
Abstract
Background
Human Malaria is transmitted by mosquitoes of the genus Anopheles. Transmission is a complex phenomenon involving biological and environmental factors of humans, parasites and mosquitoes. Among more than 500 anopheline species, only a few species from different branches of the mosquito evolutionary tree transmit malaria, suggesting that their vectorial capacity has evolved independently. Anopheles albimanus (subgenus Nyssorhynchus) is an important malaria vector in the Americas. The divergence time between Anopheles gambiae, the main malaria vector in Africa, and the Neotropical vectors has been estimated to be 100 My. To better understand the biological basis of malaria transmission and to develop novel and effective means of vector control, there is a need to explore the mosquito biology beyond the An. gambiae complex.
Results
We sequenced the transcriptome of the An. albimanus adult female. By combining Sanger, 454 and Illumina sequences from cDNA libraries derived from the midgut, cuticular fat body, dorsal vessel, salivary gland and whole body, we generated a single, high-quality assembly containing 16,669 transcripts, 92% of which mapped to the An. darlingi genome and covered 90% of the core eukaryotic genome. Bidirectional comparisons between the An. gambiae, An. darlingi and An. albimanus predicted proteomes allowed the identification of 3,772 putative orthologs. More than half of the transcripts had a match to proteins in other insect vectors and had an InterPro annotation. We identified several protein families that may be relevant to the study of Plasmodium-mosquito interaction. An open source transcript annotation browser called GDAV (Genome-Delinked Annotation Viewer) was developed to facilitate public access to the data generated by this and future transcriptome projects.
Conclusions
We have explored the adult female transcriptome of one important New World malaria vector, An. albimanus. We identified protein-coding transcripts involved in biological processes that may be relevant to the Plasmodium lifecycle and can serve as the starting point for searching targets for novel control strategies. Our data increase the available genomic information regarding An. albimanus several hundred-fold, and will facilitate molecular research in medical entomology, evolutionary biology, genomics and proteomics of anopheline mosquito vectors. The data reported in this manuscript is accessible to the community via the VectorBase website (http://www.vectorbase.org/Other/AdditionalOrganisms/).
Similar content being viewed by others


Background
Human malaria transmission is dependent on efficient development of Plasmodium parasites within anopheline mosquito vectors. Anopheline mosquitoes are a large subfamily comprising nearly five hundred species distributed in subtropical and tropical areas around the world, but only a small percentage (10-20%) are malaria vectors [1]. Intriguingly, malaria infection rates among anopheline species do not correlate with mosquito phylogenetic relationships, suggesting that genetic traits associated with vectorial capacity have quickly and independently evolved in different species [2].
Vectorial capacity is a highly complex biological phenomenon depending on mosquito behavior, lifespan and innate refractoriness or susceptibility to Plasmodium infection, which may result from the co-evolutionary forces driving the tripartite interaction between humans, mosquitoes and parasites [3]. As a result of the sequencing of the Anopheles gambiae and Plasmodium falciparum genomes, a great deal of information regarding our understanding of mosquito-pathogen interactions at a molecular level has been gained [4, 5]. Post genome research has highlighted the role of the An. gambiae innate immune response in determining mosquito refractoriness to Plasmodium[6–8].
In addition to the mosquito’s innate ability to transmit malarial parasites, critical aspects of mosquito biology, like adaptation to diverse niches, host seeking behavior, and resistance to insecticides are still unknown. Novel strategies for control, based on a deep understanding of mosquito biology and evolution, will be required to achieve the goal of eventual malaria eradication. Rapid technological advances in DNA sequencing, protein characterization by mass spectrometry and bioinformatics offer unique opportunities to generate large catalogs of genes, proteins and biological networks that may enable the identification of potential mosquito control targets beyond the An. gambiae-P. falciparum dyad [9], which can be harnessed by novel strategies such as transgenesis, mosquito-based transmission blocking vaccines [10, 11] and alternative insecticides [12].
Plasmodium vivax malaria still represents a major health and socio-economic burden in Asia, the Western Pacific and the Americas [13, 14]. Malaria in the Americas is transmitted by several anopheline species, including species belonging to the Nyssorhynchus subgenus, which is unique to the New World. Anopheles (Nyssorhynchus) albimanus is a major vector in southern México, Central America and the northern region of South America [15, 16]. The Nyssorhynchus subgenus is thought to be the earliest diverged branch of the anopheline radiation, which probably occurred more than one hundred million years (My) ago when the supercontinent Gondwana separated to give rise to the actual South American and African continents [17]. The proposed independent emergence of vectorial traits in conjunction with their rapid evolution may imply different molecular strategies involved in P. vivax and P. falciparum refractoriness and susceptibility in this subgenus. However, very little molecular information exists for any of the New World anopheline vectors, except for Anopheles (Nyssorhynchus) darlingi whose genome draft was recently released [18]. The An. albimanus genome is scheduled for genome sequencing in the near future [2].
