 156: 129-132. 10.1016/S0378-1097(97)00415-1.
Barondess JJ, Beckwith J: borgene of phage lambda, involved in serum resistance, encodes a widely conserved outer membrane lipoprotein. J Bacteriol. 1995, 177: 1247-1253.
Kropinski AM, Prangishvili D, Lavigne R: Position paper: the creation of a rational scheme for the nomenclature of viruses of Bacteria and Archaea. Environ Microbiol. 2009, 11: 2775-2777. 10.1111/j.1462-2920.2009.01970.x.
Fogg PC, Allison HE, Saunders JR, McCarthy AJ: Bacteriophage lambda: a paradigm revisited. J Virol. 2010, 84: 6876-6879. 10.1128/JVI.02177-09.
Fogg PC, Gossage SM, Smith DL, Saunders JR, McCarthy AJ, Allison HE: Identification of multiple integration sites for Stx-phage Phi24B in theEscherichia coligenome, description of a novel integrase and evidence for a functional anti-repressor. Microbiology. 2007, 153: 4098-4110. 10.1099/mic.0.2007/011205-0.
Fogg PC, Rigden DJ, Saunders JR, McCarthy AJ, Allison HE: Characterization of the relationship between integrase, excisionase and antirepressor activities associated with a superinfecting Shiga toxin encoding bacteriophage. Nucl Acids Res. 2011, 39: 2116-2129. 10.1093/nar/gkq923.
James C, Stanley K, Allison H, Flint H, Stewart C, Sharp R, Saunders J, McCarthy A: Lytic and lysogenic infection of diverseEscherichia coliand Shigella strains with a verocytotoxigenic bacteriophage. Appl Environ Microbiol. 2001, 67: 4335-4337. 10.1128/AEM.67.9.4335-4337.2001.
Johannessen GS, James CE, Allison HE, Smith DL, Saunders JR, McCarthy AJ: Survival of a Shiga toxin-encoding bacteriophage in a compost model. FEMS Microbiol Lett. 2005, 245: 369-375. 10.1016/j.femsle.2005.03.031.
Smith DL, James CE, Sergeant MJ, Yaxian Y, Saunders JR, McCarthy AJ, Allison HE: Short-tailed Stx phages exploit the conserved YaeT protein to disseminate Shiga toxin genes among enterobacteria. J Bacteriol. 2007, 189: 7223-7233. 10.1128/JB.00824-07.
Smith DL, Wareing BM, Fogg PC, Riley LM, Spencer M, Cox MJ, Saunders JR, McCarthy AJ, Allison HE: Multilocus characterization scheme for Shiga toxin-encoding bacteriophages. Appl Environ Microbiol. 2007, 73: 8032-8040. 10.1128/AEM.01278-07.
Allison HE, Sergeant MJ, James CE, Saunders JR, Smith DL, Sharp RJ, Marks TS, McCarthy AJ: Immunity profiles of wild-type and recombinant shiga-like toxin-encoding bacteriophages and characterization of novel double lysogens. Infect Immun. 2003, 71: 3409-3418. 10.1128/IAI.71.6.3409-3418.2003.
Fogg PC, Saunders JR, McCarthy AJ, Allison HE: Cumulative effect of prophage burden on Shiga toxin production inEscherichia coli. Microbiology. 2011, 158 (Pt 2): 488-497.
Mahadevan P, King JF, Seto D: CGUG: in silico proteome and genome parsing tool for the determination of "core" and unique genes in the analysis of genomes up to ca. 1.9 Mb. BMC Res Notes. 2009, 2: 168-176. 10.1186/1756-0500-2-168.
Grant JR, Stothard P: The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36: W181-184. 10.1093/nar/gkn179.
Stothard P, Wishart DS: Circular genome visualization and exploration using CGView. Bioinformatics. 2005, 21: 537-539. 10.1093/bioinformatics/bti054.
Hendrix RW, Lawrence JG, Hatfull GF, Casjens S: The origins and ongoing evolution of viruses. Trends Microbiol. 2000, 8: 504-508. 10.1016/S0966-842X(00)01863-1.
Emanuelsson O, Brunak S, von Heijne G, Nielsen H: Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007, 2: 953-971. 10.1038/nprot.2007.131.
Hofmann K, Stoffel W: TMbase - A database of membrane spanning proteins segments. Biol Chem Hoppe Seyler. 1993, 374: 166-
Lao DM, Okuno T, Shimizu T: Evaluating transmembrane topology prediction methods for the effect of signal peptide in topology prediction. In Silico Biol. 2002, 2: 485-494.
