
Abstract
Background
Avian influenza virus (AIV) outbreaks are worldwide threats to both poultry and humans. Our previous study suggested microRNAs (miRNAs) play significant roles in the regulation of host response to AIV infection in layer chickens. The objective of this study was to test the hypothesis if genetic background play essential role in the miRNA regulation of AIV infection in chickens and if miRNAs that were differentially expressed in layer with AIV infection would be modulated the same way in broiler chickens. Furthermore, by integrating with parallel mRNA expression profiling, potential molecular mechanisms of host response to AIV infection can be further exploited.
Results
Total RNA isolated from the lungs of non-infected and low pathogenic H5N3 infected broilers at four days post-infection were used for both miRNA deep sequencing and mRNA microarray analyses. A total of 2.6 M and 3.3 M filtered high quality reads were obtained from infected and non-infected chickens by Solexa GA-I Sequencer, respectively. A total of 271 miRNAs in miRBase 16.0 were identified and one potential novel miRNA was discovered. There were 121 miRNAs differentially expressed at the 5% false discovery rate by Fisher’s exact test. More miRNAs were highly expressed in infected lungs (108) than in non-infected lungs (13), which was opposite to the findings in layer chickens. This result suggested that a different regulatory mechanism of host response to AIV infection mediated by miRNAs might exist in broiler chickens. Analysis using the chicken 44 K Agilent microarray indicated that 508 mRNAs (347 down-regulated) were differentially expressed following AIV infection.
Conclusions
A comprehensive analysis combining both miRNA and targeted mRNA gene expression suggests that gga-miR-34a, 122–1, 122–2, 146a, 155, 206, 1719, 1594, 1599 and 451, and MX1, IL-8, IRF-7, TNFRS19 are strong candidate miRNAs or genes involved in regulating the host response to AIV infection in the lungs of broiler chickens. Further miRNA or gene specific knock-down assay is warranted to elucidate underlying mechanism of AIV infection regulation in the chicken.
Similar content being viewed by others

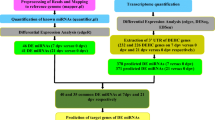
Background
Avian influenza virus (AIV) infection is a world-wide threat to both human and avian species. AIV causes an infection of the respiratory tract of the host, triggering a cascade of innate and adaptive immune responses. Great efforts have been made to develop new intervention strategies to control AIV infections in chickens[1, 2]. However, more effective measures against AIV infection in the chicken are still needed. AI outbreaks in chickens could not only cause dramatic economic losses to the poultry industry, but also threaten human health. Therefore, understanding host response to AIV infection and chicken-virus interaction is not only essential to the poultry industry, but also provides key insights into the prophylactic and therapeutic protection for other influenza hosts including humans.
miRNAs are short, 17–24 nt RNAs, which comprise a large family of regulatory molecules found in almost all multi-cellular organisms[3]. These small RNAs have been demonstrated to have important functions in a variety of biological processes and have been implicated in many diseases including influenza, hepatitis and cancer[4–10]. miRNAs are capable of regulating mammalian immune cell differentiation, the outcome of immune responses to infection, and the development of diseases of immunological origins[11]. There are multiple mechanisms of miRNA-mediated regulation of gene expression including translational repression, disruption of mRNA stability, miRNA-mediated deadenylation and inhibition of polypeptide elongation[12]. Determining how and when miRNA suppress target mRNA gene expression remains one of the greatest challenges in the field.
Through recognition of sequence-complementary target elements, miRNAs can either translationally suppress or catalytically degrade both cellular and viral RNAs[3, 13]. Host miRNAs are able to impinge on viral life cycles, viral tropism, and the pathogenesis of viral diseases[14]. miRNAs can potentially regulate different steps of a virus life cycle and abrogate toxicities of replication-competent viruses[15–18]. For example, human miR-32 represses the replication of the retrovirus primate foamy virus type 1 (PFV-1) through the down-regulation of replication-essential viral proteins encoded by open reading frame 2 (ORF2)[16]. Based on computational prediction, human miR-136 and miR-507 have potential binding sites at the polymerase basic 2 (PB2) and hemmagglutinin (HA) proteins of H5N1 AIV, and those two miRNAs may modulate AIV infection in humans[19].
Next generation sequencing (NGS, deep sequencing) has provided a powerful tool to identify differentially expressed miRNAs especially low abundance ones under conditions of physiological perturbation. We previously used a Solexa Sequencer to identify differentially expressed chicken miRNAs in AIV infected lungs and trachea of layer type birds[20]. Genetics play a significant role in host response to viral infection. We hypothesize that gene expression of host cellular miRNAs following virus infection could be different between different chicken genetic lines. There are two major types of chickens: broilers (meat type chickens) and layers (egg type chickens). In the current study, a deep sequencing approach was employed to identify differentially expressed miRNAs with AIV infection in broilers.
Identification of differentially expressed host miRNAs is just the first step towards understanding miRNA regulation of host-virus interactions, and then underlying mechanisms that how genes targeted by differentially expressed miRNAs mediate host-virus interaction would be desired. Dissection of miRNA modulation of both host and viral mRNA expression will provide insights in the cellular mechanisms of host-virus interaction. A powerful symbiosis between microarrays and NGS technologies has been witnessed[21]. Therefore, global gene expression (mRNA) profiling of host response to AIV infection was conducted to identify potential genes associated with AIV infection using a chicken 44 K Agilent microarray. We integrated predicted target genes information (based on differentially expressed miRNAs) with differentially regulated mRNA affected by AIV infection to understand how miRNAs regulate mRNA gene expression with AIV infection in the chicken.