We describe herein the results of a gene discovery based cDNA sequencing project combining conventional Sanger with Next Generation Sequencing (NGS) platforms to analyze cDNA samples derived from An. albimanus tissues including the midgut, cuticular fat body, dorsal vessel and salivary gland, which are critical organs involved in the Plasmodium life cycle [6, 19, 20] (Hernández-Martínez, Unpublished observations). The main objective of our study was to construct a reference transcriptome that will facilitate molecular and applied studies of An. albimanus refractoriness to Plasmodium infection and other biological processes relevant for disease transmission. Finally, to maximize accessibility for this and future transcriptome sequencing projects in the absence of a genome sequence, we developed an open-source sequence annotation browser called GDAV (Genome-Delinked Annotation Viewer; http://funcgen.vectorbase.org/gdav). The An. albimanus transcriptome annotations are available via the VectorBase website (http://www.vectorbase.org/Other/AdditionalOrganisms/).
Results and discussion
The An. albimanus transcriptome assembly
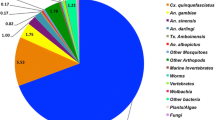
To capture as much of the transcriptome of the tissues involved in the interaction with Plasmodium spp. as possible, we combined transcriptome data generated with Sanger, 454 and Illumina sequencing platforms from several different sources of adult female An. albimanus RNA. Table 1A describes the origin of the RNA used to generate cDNA libraries sequenced by the different platforms, as well as the read number contribution of each. Owing to the inherent differences in throughput of each of the sequencing platforms used, most of the final dataset contains Illumina reads derived from the midgut transcriptome (94%), and nearly half of the transcripts were built with at least one 454 read. Only 6% of the built transcripts contained one or more Sanger reads (Table 1B). There were 8,958 (54%) transcripts expressed exclusively in the midgut, 35 (0.2%) were derived specifically from cuticular epithelium/fat body, and 80 (0.47%) were expressed only in the dorsal vessel, whereas the rest (45%) were found in two or more tissues (Figure 1).

Origin of transcripts according to mosquito organ. Distribution of transcript number (n = 16,572) according to the mosquito organ. The area of each circle is roughly a proportional approximation of the number of transcripts. A marked bias towards midgut is noted due to the enormous throughput of Illumina sequencing of a midgut library. Libraries sequenced by Sanger were left out given the low throughput of Sanger sequencing and for simplicity. Thus, not all the 16,669 transcripts are represented.
An initial transcriptome assembly was performed by combining all sequence reads described in Table 1A, which generated 15,764 contigs. Re-mapping the Sanger and 454 reads to the initial assembly revealed that a substantial number of 454 (53%) and Sanger reads (44%) were not represented in the initial transcriptome assembly, so they were re-assembled using GS Assembler, generating an additional set of 935 contigs. The total transcript dataset included 16,699 contigs with a mean contig length of 970 bp and a N50 of 1,434 bp (Table 1C). Our assembly metrics closely resemble the recently published NGS and Sanger data on the An. funestus transcriptome, which yielded 15,527 contigs with a N50 of 1,753 bp [21]. Similar results were also reported in de novo transcriptome assembly studies for the planarian worm, Schmidtea mediterranea in which 454 reads were pre-assembled to serve as scaffolds for Illumina paired-end assembly, yielding 17,465 contigs with a N50 of 1.6 kb [22], and 18,619 contigs with a mean length of 1,118 bp [23].
As a surrogate approach to estimate the contribution of each sequencing platform to the assembly quality, the transcript dataset was split into two subsets according to whether transcripts were assembled with only Illumina reads or with Illumina and 454 or Sanger reads. The Illumina-only subset was composed of 8,245 transcripts with a mean contig length of 822 bp and a N50 of 1,111 bp. The composite subset (8,445 transcripts build up with Illumina + 454 or Sanger) had a mean contig length of 1,087 bp and a N50 of 1,648 bp (Figure 2A), which is similar to the recently published Aedes albopictus transcriptome using 454 sequencing [24]. We then evaluated if there were differences in the proportion of homolog proteins in the An. gambiae predicted proteome using BLASTX (e value of 1.0 E-5) between both subsets. We did not observe a difference in the proportion of transcript matches between the Illumina-only subset and the composite ones (51% and 54%, respectively). However, the An. gambiae protein length coverage of translated transcripts was considerably improved in those transcripts belonging to the composite subset (Figure 2B) since 53% of their transcripts covered more than 70% of the An. gambiae target, whereas only 25% of the Illumina-only transcripts had an equivalent An. gambiae target coverage.

Transcript length and protein length coverage of the An. albimanus An. albimanus transcriptome. The whole transcriptome was split in two subsets according to transcripts composed of Illumina reads only or Illumina plus either of the two other platforms (composite subset). To make datasets comparable, their frequency (y axis) was expressed as a percentage. A) Transcript length distribution for both subsets and the whole dataset reveals slightly longer transcripts in the composite subset. B) An. gambiae protein length coverage obtained by BLASTX with An. albimanus transcripts. A larger fraction of the Illumina only subset covers less than one third of its respective match in An. gambiae. C) Frequency of mapped An. albimanus transcripts per An. gambiae gene. Transcripts were mapped to the An. gambiae genome using Exonerate v 2.2 [79] in the EST to genome mode (DNA vs DNA).