Reva O, Tummler B: Think big–giant genes in bacteria. Environ Microbiol. 2008, 10: 768-777. 10.1111/j.1462-2920.2007.01500.x.
Colston MJ, Davis EO: The ins and outs of protein splicing elements. Mol Microbiol. 1994, 12: 359-363. 10.1111/j.1365-2958.1994.tb01025.x.
Perler FB, Davis EO, Dean GE, Gimble FS, Jack WE, Neff N, Noren CJ, Thorner J, Belfort M: Protein splicing elements: inteins and exteins–a definition of terms and recommended nomenclature. Nucleic Acids Res. 1994, 22: 1125-1127. 10.1093/nar/22.7.1125.
Barondess JJ, Beckwith J: A bacterial virulence determinant encoded by the lysogenic coliphage λ. Nature. 1990, 346: 871-874. 10.1038/346871a0.
Zhang Y, Laing C, Zhang Z, Hallewell J, You C, Ziebell K, Johnson RP, Kropinski AM, Thomas JE, Karmali M, Gannon VP: Lineage and host source are both correlated with levels of Shiga toxin 2 production by Escherichia coli O157:H7 strains. Appl Environ Microbiol. 2010, 76: 474-482. 10.1128/AEM.01288-09.
Su LK, Lu CP, Wang Y, Cao DM, Sun JH, Yan YX: Lysogenic infection of a Shiga toxin 2-converting bacteriophage changes host gene expression, enhances host acid resistance and motility. Mol Biol. 2010, 44: 60-73.
Sato T, Shimizu T, Watarai M, Kobayashi M, Kano S, Hamabata T, Takeda Y, Yamasaki S: Genome analysis of a novel Shiga toxin 1 (Stx1)-converting phage which is closely related to Stx2-converting phages but not to other Stx1-converting phages. J Bacteriol. 2003, 185: 3966-3971. 10.1128/JB.185.13.3966-3971.2003.
Sato T, Shimizu T, Watarai M, Kobayashi M, Kano S, Hamabata T, Takeda Y, Yamasaki S: Distinctiveness of the genomic sequence of Shiga toxin 2-converting phage isolated from Escherichia coli O157:H7 Okayama strain as compared to other Shiga toxin 2-converting phages. Gene. 2003, 309: 35-48. 10.1016/S0378-1119(03)00487-6.
Sanger F, Coulson AR, Hong GF, Hill DF, Petersen GB: Nucleotide sequence of bacteriophage lambda DNA. J Mol Biol. 1982, 162: 729-773. 10.1016/0022-2836(82)90546-0.
Recktenwald J, Schmidt H: The nucleotide sequence of Shiga toxin (Stx) 2e-encoding phage phiP27 is not related to other Stx phage genomes, but the modular genetic structure is conserved. Infect Immun. 2002, 70: 1896-1908. 10.1128/IAI.70.4.1896-1908.2002.
Plunkett G, Rose DJ, Durfee TJ, Blattner FR: Sequence of Shiga toxin 2 phage 933W fromEscherichia coliO157:H7: Shiga toxin as a phage late-gene product. J Bacteriol. 1999, 181: 1767-1778.
Miyamoto H, Nakai W, Yajima N, Fujibayashi A, Higuchi T, Sato K, Matsushiro A: Sequence analysis of Stx2-converting phage VT2-Sa shows a great divergence in early regulation and replication regions. DNA Res. 1999, 6: 235-240. 10.1093/dnares/6.4.235.
Creuzburg K, Recktenwald J, Kuhle V, Herold S, Hensel M, Schmidt H: The Shiga toxin 1-converting bacteriophage BP-4795 encodes an NleA-like type III effector protein. J Bacteriol. 2005, 187: 8494-8498. 10.1128/JB.187.24.8494-8498.2005.
Gamage SD, Patton AK, Hanson JF, Weiss AA: Diversity and host range of Shiga toxin-encoding phage. Infect Immun. 2004, 72: 7131-7139. 10.1128/IAI.72.12.7131-7139.2004.
Muniesa M, Blanco JE, De Simon M, Serra-Moreno R, Blanch AR, Jofre J: Diversity of stx2 converting bacteriophages induced from Shiga-toxin-producing Escherichia coli strains isolated from cattle. Microbiology. 2004, 150: 2959-2971. 10.1099/mic.0.27188-0.
Hayashi T, Makino K, Ohnishi M, Kurokawa K, Ishii K, Yokoyama K, Han CG, Ohtsubo E, Nakayama K, Murata T, et al: Complete genome sequence of enterohemorrhagicEscherichia coliO157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001, 8: 11-22. 10.1093/dnares/8.1.11.