Results
Virus titers in lungs
Virus replication in lungs was examined using real-time RT-PCR by measuring influenza virus matrix gene from total RNAs at 4 days post inoculation (dpi). Virus titers in the four infected chicken, determined by extrapolation of real-time RT-PCR data, were 1.69, 3.41, 3.81, and 4.52 log10 EID50/ml. Lung samples from all 4 non-infected chickens were negative.
miRNA sequences from small RNA libraries
A total of 2,672,582 and 3,318,307 filtered high quality reads were obtained from infected and non-infected chicken, respectively (Table1). In the library from infected chicken lungs, 2,314,793 of these reads were exact matches and another 357,789 reads were loose matches to known chicken miRNAs. In the library of non-infected chicken lungs, 2,875,366 of these reads were exact matches and another 442,941 reads were loose matches to known chicken miRNAs. All reads with a perfect match to mature miRNA sequences from chicken deposited in miRBase version 16.0 (http://microrna.sanger.ac.uk/)[22–24] with insertions or deletions of 1–4 nucleotides at the 5’ and 3’ ends of miRNAs were considered to represent Dicer-processing products from each of the chicken miRNA precursors[20]. The loose match reads were defined as no more than 4 nt differences comparing to the known chicken miRNA sequences determined as we did previously[20]. The sum of exact and loose match reads was used as the total number of reads for each miRNA.
Of the 499 distinct Gallus gallus (gga) miRNA entries in miRBase[22–24], 272 miRNAs were identified in the current chicken lung small RNA library. Also, one potential novel miRNA was identified (Additional file1: Table S1). Differential expression of this novel miRNA “N1” and chicken miRNA miR-1711 were confirmed by Northern blot analysis (Figure1).

Confirmation of miRNAs. Northern blot analysis was performed to confirm the presence of a novel miRNA (N1) and another known chicken miRNA (gga-miR-1711) in infected and non-infected chicken lungs. U6 probe was used as a control.
miRNA expression profiling analysis
miRNA expression profiles of infected and non-infected chicken lungs were compared. Differentially expressed miRNAs were identified (P < 0.05, Q < 0.05 and fold change > 2) by Fisher’s exact test. Between infected and non-infected lungs, 121 miRNAs were differentially expressed. Of those, 43 miRNAs were unique to infected lung and 8 miRNAs were unique to uninfected lung. Sixty-five miRNAs were more highly expressed in infected lungs, while 5 miRNAs were more highly expressed in non-infected lungs (Table2). With limited biological replicates for the nature of next generation sequencing technology, there is no standard method available for this type of analysis. Two newly developed methods (more conservative): DESeq and edgeR, were implemented in this study. The differentially expressed miRNAs are presented in Tables3 and4, there were 8 and 12 miRNAs differentially expressed (P < 0.05, fold change > 2) by DESeq and edgeR, respectively, and all these miRNAs were identified by the Fisher’s exact test (Figure2). Three out of eight miRNAs by DESeq analysis were also identified by edgeR. All miRNAs identified by DESeq were up-regulated with AIV infection. For the 12 miRNAs identified by the edgeR program, nine miRNAs specifically expressed in the infected chickens and one miRNA in non-infected birds; two miRNAs were up-regulated with AIV infection.

Integration of numbers of differentially expressed miRNAs by different statistical methods. Differentially expressed miRNAs were identified by Fisher exact test, DESeq and edgeR, respectively (P < 0.05, fold change > 2).
TaqMan miRNA assays were used to confirm two differentially expressed miRNAs identified by deep sequencing. There was general consistency between the TaqMan assays and deep sequence analysis of miR-451 and miR-206 in terms of direction of regulation and statistical significance (Figure3). Specifically, there was a 2.05 fold up-regulation (7.25 fold in deep sequencing analysis) in miR-451, and 4.71 fold down regulation (2.86 fold in deep sequencing analysis) in miR-206 with AIV infection in lungs (P < 0.05).

Validation of two differentially expressed miRNAs by TaqMan miRNA assays. Two differentially expressed miRNAs (gga-miR-206 and 451) identified by deep sequencing were confirmed by using TaqMan miRNA assays.* P <0.05.
miRNA target identification and validation
Potential targets of differentially expressed miRNAs were predicted by the miRNA target prediction algorithm miRanda 3.1[25]. The entire list of immune target genes related to AIV infection is listed in Additional file2: Table S2. One hundred and seventy-one immune related genes were predicted to be targets of 35 differentially expressed miRNAs. Each miRNA can target hundreds of genes. We are especially interested in the targeted immune-related genes; some immune related genes each had several predicted miRNA binding sites. For example, IL-17 receptor D (Accession No.: AY278204), has predicted binding sites for seven differentially expressed miRNAs: gga-miR-30b, 34a, 142-5p, 202, 460b-5p, 449b, and 460a. Interestingly, first five miRNAs were up-regulated, while last two miRNAs were down-regulated following AIV infection. This might explain why IL-17 receptor D was not differentially expressed if these differentially expressed miRNA regulate in different direction.
Of particular note, gga-miR-146a is one of differentially expressed miRNAs that was associated with virus infection in both broiler (current study) and layer chickens[20]. Seven potential target genes (Table5) of gga-miR-146a were picked for the validation by a dual luciferase reporter assay. The results are shown in Figure4. The 3’ UTR of five (ARL11, CHMP2B, POU1F1, PDHB and HIF1AN) out of the seven genes targeted by miR-146a showed significant suppression of Renilla luciferase activity in RCAS-miR-146a infected cells relative to those infected with RCAS-SC (P < 0.05). Inhibition of the luciferase activity of significant targets varied between 65–85% amongst target sites.