Genome mapping results to An. gambiae and An. darlingi
Since there is no genome sequence available for An. albimanus, unambiguous sequence alignment of An. albimanus transcripts to a reference anopheline genome could provide additional measures of the transcriptome assembly accuracy and completeness. Moreover, it could provide the means for refining reference genome annotation and provide evidence of functionally conserved genes that were missed by current gene finding algorithms [25]. The final cDNA assembly was mapped to the An. gambiae (100 My divergence, subgenus Cellia) and An. darlingi genomes (closer relative from subgenus Nyssorynchus). The current version of the An. gambiae PEST strain genome (AgamP3.6) is 278.2 Mbp long and it is in an advanced stage of assembly (oriented scaffolds) [26] and annotation (13,320 predicted genes) [27]. The An. darlingi genome was sequenced using 454 sequencing and is in an early stage of assembly (3,990 un-oriented contigs) and annotation (11,430 predicted protein coding genes) [18]. Using transcript to genome DNA alignment (see methods), 15,441 An. albimanus transcripts (92%) aligned (Exonerate bestn score >300) to the An. darlingi genome. Expressed as aligned base pairs, 13.8 of 16.1 Mbp (85%) of the An. albimanus transcriptome aligned to the An. darlingi genome (Table 2). Transcriptome mapping with the same parameters to the An. gambiae genome resulted in 9,648 aligned contigs (58%) with 7.1 Mbp (46%) aligned. Contigs that aligned to An. darlingi but not to the An. gambiae genome were predominantly short contigs (0.3-1.5 Kb) (Table 2. Figure 3A). As expected from the overall transcriptome aligned fraction, the fraction of An. albimanus transcript sequences that aligned to An. darlingi (transcript coverage) was considerably higher than for the An. gambiae genome, such that 76% of An. albimanus transcripts that were mapped to the An. darlingi genome aligned with more than 90% of their respective length (Figure 3B). Our mapping results using An. darlingi as the reference genome are significantly better than those described for the planarian worm S. mediterranea, which were mapped to its own genome [23], suggesting that despite its early stage of annotation, the An. darlingi genome is an appropriate surrogate genome for An. albimanus, and supports the accuracy and quality of our assembly. Transcripts that did not map to the An. darlingi genome (8%), as well as partial alignments may represent mis-assemblies in the transcriptome or the genome, rapidly evolving genes or the rapid evolution of untranslated regions (UTRs). Conversely, poor mapping to the An. gambiae reference genome may be result of all the above, further confounded by the increased evolutionary distance separating the two species.

Reference genome mapping of the An. albimanus transcriptome. Transcripts were mapped to the An. gambiae (blue line) or the An. darlingi (red line) genomes using Exonerate v 2.2 as in Figure2C. Mapped transcripts are expressed as a percentage of the total An. albimanus dataset. A) Aligned length plot; B) Fraction of the An. albimanus transcripts that aligned to either genome; C) Putative exon number relative to the proportion of An. albimanus mapped transcripts. For comparison, distribution of exon frequency in An. gambiae is shown (dotted black line).
An analysis of the protein length coverage for An. albimanus-An.gambiae BLASTX alignments showed a considerable proportion of transcripts with only partial coverage of their corresponding An. gambiae match (Figure 2B), indicating the possibility of multiple hits per An. gambiae gene. The extent of multiple transcripts mapping to a single An. gambiae gene was estimated by comparing transcript to genome and An. gambiae gene coordinates. Anopheles albimanus transcripts mapped partially or fully within the sequences of 6,305 An. gambiae genes, and 62% of these transcripts did so within a single An. gambiae gene (Table 2. Figure 2C). Based on our protein length coverage observations (Figure 2B), we noted a higher proportion of single gene hits in the composite subset (80%) than in the Illumina-only subset (68%) (Figure 2C).
As an additional approach to estimate transcript completeness, we used reference genome mapping to analyze exon coverage. As expected from the phylogenetic relationships, transcript mapping to the An. darlingi genome showed higher average exon coverage (2.1 exons per transcript) than to the An. gambiae genome (1.9 exons per transcript), which falls below the 4.4 exons per transcript in An. gambiae[28] (Figure 3C). However, there was no proportional difference in the exon coverage using An. gambiae or An. darlingi relative to the number of significant alignments (data not shown). More than half (54.1%) of our mapped transcript dataset covers at least two exons, whereas in An. gambiae, more than 86% of the genes are composed of at least two exons (Figure 3C). Only ten and thirty-one transcripts that mapped to An. gambiae and An. darlingi, respectively, had more than 10 exons, compared to 742 for An. gambiae. The An. albimanus transcript that covered most exons (23) when mapped to the An. darlingi genome was Locus_3073_Length_3,997, and corresponds to the An. gambiae AGAP000009 homolog, which is predicted to have 25 exons (data not shown).