Perna NT, Plunkett G: Burland V, Mau B, Glasner JD, Rose DJ, Mayhew GF, Evans PS, Gregor J, Kirkpatrick HA, et al: Genome sequence of enterohaemorrhagicEscherichia coliO157:H7. Nature. 2001, 409: 529-533. 10.1038/35054089.
Serra-Moreno R, Jofre J, Muniesa M: The CI repressors of Shiga toxin-converting prophages are involved in coinfection ofEscherichia colistrains, which causes a down regulation in the production of Shiga toxin 2. J Bacteriol. 2008, 190: 4722-4735. 10.1128/JB.00069-08.
Strauch E, Hammerl JA, Konietzny A, Schneiker-Bekel S, Arnold W, Goesmann A, Puhler A, Beutin L: Bacteriophage 2851 is a prototype phage for dissemination of the Shiga toxin variant gene 2c in Escherichia coli O157:H7. Infect Immun. 2008, 76: 5466-5477. 10.1128/IAI.00875-08.
Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL: An efficient recombination system for chromosome engineering inEscherichia coli. PNAS USA. 2000, 97: 5978-5983. 10.1073/pnas.100127597.
King G, Murray NE: Restriction alleviation and modification enhancement by the Rac prophage ofEscherichia coliK-12. Mol Microbiol. 1995, 16: 769-777. 10.1111/j.1365-2958.1995.tb02438.x.
Labrie SJ, Samson JE, Moineau S: Bacteriophage resistance mechanisms. Nature Rev Microbiol. 2010, 8: 317-327. 10.1038/nrmicro2315.
Tyler JS, Friedman DI: Characterization of a eukaryotic-like tyrosine protein kinase expressed by the Shiga toxin-encoding bacteriophage 933W. J Bacteriol. 2004, 186: 3472-3479. 10.1128/JB.186.11.3472-3479.2004.
Riley LM, Veses-Garcia M, Hillman JD, Handfield M, McCarthy AJ, Allison HE: Identification of genes expressed in cultures of E. coli lysogens carrying the Shiga toxin-encoding prophage Phi24B. BMC Microbiol. 2012, 12: 42-56. 10.1186/1471-2180-12-42.
Fogg PC, Rigden DJ, Saunders JR, McCarthy AJ, Allison HE: Characterization of the relationship between integrase, excisionase and antirepressor activities associated with a superinfecting Shiga toxin encoding bacteriophage. Nucleic Acids Res. 2010.
Hochschild A, Douhan J: Ptashne M: How lambda repressor and lambda Cro distinguish between OR1 and OR3. Cell. 1986, 47: 807-816. 10.1016/0092-8674(86)90523-4.
Makino K, Yokoyama K, Kubota Y, Yutsudo CH, Kimura S, Kurokawa K, Ishii K, Hattori M, Tatsuno I, Abe H, et al: Complete nucleotide sequence of the prophage VT2-Sakai carrying the verotoxin 2 genes of the enterohemorrhagic Escherichia coli O157:H7 derived from the Sakai outbreak. Genes Genet Sys. 1999, 74: 227-239. 10.1266/ggs.74.227.
Berngruber TW, Weissing FJ, Gandon S: Inhibition of superinfection and the evolution of viral latency. J Virol. 2010, 84: 10200-10208. 10.1128/JVI.00865-10.
Clerget M, Boccard F: Phage HK022 Roi protein inhibits phage lytic growth in Escherichia coli integration host factor mutants. J Bacteriol. 1996, 178: 4077-4083.
Rooks DJ, Yan Y, McDonald JE, Woodward MJ, McCarthy AJ, Allison HE: Development and validation of a qPCR-based method for quantifying Shiga toxin-encoding and other lambdoid bacteriophages. Environ Microbiol. 2010, 12: 1194-1204. 10.1111/j.1462-2920.2010.02162.x.
Ford ME, Sarkis GJ, Belanger AE, Hendrix RW, Hatfull GF: Genome structure of mycobacteriophage D29: implications for phage evolution. J Mol Biol. 1998, 279: 143-164. 10.1006/jmbi.1997.1610.
Ford ME, Stenstrom C, Hendrix RW, Hatfull GF: Mycobacteriophage TM4: genome structure and gene expression. Tuber Lung Dis. 1998, 79: 63-73. 10.1054/tuld.1998.0007.
Hatfull GF, Barsom L, Chang L, Donnelly-Wu M, Lee MH, Levin M, Nesbit C, Sarkis GJ: Bacteriophages as tools for vaccine development. Dev Biol Stand. 1994, 82: 43-47.