Validation of miR-146a target genes in the Renilla luciferase reporter system. Seven potential miR-146a target genes predicted by the miRanda algorithm were chosen for validation. For each predicted target gene a luciferase reporter vector was constructed in which the predicted miR-146a binding site was cloned into the 3’ UTR of a Renilla luciferase reporter gene. The Renilla luciferase activities were normalized to Firefly luciferase activities (under the control of an independent promoter). The relative expression of each Renilla luciferase target construct was compared between cells expressing miR-146a and those expressing the scrambled control sequence (SC) using a t-test for statistical significance (p < 0.05). Error bars indicate standard deviation. CASP6, a gene containing no binding site for miR-146a but predicted to contain a miR-143 target site was used as negative control.
Recent evidence indicates that cellular miRNAs can also target viral genes[16]. Potential viral genes targeted by differentially expressed miRNAs were predicted using Vita program[26]. All of the AIV genes were predicted to be targeted by at least one of up or down regulated miRNAs (Table6). For example, the gga-miR-34a, which was only expressed in infected chicken lungs, not only had 14 immune related target genes (Additional file2: Table S2), but also targeted the AIV HA, NA, PA, PB1 and PB2 genes. In general, more AIV genes were targeted by induced host miRNAs than repressed miRNAs (8.33 times higher). Some miRNAs had only two viral targets, such as gga-mir-32 (targeting HA and NS genes) and gga-miR-30b (targeting M and NA genes). Some differentially expressed miRNAs had multiple predicted viral targets, such as gga-miR-202 which is predicted to target all nine AIV genes.
Host mRNA profile analysis
The genome-wide expression profiling of host response to AIV infection was carried out using chicken 44 K Agilent microarray. There were 508 genes differentially expressed (161 up-regulated vs. 347 down-regulated) between AIV infected vs. non-infected chickens (P < 0.05, Fold-change > 1.5). The fold-change of gene expression between infected and non-infected group ranged from 34.33 to −10.10.
The integrative differentially expressed miRNAs and mRNA expression of its potential targeted immune-related genes are presented in Table7. Eight immune related host genes were significantly up or down regulated with AIV infection. Four genes were significantly up-regulated, while the rest were significantly down-regulated. Chicken MX1 gene, which was reported to be associated with influenza virus resistance[27], had the highest fold-change (11.46 fold) followed by interleukin 8[28] (11.03 fold) and interferon regulatory factory 7[29] (2.11 fold). Tumor necrosis factor receptor superfamily member 19 was down-regulated[30] (1.85 fold). In general, positive correlations between miRNAs and mRNA expression were observed.
Gene ontology (GO) analysis
The significantly enriched functional terms in biological processes from differentially expressed host genes and predicted target genes of differentially expressed miRNAs with AIV infection, respectively, are presented in Additional file3: Table S3. The immune related GO terms are presented in Figure5. Developmental process (1.95 fold) was enriched by both target genes of repressed miRNAs and induced mRNAs. Two immune related GO terms, immune system process and immune response, were only enriched by induced host genes.

Gene ontology (GO) annotation of differentially expressed genes and target genes of differentially expressed miRNAs between lungs of AIV infected and non-infected chicken in biological process category (P < 0.05). Fold enrichment is a ratio obtained by dividing user’s percentage by the percentage of each category of the whole genome.
GO terms significantly enriched in repressed genes included four clusters: cytoskeleton-dependent intracellular transport, DNA unwinding during replication microtubule-based movement and response to DNA damage stimulus. For GO terms significantly enriched in targets of induced miRNAs, five GO terms were enriched. These included calcium ion transport; homophilic cell adhesion; T cell activation; gamma-aminobutyric acid signalling pathway; and Wnt receptor signalling pathway.
Discussion
Integration of global profiling of miRNAs and mRNA expression may provide a unique opportunity in enhancing our understanding of regulatory mechanisms in many biological processes including virus infection. Identification of differentially expressed miRNAs associated with AIV infection is the first critical step of this comprehensive analysis. With limited number of biological replicates, and the nature of distribution of discrete measurement data other than continuous variable in microarray data, no single statistical method currently available can meet all conditions of this type of data[31]. As the main purpose of this step was to narrow down a small set of differentially expressed miRNAs that are potentially associated with AIV infection, our strategy was to include any potential important miRNAs (relative loose criteria with potential high false positive rate) for global enrichment analysis, but more conservative for further analysis for individual miRNA. Therefore, we chose three methods that have been intensively used for next generation sequencing data analysis[32–34], including Fisher’s exact test, DESeq and edgeR. Based on the results, Fisher’s exact test is less stringent than both DESeq and edgeR. On the other hand, both DESeq and edgeR might be too conservative. For example, gga-miR-206 was not identified by DESeq, neither by edgeR, but it was confirmed by real-time RT-PCR. This did suggest that the criteria used in the DESeq and edgeR analyses might miss some true positive, although this may exclude potential false positives. Thus, for the following global functional term enrichment analysis, our analysis was primarily based on the results from Fisher’s exact test. However, we are more interested in the 17 miRNAs (Tables2,3 and4) that were identified as significant in at least two of the three methods of analysis, and especially the three miRNAs (gga-mir-1719, 1594 and 1599) identified across all three analysis. Of particular note, these three miRNAs are chicken specific miRNAs. To our knowledge, this is the first report of potential function study of these three miRNAs in animals. Further investigation of underlying mechanisms of these miRNAs in regulating AIV replication will expand our knowledge in host-pathogen interaction in animals.