In summary, transcript mapping to reference genomes and the derived analysis of exon structure of our transcript dataset revealed a degree of incompleteness when using An. gambiae as our reference. The increased proportion of genome alignments without a spanning intron (single exon transcripts) observed in An. albimanus could result from incorrect splitting of the transcript during the assembly of transcriptional units having more than one exon. Correct exon representation is relevant because alternative splicing is a means to increase proteome diversity and this phenomenon has been observed frequently among dipterans [29, 30]. Incomplete exon-exon structure across An. albimanus transcripts could underestimate the diversity of protein configurations and thus, may limit protein identification by proteomic approaches in the absence of the genome, and empirical studies to assess this possibility are required to fill in this gap in knowledge. Despite the limitations in our dataset, information regarding exon-exon structure may be useful for experimentalists when designing primers and probes for one gene-targeted analysis.
Estimated proteome coverage
As a starting point for transcript annotation, the proportion of the An. albimanus transcriptome that was homologous to a predicted protein sequence in other genomes was analyzed. Protein similarity to other insect proteomes and the NCBI nr databases was assessed using BLASTX (using an e-value threshold of 1.0E-5). A total of 10,000 sequences (62%) in our dataset had a significant match with at least one species. However, we note that probably due to methodological differences, this proportion is lower than the 84% match described by Crawford, when the An. funestus transcriptome was compared with the An. gambiae proteome [21].
Contrary to what would be expected based on degree of evolutionary relationships, and the results observed in transcript to genome alignments, we observed that a higher proportion of An. albimanus transcripts (56%) had a match with An. gambiae than with An. darlingi (54%) (Table 3. Figure 4B). However, a comparison between An. darlingi and An. gambiae revealed that 80% of the An. darlingi proteins matched the An. gambiae proteome. This can be partially explained because the An. albimanus predicted proteome is considerably larger (16,699) than the An. darlingi predicted proteome.

Proteome comparison to other insect proteomes. The An. albimanus transcriptome was compared to insect proteomes using BLASTX (e val. 1.E-05). A) Distribution of protein identity (%) of all high scoring pairs (HSP) for An. darlingi (Adar, red), An. gambiae (Agam, blue), Ae. aegypti (Aea, light green), C. quinquefasciatus (Cqf, dark green), D. melanogaster (Dmel, pink); I. scapularis (Isca, yellow) and P. humanus (Phum, orange). B) Average protein identity of An. albimanus best hits to insect proteomes. Color codes for each species as in A.
Considering the total amount of protein coding genes in each of the sequenced vector genomes, 51% of An. gambiae and 48% of the An. darlingi protein coding genes matched at least one transcript in An. albimanus, whereas An. darlingi covered 62% of the An. gambiae proteome. Proteome coverage tended to decrease according to phylogenetic distance (Figure 4B). However, the proportion of significant best hits decreased according to phylogenetic relatedness, so that protein percent identity among best BLASTX hits was the highest with An. darlingi (average identity 85%), and decreased according to phylogenetic distance (Table 3. Figure 4A-B).
The lower number of transcript matches observed between An. albimanus and An. darlingi than between An. albimanus and An. gambiae is likely the consequence of both the incompleteness of the An. albimanus dataset due to sampling of only a limited set of tissues from adult mosquitoes, and the current incomplete assembly status of the An. darlingi genome (Figure 4B). Moreover, the An. gambiae genome annotation relied heavily on homology based annotation approaches [5], and thus is very conservative. This implies that rapidly evolving genes may escape identification by such conservative approaches. Combining conservative and ab initio gene prediction strategies increased considerably the amount and quality of the An. gambiae protein coding genes [25]. Given the fact that most of the An. albimanus transcripts mapped to the An. darlingi genome, many transcripts may in fact be derived from true protein coding genes. Additionally, many of the sequences could represent non-protein coding transcripts of potential biological significance [31].
Orthologs
The proportion of the core eukaryotic genome covered by the An. albimanus transcriptome was investigated by searching for the 458 core eukaryotic protein models [32] in the An. albimanus predicted proteome, as well as the predicted proteomes of An. darlingi An. gambiae Ae. aegypti C. quinquefasciatus D. melanogaster, I. scapularis, P. humanus and Rhodnius prolixus. As expected, we observed almost complete coverage for D. melanogaster (99%) and An. gambiae genomes (98%). We identified 415 core eukaryotic genes (CEGs) in the An. albimanus dataset corresponding to 90% coverage. Coverage ranged from 95-98% in the other genome sequenced species. The lowest coverage was observed for An. darlingi (88%), which as discussed in the previous section, may reflect the early stage of that sequencing project (Table 4).
An important goal of the Anopheles genome cluster will be to define a “core anopheline genome”. Due to the incompleteness of the predicted proteome for An. albimanus, we reasoned that an An. albimanus - An. gambiae comparison could be very inaccurate in terms of ortholog prediction. However, using An. darlingi as an additional species for bidirectional comparisons, specificity could be increased but at the expense of sensitivity. To identify putative orthologs we used BLASTX and TBLASTN for bidirectional comparisons among the An. gambiae An. darlingi and An. albimanus proteomes. Anopheles albimanus - An. darlingi An. albimanus - An. gambiae and An. darlingi - An. gambiae bidirectional comparisons revealed 5,029, 5,556 and 7,609 best reciprocal hits, respectively. The three species comparison yielded a set of 3,772 1:1:1 putative orthologs (32% and 29% of the An. darlingi and An. gambiae predicted proteome, respectively) (Figure 5A). The 1:1 ortholog dataset between An. darlingi and An. gambiae comprised 64% and 60% of their proteome, respectively. The proportion of orthologs between An. gambiae and D. melanogaster is 47 and 44%, respectively [28], and ranged between 73% (D. melanogaster-D. grimshawi) to 93% (D. melanogaster-D. yakuba) within the Drosophila cluster [33].