Hatfull GF, Pedulla ML, Jacobs-Sera D, Cichon PM, Foley A, Ford ME, Gonda RM, Houtz JM, Hryckowian AJ, Kelchner VA, et al: Exploring the mycobacteriophage metaproteome: phage genomics as an educational platform. PLoS Genet. 2006, 2: e92-10.1371/journal.pgen.0020092.
Hatfull GF: Bacteriophage genomics. Curr Opin Microbiol. 2008, 11: 447-453. 10.1016/j.mib.2008.09.004.
Lawrence JG, Hatfull GF, Hendrix RW: Imbroglios of viral taxonomy: genetic exchange and failings of phenetic approaches. J Bacteriol. 2002, 184: 4891-4905. 10.1128/JB.184.17.4891-4905.2002.
Payne K, Sun Q, Sacchettini J, Hatfull GF: Mycobacteriophage lysin B is a novel mycolylarabinogalactan esterase. Mol Microbiol. 2009, 73: 367-381. 10.1111/j.1365-2958.2009.06775.x.
Pedulla ML, Ford ME, Houtz JM, Karthikeyan T, Wadsworth C, Lewis JA, Jacobs-Sera D, Falbo J, Gross J, Pannunzio NR, et al: Origins of highly mosaic mycobacteriophage genomes. Cell. 2003, 113: 171-182. 10.1016/S0092-8674(03)00233-2.
Pitcher RS, Tonkin LM, Daley JM, Palmbos PL, Green AJ, Velting TL, Brzostek A, Korycka-Machala M, Cresawn S, Dziadek J, et al: Mycobacteriophage exploit NHEJ to facilitate genome circularization. Mol Cell. 2006, 23: 743-748. 10.1016/j.molcel.2006.07.009.
Saunders JR, Allison HE, James CE, McCarthy AJ, Sharp R: Phage-mediated transfer of virulence genes. J Chem Tech Biotech. 2001, 76: 662-666. 10.1002/jctb.437.
Rooks D, Smith D, McDonald J, Woodward M, McCarthy A, Allison H: 454-pyrosequencing: a molecular battiscope for freshwater viral ecology. Genes. 2010, 1: 210-226. 10.3390/genes1020210.
Tettelin H, Riley D, Cattuto C, Medini D: Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol. 2008, 11: 472-477. 10.1016/j.mib.2008.09.006.
McDonald JE, Smith DL, Fogg PC, McCarthy AJ, Allison HE: High-throughput method for rapid induction of prophages from lysogens and its application in the study of Shiga Toxin-encoding Escherichia coli strains. Appl Environ Microbiol. 2010, 76: 2360-2365. 10.1128/AEM.02923-09.
Sambrook J, Fritsch EF, Maniatis T: Molecular Cloning: a Laboratory Manual. 1989, Cold Spring Harbour Laboratory, Cold Spring Harbour, NY, 2
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream M, Barrell B: Artemis: sequence visualization and annotation. Bioinformatics. 2000, 16: 944-945. 10.1093/bioinformatics/16.10.944.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL: Versatile and open software for comparing large genomes. Genome Biol. 2004, 5: R12-10.1186/gb-2004-5-2-r12.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones S, Marra M: Circos: an information aesthetic for comparative genomics. Genome Res. 2009, 19: 1639-1645. 10.1101/gr.092759.109.
Acknowledgements
Funding for this work was received from the Biotechnology and Biological Sciences Research Council (BBSRC, UK, grant: BBS/B/05265) and the Department of the Environment, Food and Rural Affairs and the Higher Education Funding Council for England (DEFRA/HEFCE, UK, grant: VT0103/CSA 6602). The authors would also like to extend their sincere gratitude to the Welcome Trust Sanger Sequencing Centre for funding the sequencing of Ф24B::Cat.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors have been involved in the writing of this manuscript and have approved the final version. PCMF prepared the phage genomic DNA and NRT sequenced and assembled the genome. HEA and DLS annotated the genome and DLS and ACD undertook the comparative genome analyses. DJR performed and analysed the multi locus characterisation. AJM and HEA conceived and directed this research and the ensuing manuscript. DLS and DJR are joint first authors and HEA is the corresponding author.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Smith, D.L., Rooks, D.J., Fogg, P.C. et al. Comparative genomics of Shiga toxin encoding bacteriophages. BMC Genomics 13, 311 (2012). https://doi.org/10.1186/1471-2164-13-311
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-13-311