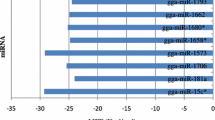
Genetics play an important role in the regulation of miRNAs expression in animals[35]. We hypothesize that genetics affect the regulation of miRNA expression during AIV infection in the chicken. In the present study with broiler chickens, there were more miRNAs up-regulated than down-regulated with virus infection, which was the opposite with our previous miRNA profiling in layer chickens with AIV infection (more down-regulated than up-regulated)[20]. Table8 lists differentially expressed miRNAs in both current broiler and previous layer studies. Only two (miR-1599 and miR-1416) of eighteen miRNAs had consistent directions of regulation following AIV infection, which suggests that these two miRNAs were conserved responses to AIV infection across diverse genetic lines. The major discrepancies between two studies might be due to: broilers and layers being genetically distinct chicken breeds with long-term diverse selection targeting on growth and egg production, respectively. Specifically, broilers are specialized in the production of a short term humoral response, while layers have a long-term humoral response in combination with a strong cellular mediated response[36]. The differential expression of miRNAs between them may reflect different miRNA-mediated host responses to virus infection. For example, differential expression of miR-142-3p in conventional CD4+ T cells and CD25+ TREG cells in mice control the functions of both effector and suppressor cells. In the current study, gga-miR-142-3p was down-regulated in layers and up-regulated in broilers, which indicate that host immune response to AIV infection mediated by gga-miR-142-3p in broiler chickens may be different from layer. In summary, although other factors such as time of challenge may contribute to great miRNA difference between broilers and layers, these results strongly suggest that genetic backgrounds play a vital role in the regulation of miRNAs during AIV infection in chickens.
The miR-155 has been reported to play important roles in both innate and adaptive immunity in mammals[7, 37, 38]. miR-155 knock-out mice are not capable of generating defensive immune responses, developing lymphocytes, or antigen-presenting cell functions[39]. The up-regulation of miR-155 with poly (I:C) and IFNβ stimulation in mouse macrophages suggest an important role of miR-155 in the regulation of viral infection[37]. In the current study, gga-miR-155 was significantly induced by AIV infection, which was consistent with other studies[40]. Based on target prediction, miR-155 could target the chicken anti-influenza gene MX1, therefore playing a role in host and AIV interactions in chickens. The activation of c-Jun NH2-terminal kinases (JNK) pathway can eliminate virus-infected cells by apoptosis. The inhibition of JNK pathway blocked the expression of miR-155 in murine macrophages[37, 40]. Down-regulation of TNFRSF19 (TNF receptor superfamily member 19), one of key genes in JNK pathway, indicate that antiviral activities through JNK pathway might be inhibited. Therefore, up-regulated miR-155 might also activate JNK pathway, and subsequently induce apoptosis to eliminate virus infected cells[37, 40].
Most of the up-regulated miRNAs were predicted to target the hemaglutinin (HA) and neuraminidase (NA) mRNAs such as miR-34a and miR-155. Both HA and NA are major surface glycoproteins. HA is responsible for receptor binding and virus fusion[41], while NA is responsible for receptor destruction and virion release[41]. Therefore, induction of these miRNAs might affect virus attachment and release and therefore the formation of new infectious viral particles. In addition, three down-regulated miRNAs (miR-206, miR-301 and miR-187) also were predicated to target on AIV genome. The first line of evidence from this integrative analysis strongly indicates that the importance of several candidate miRNAs including miR-34a, 146a, 155 and 206 warrant further investigation to understand the mechanisms of miRNA regulation of AIV infection in chickens.
Conclusions
In summary, this comprehensive analysis has provided several lines of new evidence on how host miRNA might regulate host response to AIV replication in broilers. Specifically, this study generated a list of strong candidate miRNAs including miR-34a, 122–1, 122–2, 146a, 155, 206, 1719, 1594, 1599 and 460 that potentially regulate AIV infection in chickens. In addition, several candidate genes including MX1, IL-8, IRF-7, TNFRS19 have been identified to be associated with AIV infection in broilers. Finally, comparison with our previous layer miRNA profiling, this study strongly indicates that genetic background is a critical factor in determining miRNA abundance and regulation during AIV infection. As main focus of this global profiling analysis was to generate new hypothesis by screening whole genome miRNAs and mRNAs, our on-going effort using experimental approach such as knock-down or over-express candidate miRNAs and mRNAs in vitro is expected to provide new evidence in understanding these regulatory mechanisms of AIV infection in chickens.
Methods
Sample collection and RNA isolation
Day old broilers (Cobb-Vantress, Inc.) were randomly divided into two groups (4 chickens per group), housed in negative pressure Horsfall-Bauer, temperature control isolation units and provided with water and commercial feed ad libitum. At one week of age, one group was inoculated with 0.1 ml of CK/TX/02/H5N3 virus containing107.5 EID50/ml and the remaining chickens were inoculated with PBS (mock treatment) by the intra-choanal cleft route. At 4 days post-inoculation (dpi), depression and severely congested lungs were observed in the treated chickens. Therefore, all chickens were humanely euthanized at 4 dpi, and lungs were collected for RNA isolation. The animal experiment was performed according to the guidelines approved by the Institutional Animal Care and Use Committee, Texas A&M University.
Two pools of total RNA samples (2 random chickens per pool) were generated from the infected and non-infected group. Total RNAs were isolated using Trizol (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol. Dnase I (Ambion, Austin, TX) digestion was carried out after RNA isolation according to manufacturer’s instructions. RNA concentration and purity were determined by measuring absorbance at 260 nm and A260/A280 ratio using a NanoDrop ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE). RNA samples were stored at −80°C until further use.