Ortholog prediction in An. albimanus . Best reciprocal hits were identified by bidirectional BLASTX, BLASTP and TBLASTN comparisons between An. albimanus transcriptome, An. darlingi and An. gambiae genomes. A) Venn diagram depicting the numbers of entries for each comparison. The area of each circle is a proportional approximation of the number of transcripts in each. B) Protein length coverage frequency distribution in total An. albimanus translated transcripts matching the An. gambiae proteome (n = 9,445. Green line) and the 1:1:1 putative ortholog dataset (n = 3,772. Red line).
To further validate the accuracy of our ortholog assignment, we compared protein length coverage, using pair-wise alignments between translated An. albimanus proteins and their corresponding best match in An. gambiae. A considerable improvement of protein length coverage was observed in orthologs (average protein length coverage was 74%), when compared to the protein coverage of the overall dataset (average protein length coverage of 53%). As described previously (Figure 2B), protein coverage displayed a bimodal distribution where 40% of the translated An. albimanus products covered at least two thirds of their corresponding An. gambiae matches and another 40% covered less than one third (Figure 5B). A significant and expected improvement was observed in the ortholog dataset, which was biased toward higher coverage, resulting in 66% of the orthologs covering more than two thirds of their corresponding An. gambiae matches (Figure 5B).
If our BLAST best reciprocal hit ortholog prediction strategy was accurate, we would expect that all the core eukaryotic genes (CEGs) found would be represented in the 1:1:1 ortholog dataset. As shown in Table 4, only 283 of the identified An. albimanus CEGs were included in the three species ortholog dataset, which corresponds to 61% of the core eukaryotic genome. However, the proportion of CEGs found between two species best reciprocal hits was higher and ranged from 70 to 83%. Together, our data indicate that although there is a relatively high coverage of the core eukaryotic genome, the anopheline ortholog assignment remains incomplete (i.e., it is missing a considerable amount of true orthologs), and may be inaccurate (i.e., includes a significant proportion of paralogs).
Functional annotation of the An. albimanus transcriptome
To gain insight into the predicted functional characteristics of the current dataset, translated products were functionally annotated using the Gene Ontology (GO) classification as implemented in the Blast2GO software [34], as well as using the InterPro classification [35]. A total of 5,086 transcripts were annotated according to biological process, and 5,792 transcripts were annotated according to molecular function, which corresponds roughly to one third of all the transcripts in the dataset for both categories, and to one half of the total BLASTX matches in the vectors’ predicted proteomes (Table 3).
To gain insight into the protein evolution rate according to biological process and molecular function, the annotated transcriptome dataset was partitioned according to the most abundant biological process and molecular function GO slim categories. Based on BLASTX matches, average protein percent identity was estimated for each GO slim category, either in the 1:1:1 ortholog dataset, or as in all best hits (homologs) against the An. gambiae predicted proteome dataset (data not shown). For both the homologous and orthologous subsets, most conserved proteins belonged to general biological processes such as cytoskeletal organization, ion transport, translation and protein transport (Figure 6A), in agreement with other comparative studies in mosquitoes and D. melanogaster[28, 36]. Putative homologous and orthologous proteins assigned as having stress response and transcription functions were the least conserved (P < 0.05), consistent with the notion of higher evolutionary rates in genes involved in immune response [28, 33, 37, 38], which are a subset of stress response genes. Although the average protein identity observed in the response to stress genes category was lower than the average protein identity in the entire annotated ortholog dataset, such differences were not statistically significant (P >0.05). Also, the average protein identity observed in the response to stress genes category was significantly higher relative to the average protein identity in the entire transcriptome (matching proteins in An. gambiae) (P < 0.5). This may be the result of the high proportion of genes present in our dataset that have no GO annotation and that, not surprisingly, are the least conserved (Figure 6A).

Protein identity according to Gene Ontology. An. albimanus transcriptome was GO annotated using Blast2GO and partitioned according to biological process or molecular function. A) Average percent identity of biological function classes with respect to An. gambiae in the ortholog dataset. Average protein identity of response to stress and transcription genes was significantly lower than cytoskeleton organization and protein transport genes (*** P < 0.001. Kruskal-Wallis test and Dunn’s multiple comparison test). B) Average protein identity of best hit matches with respect to An. gambiae categorized according to molecular function. Average identity of electron carrier function and Peptidases were significantly lower than calcium and actin binding, translation factor and structural function genes (*** P < 0.001. Kruskal-Wallis test and Dunn’s multiple comparison test). For A and B, red lines indicate the average protein identity of the corresponding GO annotated subset. Blue lines indicate overall protein identity.