Viral titration
Virus titers in lungs of inoculated chickens were determined at 4 dpi by real-time RT-PCR of influenza virus matrix gene using AgPath-ID™ AIV- M kit (Ambion, Austin, TX) following the manufacturer’s instructions. For quantitation of virus load, RNA was extracted from serially diluted H5N3 virus stock (101.5–105.5 log10 EID50/ml) and used to generate a standard curve. The amount of RNA in the samples was converted into log10 EID50/ml by interpolation as described previously[42].
Small RNA sequencing and analysis
For small RNA library construction, total RNA samples from lungs of infected and non-infected broiler chickens were prepared using the DGE-Small RNA Sample Prep Kit (Illumina, San Diego, CA) as previously described[20]. A total of two Solexa-ready small RNA templates were analyzed on an Illumina 1 G Genome Analyzer at the University of Houston. Cluster generation was performed and clusters were sequenced. Initial sequence process and analysis was done as previously described[20]. All unique sequence reads with a minimum read count of 5 were aligned with precursor chicken miRNA sequences from miRBase version 16[22–24]. Reads of each miRNA were the sum of exact and loose matches (± 4 bp) to known miRNAs. For each sample, counts were normalized to the total number of small RNA sequences, and then for each miRNA, the normalized number of counts was compared between groups. False discovery rate (FDR) (Q values) was calculated by R program according to Benjamin’s method[43]. Fisher’s exact test, DESeq[33] and edgeR[32] were used to identify differentially expressed miRNAs (P < 5%). Fold changes for Fisher’s exact test were calculated as the ratio of normalized reads of infected over non-infected group. Statistics related to over representation of functional categories were performed using DAVID[44–46]. A P < 0.05 was considered significant. Novel miRNAs were identified using the methods of Creighton et al. and Wang et al.[20, 47].
Confirmation of miRNA expression by northern-blot
Expression of one potential novel miRNA was confirmed by Northern blot analysis using the same total RNA samples as those used for small RNA library construction. Total RNA of infected and non-infected lung samples (15 μg each) were separated on a 15% denaturing acrylamide gel and transferred onto a GeneScreen Plus nylon membrane (GE Healthcare, Piscataway, NJ). Membranes were fixed by UV cross-linking at 1200 μJ and baking at 80°C for 1 hour. DNA probes (antisense to two mature miRNA sequences) were end-labeled with [γ-32P] ATP (GE Healthcare, Piscataway, NJ) using a mirVana Probe & Marker Kit (Ambion, Austin, TX). Pre-hybridization, hybridization and washes were carried out at 42°C using ULTRAhyb-Oligo hybridization buffer according to the manufacturer’s instructions (Ambion, Austin, TX). Chicken U6 small nuclear RNA was used as an internal control to account for loading differences between samples.
Confirmation of differentially expressed miRNAs by TaqMan miRNA assay
To determine the expression of miRNAs by quantitative RT-PCR (qRT-PCR), TaqMan miRNA assays were performed. The specific stem-loop RT primers for miR-206, miR-451 and U6 were obtained commercially from Applied Biosystems (Foster City, CA). In brief, cDNA was synthesized from total RNA by using the miRNA specific primers according to the protocol of TaqMan Micro RNA Assays (Applied Biosystems, CA). Reverse transcriptase reactions contained 10 ng of RNA samples, 3 μl of 50 nM stem loop RT primer and reagents from the TaqMan MiRNA Reverse Transcription Kit (Applied Biosystems, CA). The 15 μl reactions were incubated for 30 min at 16°C, 30 min at 42°C and 5 min at 85°C, and then held at 4°C. Real-time PCR was performed using gene specific probes and a pair of primers and TaqMan 2X Universal PCR Master Mix (No AmpErase UNG) (Applied Biosystems, CA). The 20 μl PCR reactions included 1.33 μl cDNA products, 10 μl PCR master mix, and 1 μl 20X TaqMan MiRNA Assay mix (Applied Biosystems, CA). These reactions were incubated at 95°C for 10 min, followed by 40 cycles at 95°C for 10 s, 60°C for 40 s and 72°C for 1 s using an ABI 7900 Realtime PCR instrument (Applied Biosystems, CA). All reactions were run in triplicate. The threshold cycle was defined as the fractional cycle number at which the fluorescence passes the fixed threshold. The expression levels of miR-206 and miR-451 in each sample were measured in terms of threshold cycle value and normalized to U6 expression using 2-∆∆CT[48].
miRNA prediction and validation
Target prediction
The chicken (Gallus gallus) Unigene database (NCBI) and the miRNA target prediction algorithm miRanda 3.1 (http://www.microran.org/microrna/getDownloads.do) were employed to predict potential targets of all the differentially expressed miRNAs. For miRanda, default parameters were used with the following exceptions: the score was set to ≥ 130 and the free energy was set to ≤ −16 kCal/mol. The predicted targets were further filtered using more stringent criteria in which they must contain either (1) a match between nucleotides 2–8 of the miRNA with the target sequence or (2) a match between nucleotides 2–7 and 13–16 of the miRNA with the target sequence (G:U base-pairing was tolerated). A set of target genes containing miR-146a binding sites within their 3´UTRs were selected for further analysis using a dual luciferase reporter assay.
Insertion of target sequences into psiCHECK-2
For each potential target gene, the region of 3´UTR flanking the miR-146a binding sites was PCR amplified from Red Jungle Fowl genomic DNA using gene specific primers (Additional file4: Table S4). Each PCR product was cloned into the 3´UTR of the Renilla reporter gene in the psiCHECK-2 vector (Promega, WI) using NotI and XhoI restriction sites from the multicloning site.
Construction of RCAS viruses expressing chicken miR-146a
The previous described RCASBP(A)-miR vector[49] was used to ectopically express miR-146a. In order to produce RCAS viruses expressing chicken miR-146a, an entry vector was constructed using PAGE purified 76-nt forward and 68-nt reverse oligos (Invitrogen). Restriction sites for SphI and NgoMIV were introduced at the 5´- and 3´-ends, respectively. Forward and reverse oligos were mixed at a final concentration of 1 μM, denatured at 95°C for 20 sec and annealed at RT to generate a short double-stranded DNA fragment. The fragment was then cloned into the pENTR3C-miR-SphNgo vector at the SphI and NgoMIV restriction sites. The RCASBP(A)- miR-146a vector was generated via a recombination between the pENTR3C- miR-146a entry vector and RCASBP(A)-YDV gateway destination vector using a LR clonase kit (Invitrogen, CA). To produce miR-146a expressing viruses (RCAS- miR-146a), the RCASBP(A)-miR146a plasmid vector was transfected into DF-1 cells, a chicken embryo fibroblast continuous cell line, using FuGENE 6 (Promega, WI). Virus stock was harvested at day 6 post transfection and titer was determined using immunofluorescence staining with the monoclonal 3 C2 antibody against the RSV/ALV gag protein (Developmental Studies Hybridoma Bank, University of Iowa) and FITC-conjugated goat anti mouse IgG (Invitrogen, CA). In addition, RCAS viruses (RCAS-SC) expressing a scrambled control sequences were produced to serve as a negative control. Ectopic expression of the miR-146a was validated using a miScript Reverse Transcription kit and a miScript SYBR Green PCR kit (Qiagen, CA).
Dual luciferase reporter assay
DF1 cells were infected with either RCAS-miR-146a or RCAS-SC at a multiplicity of infection of 1 and maintained for 6 days in a 96-well plate in RPMI 1640 medium supplemented with 1% heat-inactivated FBS, L-glutamine, penicillin (100 U/ml), streptomycin (100 μg/ml), and fungizone (4 μg/ml), at 37°C with 5% CO2. The psiCHECK-2 construct (100 ng) for each potential target gene, as well as the scramble control, were then transfected into both RCAS-miR-146a or RCAS-SC infected DF-1 cells using FuGENE 6 (Promega, WI). Forty-eight hours post-transfection, cells were washed with PBS and lysed in Passive Lysis Buffer (Promega, WI). For each transfection, firefly and Renilla luciferase activities were determined using the Dual-Luciferase Reporter Assay System (Promega, WI) and a VictorLight 1420 luminescence counter (PerkinElmer, MA). The Renilla luciferase signal was normalized to the firefly luciferase signal. The normalized Renilla luciferase activity was compared between the RCAS-miR-146a and the RCAS-SC using student’s t-test (P < 0.05). Triplicates for each target construct were performed and the assay was repeated to confirm the results.
Microarray analysis
Microarray experiment design: Four biological replicates from infected and non-infected groups were used with dye balance in order to prevent dye-bias during sample labeling.
Labeling and hybridization: The integrity of total RNA samples was confirmed using Agilent Bioanalyzer 2100 Lab-on-chip system (Agilent Technologies, Palo Alto, CA). Four hundred nano-grams (ng) of total RNA were reverse-transcribed to cDNA during which a T7 promoter sequence was introduced into the cDNA. T7 RNA polymerase-driven RNA synthesis was used for preparation and labeling of RNA with Cy3 (or Cy5) dye. Fluorescent cRNA probes were purified using Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA), and an equal amount (825 ng) of Cy3 and Cy5 labeled cRNA probes were hybridized to a 44 K chicken Agilent array (GEO accession: GSE9416). The hybridized slides were washed using a commercial kit package (Agilent Technol-ogies, CA) and then scanned using a Genepix 4100A scanner (Molecular Devices Corporation, Sunnyvale, CA) with a tolerance of saturation setting of 0.005%.
Microarray data collection and analysis: For each channel, the median of the signal intensity and local background values were used. A Locally Weighted Linear Regression (LOWESS) normalization was applied to remove signal intensity-dependent dye bias for each array using R program. A mixed model that included the fixed effects of dye (cy3 and cy5) and random effect of slide and array was used to analyze the normalized data by SAS (SAS institute, Cary, NC). P < 0.05 was considered significant. These data have been deposited in GEO (Accession numbers: GSM879919, GSM879925, GSM879936, GSM879937).
Gene ontology
Functional annotations for differentially expressed genes were performed through the use of the Database for Annotation, Visualization and Integrated Discovery (DAVID)[44–46]. Statistics related to over representation of functional categories was based upon a Fisher’s Exact statistic methodology similar to that described by Al-Shahrour et al.[50]. A P < 0.05 was considered as significant.
References
Shahzad MI, Naeem K, Mukhtar M, Khanum A: Passive immunization against highly pathogenic Avian Influenza Virus (AIV) strain H7N3 with antiserum generated from viral polypeptides protect poultry birds from lethal viral infection. Virol J. 2008, 5: 144-10.1186/1743-422X-5-144.
Li DY, Xue MY, Wang C, Wang JB, Chen PY: Bursopentine as a Novel Immunoadjuvant Enhances both Humoral and Cell-Mediated Immune Responses to Inactivated H9N2 Avian Influenza Virus in Chickens. Clin Vaccine Immunol. 2011, 18 (9): 1497-1502. 10.1128/CVI.05133-11.
Bartel DP: MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004, 116 (2): 281-297. 10.1016/S0092-8674(04)00045-5.
Guo HS, Xie Q, Fei JF, Chua NH: MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for arabidopsis lateral root development. Plant Cell. 2005, 17 (5): 1376-1386. 10.1105/tpc.105.030841.