According to molecular function, the best protein matches, those that were involved in translation, structural functions, calcium and actin binding were the most conserved, which agrees with the initial biological process categorization. The least conserved putative proteins were categorized as electron carrier and proteases, whose conservation was significantly lower than that observed for the translation, actin binding and calcium binding (P < 0.05) ontologies. Proteases were significantly less conserved than the whole set of annotated predicted proteins (Figure 6B). Proteases have been reported as being subjected to positive selection in the Drosophila genus [36]. Also, trypsin family proteases have been significantly expanded in An. gambiae compared to D. melanogaster, suggesting faster evolution in anophelines that may be related to hematophagy [28, 39]. Similarly, electron carrier activity is related to cytochrome function and is overrepresented in our dataset. The cytochrome P450 family, involved in insecticide resistance, is also expanded in An. gambiae[40] and may be also evolving rapidly in New World anophelines.
The transcript dataset was also annotated using InterProScan, which yielded 17,850 InterPro annotations. We noted that 7,154 (42%) transcripts have at least one InterPro annotation with an average of 2.4 annotations per annotated transcript. Annotation distribution was similar to An. gambiae (Table 5). The most abundant annotations, such as zinc-fingers (IPR007087), WD-40 repeat (IPR001680), Protein kinase domain (IPR011009), Armadillo-like fold (IPR016024) and Serine/Cysteine proteases (IPR009003) and the general substrate transporters of the Major facilitator superfamily (IPR016196), were among the top most frequent annotations for both the An. albimanus transcriptome and the An. gambiae predicted proteome [41]. This similarity suggests that in general terms and despite being derived from a limited set of adult tissues, the An. albimanus dataset exhibits a similar representation to that of An. gambiae. However, certain InterPro annotations were more abundant in the An. albimanus dataset than in An. gambiae. For example, the C-terminal-like Glutathione S-transferase (GST) (IPR010987) was ranked in the 26th place versus 79th in An. gambiae and contained almost the complete set (32 out of 38 in An. gambiae). Other highly ranked annotations that contained near-complete expected sets were E2 Ubiquitin-conjugating enzymes (IPR000608) (24 of 26); and Peptidase M1, membrane alanine aminopeptidase, (IPR014782) (20 out of 25 in An. gambiae). Finally, 41 An. albimanus transcripts were annotated as Protein of Unknown Function DUF227 (IPR004119), which ranked among the top 20 most abundant annotated transcripts, whereas in An. gambiae this classification was ranked in the 69th place with 43 annotated proteins. Considering the over-representation of midgut-derived transcripts in our dataset (Figure 1), aminopeptidase enrichment is expected due to their proteolytic role in blood digestion [42]. GSTs and other detoxification-related gene expression is particularly enriched in the anterior midgut of An. gambiae larvae [43], suggesting that detoxification may also be maintained in the adult midgut. E2 ubiquitin conjugating enzymes are part of a general housekeeping homeostatic mechanism [44], and to our knowledge, they do not play a particular role in midgut physiology. However, in D. melanogaster, up- regulation of the ubiquitin-conjugating enzyme homolog coded by vihar and the 26 S proteosomal subunit RPN9 in response to dietary Bowman–Birk inhibitor (BBI) intoxication has been described [45]. It can be speculated that overrepresentation of E2 ubiquitin conjugating enzymes in the mosquito midgut may be part of a general stress response to different xenobiotics.
Structurally, the DUF227 domain overlaps with the SCOP Protein kinase-like (PK-like) superfamily (IPR011009). The presence of DUF227 containing predicted homologs was investigated by querying electronically inferred orthology in the BioMart database [46] and found that DUF227 is present in a family of proteins well represented in dipterans, nematodes, and to a lesser extent in other insects, but absent in other invertebrates such as the sea urchin. The number of DUF227 containing orthologs according to BioMart decreased with phylogenetic distance so that between An. gambiae and Ae. aegypti there were sixteen 1:1 orthologs and between An. gambiae and D. melanogaster or Pediculus humanus there were only nine and five, respectively. Using VectorBase and MozAtlas for microarray data mining to identify significant changes in gene expression (P < 1.E-5) for those An. gambiae genes containing the DUF227 domain, we found predominant expression in the Malpighian tubules, midgut and head of adult mosquitoes, with expression levels higher in males than in females [47]. In the midgut, 34 out of 43 genes had a significantly modified expression pattern after a blood meal [42]. Five genes were enriched in hemocytes and another five were enriched in the carcass [48]. Two genes were induced during P. berghei midgut invasion [49].
Conversely, certain annotations that are abundantly represented in An. gambiae such as Insect cuticle proteins (IPR000618); Tropomyosin (IPR000533); 7TM chemoreceptor (IPR013604); Mitochondrial Rho-like (IPR013684); Olfactory receptor, Drosophila (IPR004117); Pollen allergen Poa pIX/Phl pVI, C-terminal (IPR001778) were substantially scarce in our dataset (Table 5). As expected, olfactory receptors, 7TM chemoreceptors, and pollen allergen were underrepresented protein annotations since they are not expressed in the midgut [47]. Similarly, insect cuticle proteins that are involved in adult cuticle synthesis show a peak during metamorphosis and are under-expressed in adults [50].