Hatfield SD, Shcherbata HR, Fischer KA, Nakahara K, Carthew RW, Ruohola-Baker H: Stem cell division is regulated by the microRNA pathway. Nature. 2005, 435 (7044): 974-978. 10.1038/nature03816.
Lee RC, Feinbaum RL, Ambros V: The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993, 75 (5): 843-854. 10.1016/0092-8674(93)90529-Y.
Lindsay MA: microRNAs and the immune response. Trends Immunol. 2008, 29 (7): 343-351. 10.1016/j.it.2008.04.004.
Sassen S, Miska EA, Caldas C: MicroRNA: implications for cancer. Virchows Arch. 2008, 452 (1): 1-10. 10.1007/s00428-007-0532-2.
Jopling CL, Schutz S, Sarnow P: Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe. 2008, 4 (1): 77-85. 10.1016/j.chom.2008.05.013.
Song L, Liu H, Gao S, Jiang W, Huang W: Cellular microRNAs inhibit replication of the H1N1 influenza A virus in infected cells. J Virol. 84 (17): 8849-8860.
Baltimore D, Boldin MP, O'Connell RM, Rao DS, Taganov KD: MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008, 9 (8): 839-845. 10.1038/ni.f.209.
MicroRNAs: from basic science to disease biology. Edited by: Appasani K. 2008, Cambridge University Press: New York, 1
Sullivan CS, Grundhoff AT, Tevethia S, Pipas JM, Ganem D: SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature. 2005, 435 (7042): 682-686. 10.1038/nature03576.
Cullen BR: Viruses and microRNAs. Nat Genet. 2006, 38 (Suppl): S25-30.
Kelly EJ, Russell SJ: MicroRNAs and the regulation of vector tropism. Mol Ther. 2009, 17 (3): 409-416. 10.1038/mt.2008.288.
Lecellier CH, Dunoyer P, Arar K, Lehmann-Che J, Eyquem S, Himber C, Saib A, Voinnet O: A cellular microRNA mediates antiviral defense in human cells. Science. 2005, 308 (5721): 557-560. 10.1126/science.1108784.
Nathans R, Chu CY, Serquina AK, Lu CC, Cao H, Rana TM: Cellular microRNA and P bodies modulate host-HIV-1 interactions. Mol Cell. 2009, 34 (6): 696-709. 10.1016/j.molcel.2009.06.003.
Otsuka MJQ, Georgel P, New L, Chen J, Mols J, Kang YJ, Jiang Z, Du X, Cook R, Das SC, Pattnaik AK, Beutler B, Han J: Hypersusceptibility to vesicular stomatitis virus infection in Dicer1-deficient mice is due to impaired miR24 and miR93 expression. Immunity. 2007, 1: 12-
Scaria V, Hariharan M, Maiti S, Pillai B, Brahmachari SK: Host-virus interaction: a new role for microRNAs. Retrovirology. 2006, 3: 68-10.1186/1742-4690-3-68.
Wang Y, Brahmakshatriya V, Zhu H, Lupiani B, Reddy SM, Yoon BJ, Gunaratne PH, Kim JH, Chen R, Wang J: Identification of differentially expressed miRNAs in chicken lung and trachea with avian influenza virus infection by a deep sequencing approach. BMC Genomics. 2009, 10: 512-10.1186/1471-2164-10-512.
Hurd PJ, Nelson CJ: Advantages of next-generation sequencing versus the microarray in epigenetic research. Brief Funct Genomic Proteomic. 2009, 8 (3): 174-183. 10.1093/bfgp/elp013.
Griffiths-Jones S: The microRNA Registry. Nucleic Acids Res. 2004, 32 (Database issue): D109-D111.
Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ: miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34 (Database issue): D140-D144.
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ: miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008, 36 (Database issue): D154-D158.
John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS: Human MicroRNA targets. PLoS Biol. 2004, 2 (11): e363-10.1371/journal.pbio.0020363.
Hsu PW, Lin LZ, Hsu SD, Hsu JB, Huang HD: ViTa: prediction of host microRNAs targets on viruses. Nucleic Acids Res. 2007, 35 (Database issue): D381-D385.
Ko JH, Jin HK, Asano A, Takada A, Ninomiya A, Kida H, Hokiyama H, Ohara M, Tsuzuki M, Nishibori M: Polymorphisms and the differential antiviral activity of the chicken Mx gene. Genome Res. 2002, 12 (4): 595-601.
Medin CL, Fitzgerald KA, Rothman AL: Dengue virus nonstructural protein NS5 induces interleukin-8 transcription and secretion. J Virol. 2005, 79 (17): 11053-11061. 10.1128/JVI.79.17.11053-11061.2005.
Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N: IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005, 434 (7034): 772-777. 10.1038/nature03464.
Qiu WM, Hu YH, Andersen TE, Jafari A, Li N, Chen W, Kassem M: Tumor Necrosis Factor Receptor Superfamily Member 19 (TNFRSF19) Regulates Differentiation Fate of Human Mesenchymal (Stromal) Stem Cells through Canonical Wnt Signaling and C/EBP. J Biol Chem. 2010, 285 (19): 14438-14449. 10.1074/jbc.M109.052001.
Kvam VM, Liu P, Si Y: A comparison of statistical methods for detecting differentially expressed genes from RNA-seq data. Am J Bot. 2012, 99 (2): 248-256. 10.3732/ajb.1100340.
Robinson MD, McCarthy DJ, Smyth GK: edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010, 26 (1): 139-140. 10.1093/bioinformatics/btp616.