Immunity related genes
Mosquito immune responses are thought to play an important role in influencing vectorial competence. Higher protein divergence in genes implicated in stress response (Figure 6) is in agreement with the observation of higher divergence rate in immunity related genes (IRGs) than in genes involved in other core cellular processes [33, 36–38]. Thus, we searched for potential immunity related genes by comparing our transcript sequences to a manually curated dipteran IRG dataset, which includes 385 genes classified into 27 families implicated in recognition, regulation, signal transduction and effector phases of the immune response [38, 51]. We found 413 best BLASTX reciprocal matches between our dataset and the ImmunoDB, representing all 27 families. However, this number may be an overestimation of the true number of IRGs in our dataset, because it includes a large number of structurally homologous proteins that may not be involved in insect immune responses (for example many proteases and tyrosine kinases). To refine our IRG search, we considered a rather conservative approach that contemplated the putative orthology as described in the previous section (Figure 5A), as well as structural signatures derived from the InterPro annotation. Among our 3,772 ortholog 1:1:1 dataset, 79 ortholog groups have matches in the ImmunoDB (Additional file 1) and 73 were consistently supported by InterPro annotation or other protein family classification system. This set was further increased by the inclusion of three unannotated genes of the Toll and Imd pathways (TOLLPATH1 IMDPATH5 and IMDPATH8) with supporting structural evidence based on InterPro annotation. This 82 IRG putative ortholog dataset exhibited 75% average identity to their corresponding An. gambiae proteins, which is very similar to the average identity of all An. albimanus: An. gambiae matches. However, average identity was lower than the average identity of GOslim annotated ortholog proteins (84%).
Gene duplication has been a major evolutionary force shaping immunity related genes in dipterans [38]. Thus, the identification of putative IRGs in our dataset based solely on the inclusion of ortholog groups may be missing putative IRGs. Thus, by considering InterPro annotations, we found representative matches in 25 out of 27 families, representing the recognition, signaling, regulation and effector immune response processes.
Among the ‘recognition phase’ genes, different pathogen recognition molecules were identified such as six Peptidoglycan Recognition Proteins (PGRPs), including the putative PGRP-LD ortholog [52]; three 1-3 β-glucan binding proteins (BGBPs), including the GNBPB2 and GNBPA1 orthologs [53]; six C-type lectins (CTLs), including the CTL6 ortholog [54] and 19 Fibrinogen Related Proteins (FREPs), including the An. gambiae FREP3 ortholog (Locus_25924_Length_958), but not An. gambiae FREP9 ortholog which has been implicated in the anti-Plasmodium response [55] (Figure 7). We further identified six thioester containing proteins (TEPs) based on InterPro annotation (IPR009048) [56] that were missed by the orthology approach. Proteins containing Leucine-Rich Repeats (LRR) are highly abundant in metazoans and are involved in molecular recognition in a wide variety of biological processes. As mentioned in the previous section, we identified 38 transcripts containing LRR domains (IPR001611). However, mosquitoes possess a unique type of LRR-domain among proteins that are involved in immune responses [57]. Apart from the LRR, these proteins share structural features including a conserved pattern of cysteine residues and coiled-coil domains. We found one putative Leucine Rich Repeat immune protein (LRIM) that displays structural features of the Long LRIM subfamily and two that show compatible features with the short LRIM subfamily, including the LRIM6 putative ortholog (Locus_11561_Length_1221) [57].

Genome de-linked annotation viewer. Screen shots of the An. albimanus transcript Locus_25924_Length_958 corresponding to the FREP3 ortholog. A) Sequence pane. B) Alignment pane. An exonerate alignment to the An. gambiae reference genome can be displayed via the Distributed Annotation System (DAS) in the VectorBase genome browser, by following the link. The lower panes correspond to annotations which include InterPro annotation (C), and BLASTX comparisons (D) to An. gambiae, An. darlingi, D. melanogaster, Ae. aegypti, C. quinquefasciatus, I. scapularis and P. humanus. For A, C and D only partial information is shown.
The major immune response signal transduction pathways in dipterans are the Toll and the Imd pathways. Gene members of these pathways tend to be better conserved in different mosquito species than genes implicated in the recognition or effector phases [38, 58]. Although transcripts containing LRR or Toll/interleukin-1 receptor homology (TIR) domain (IPR000157) were found, a clear Toll receptor homolog was not. However, we found important members of the Toll pathway such as PELLE MYD88 and TRAF6; the Imd pathway such as CASPAR IKK1 and IKK2[38] and STAT pathway such as DOME SOCS and STAT2[59, 60] (Additional file 1), as well as REL2 transcription factor [61].