Anders S, Huber W: Differential expression analysis for sequence count data. Genome Biol. 2010, 11 (10): R106-10.1186/gb-2010-11-10-r106.
Auer PL, Doerge RW: Statistical design and analysis of RNA sequencing data. Genetics. 2010, 185 (2): 405-416. 10.1534/genetics.110.114983.
Bueno MJ, Perez de Castro I, Gomez de Cedron M, Santos J, Calin GA, Cigudosa JC, Croce CM, Fernandez-Piqueras J, Malumbres M: Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell. 2008, 13 (6): 496-506. 10.1016/j.ccr.2008.04.018.
Koenen ME, Boonstra-Blom AG, Jeurissen SH: Immunological differences between layer- and broiler-type chickens. Vet Immunol Immunopathol. 2002, 89 (1–2): 47-56.
O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D: MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007, 104 (5): 1604-1609. 10.1073/pnas.0610731104.
Romania P, Lulli V, Pelosi E, Biffoni M, Peschle C, Marziali G: MicroRNA 155 modulates megakaryopoiesis at progenitor and precursor level by targeting Ets-1 and Meis1 transcription factors. Br J Haematol. 2008, 143 (4): 570-580.
Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL: Regulation of the germinal center response by microRNA-155. Science. 2007, 316 (5824): 604-608. 10.1126/science.1141229.
Taganov KD, Boldin MP, Chang KJ, Baltimore D: NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006, 103 (33): 12481-12486. 10.1073/pnas.0605298103.
Fields of virology. Edited by: Knipe DM, Howley PM. 2007, Lippincott Williams & Wilkins, Ith edn, Fifth Edition edition (2006)Ith
Lee CW, Suarez DL: Application of real-time RT-PCR for the quantitation and competitive replication study of H5 and H7 subtype avian influenza virus. J Virol Methods. 2004, 119 (2): 151-158. 10.1016/j.jviromet.2004.03.014.
Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I: Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001, 125 (1–2): 279-284.
da Huang WSB, Lempicki RA: Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009, 4 (1): 13-10.1038/nnano.2008.387.
Huang D, Chang TR, Aggarwal A, Lee RC, Ehrlich HP: Mechanisms and dynamics of mechanical strengthening in ligament-equivalent fibroblast-populated collagen matrices. Ann Biomed Eng. 1993, 21 (3): 289-305. 10.1007/BF02368184.
Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA: DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4 (5): P3-10.1186/gb-2003-4-5-p3.
Creighton CJ, Nagaraja AK, Hanash SM, Matzuk MM, Gunaratne PH: A bioinformatics tool for linking gene expression profiling results with public databases of microRNA target predictions. RNA. 2008, 14 (11): 2290-2296. 10.1261/rna.1188208.
Schmittgen TD, Livak KJ: Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008, 3 (6): 1101-1108. 10.1038/nprot.2008.73.
Chen M, Payne WS, Hunt H, Zhang H, Holmen SL, Dodgson JB: Inhibition of Marek's disease virus replication by retroviral vector-based RNA interference. Virology. 2008, 377 (2): 265-272. 10.1016/j.virol.2008.03.019.
Al-Shahrour F, Diaz-Uriarte R, Dopazo J: FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics. 2004, 20 (4): 578-580. 10.1093/bioinformatics/btg455.
Savenije OEM, Kerkhof M, Reijmerink NE, Brunekreef B, de Jongste JC, Smit HA, Wijga AH, Postma DS, Koppelman GH: Interleukin-1 receptor-like 1 polymorphisms are associated with serum IL1RL1-a, eosinophils, and asthma in childhood. J Allergy Clin Immun. 2011, 127 (3): 750-U394. 10.1016/j.jaci.2010.12.014.
Chaves LD, Krueth SB, Reed KM: Defining the Turkey MHC: Sequence and Genes of the B Locus. J Immunol. 2009, 183 (10): 6530-6537. 10.4049/jimmunol.0901310.
Akram A, Han B, Masoom H, Peng C, Lam E, Litvack ML, Bai XH, Shan YX, Hai T, Batt J: Activating Transcription Factor 3 Confers Protection against Ventilator-induced Lung Injury. Am J Resp Crit Care. 2010, 182 (4): 489-500. 10.1164/rccm.200906-0925OC.
Sarson AJ, Read LR, Haghighi HR, Lambourne MD, Brisbin JT, Zhou HJ, Sharif S: Construction of a microarray specific to the chicken immune system: profiling gene expression in B cells after lipopolysaccharide stimulation. Can J Vet Res. 2007, 71 (2): 108-118.
Acknowledgements
This project was supported by the Formula Animal Health Grant no. 07–024 and partially supported by the Formula Animal Health Grant no. TEX09244 from AFRI/NIFA. We thank Drs. Xianyao Li and Hsin-I Chiang for their help with the animal trial and thank Xianbo Jia for his help with data analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that there are no competing interests.
Authors' contributions
YW carried out the RNA isolation, small RNA library construction preparation, microarray analysis, analyzed data and drafted the manuscript. VB was responsible for the animal trial. BL and SR contribute to experiment design. BY, AB and PG contributed to the running of miRNA deep sequencing and analysis of miRNA. HL and NT developed the miRNA target confirmation; NI contributed to miRNA northern-blotting; RO provided experimental animals; HZ provided the concepts of the study, designed the experiment and revised the manuscript. All authors submitted comments, read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wang, Y., Brahmakshatriya, V., Lupiani, B. et al. Integrated analysis of microRNA expression and mRNA transcriptome in lungs of avian influenza virus infected broilers. BMC Genomics 13, 278 (2012). https://doi.org/10.1186/1471-2164-13-278
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-13-278