Autophagy was originally described as a cellular response to starvation. However, it has recently been shown to be a critical process related to immune and stress responses, clearance of intracellular pathogens and damaged organelles, as well as cell survival. There are several genes involved in the induction of autophagy, autophagosome nucleation, autophagosome expansion and autophagosome recycling [62]. There are 20 An. gambiae entries in the ImmunoDB related to autophagy. Given the importance of all the mentioned processes in malaria transmission, we searched the ImmunoDB for orthologs and found 12. In the induction of the pathway, only the TOR homolog was found. Similarly, the only homolog involved in the nucleation phase that we found was the BCL-2 homolog DEBCL (buffy in D. melanogaster) [63]. The remaining ten corresponded to genes involved only in autophagosome expansion such as APG3 APG4A APG4B APG7A APG7B APG8 and APG16 orthologs or autophagosome recycling such as APG2 APG9 and APG18 (Additional file 1). In contrast, we found a relative depletion of caspases (only CASPS7 and CASPL1, of 14 caspases in An. gambiae) and the absence of caspase activators (AKR and Michelob_x), suggesting that, as in larvae midgut [64], autophagy could be a more active homeostatic tissue process than apoptosis in adult mosquitoes.
Conclusions
We have explored the adult female transcriptome of an important New World malaria vector, An. albimanus, by sequencing cDNA libraries generated from different tissue sources related to the Plasmodium life cycle such as midgut, cuticular fat body, dorsal vessel and salivary gland. Merging Sanger and NGS data into a single assembly generated a robust dataset with adequate transcript lengths that could be effectively mapped to the An. darlingi genome, covered 90% of the core eukaryotic genome and half of the predicted proteome of other mosquito vectors. We identified protein-coding transcripts involved in biological processes such as immune recognition, immune signaling pathways, insecticide resistance and autophagy that may be relevant to the Plasmodium cycle or may represent targets for novel control strategies. As a result of this work, the genomic information available for An. albimanus has increased several hundred-fold, thus providing molecular inputs for research in this species: 1) from a single gene perspective; 2) to gain insight into the anopheline radiations in the New World; 3) facilitating further genomic and proteomic approaches; and 4) assisting gene finding and validation of the An. albimanus genome in the context of the Anopheles cluster genome sequencing project [2]. Sequence information, predicted proteome comparisons, transcript mapping to the An. gambiae genome and InterPro annotations described in this manuscript are accessible to the community through the VectorBase website (http://www.vectorbase.org/Other/AdditionalOrganisms/).
Materials and methods
Mosquitoes and mosquito infections with P. vivax
All the mosquito samples used in the present work were 3-5 day post-emergence female An. albimanus of the white-stripe strain [65] obtained from the insectary of the National Institute of Public Health (INSP) in Cuernavaca, México. Mosquitoes were fed with 10% sucrose ad libitum and reared in a 12:12 h light cycle maintained at 28 °C and 80% relative humidity.
Mosquito infections with P. vivax CSP-VK210 and extraction of midgut epithelium of 24 h and seven days after an infectious blood meal were performed as described [66] in the insectary of the Centro Regional de Investigación en Salud Pública (CRISP) in Tapachula, Chiapas, México, according to Institute ethical guidelines and approval.
cDNA libraries for Sanger sequencing
To capture transcripts from mosquito organs that are relevant for the Plasmodium sp life cycle, three cDNA libraries were generated for Sanger sequencing: A sucrose-fed female midgut (site of invasion) and a salivary gland (site of sporozoite maturation and inoculation to vertebrate host) cDNA libraries were constructed from 5 μg of total RNA. cDNA was ligated to Uni-ZAP XR vector (Stratagene). The phage library was mass excised and plated into LB-agar plates. Individual colonies were replicated in 384-well plates. A third cDNA library was constructed from 0.5 μg of Poly A + RNA extracted from whole female An. albimanus 12 h after inoculation with 0.0004 OD of Serratia marcescens in the hemocoel, which elicits an immune response that limits the development of P. vivax[67]. cDNA synthesis and library construction was done using the Creator SMART cDNA Library Construction Kit (Clontech) according to the manufacturer instructions. The library was transformed in Escherichia coli by electroporation and plated in LB-Agar with chloramphenicol (30 μg/mL). Individual colonies were replicated in 384-well plates.
Template preparation and Sanger sequencing
E. coli clones were inoculated in CIRCLEGROW® (Krackler scientific) liquid media with either chloramphenicol (pDNA-lib) or ampicilin (pBluescript) at 37 °C in 96-well plates for 16 h. Plasmid DNA was prepared by alkaline lysis in Millipore filters and ethanol-precipitated and suspended in sterile deionized water. Sequencing was performed with fluorescent dye terminators in a 3100 Genetic Analyzer (Applied Biosystems).
EST processing
Raw chromatogram files were quality assessed and trimmed with Phred using the trim_alt command with default parameters [68] and then converted to FASTA files using PH2FASTA. Vector sequence and linker sequences were removed using CrossMatch [69] and SeqClean [70]. Identification of mitochondrial and ribosomal protein transcripts was done by BLASTn searches to mosquito mitochondrial genomes or ribosomal protein sequence databases and filtered out. ESTs were submitted to dbEST at NCBI (Accession Numbers EV406110.1 - EV410194.1).
454 sequencing
Abdominal cuticles and the underlying fat body were obtained from 20 female adult mosquitoes and total RNA was extracted with TRIzol (Life Technologies). Integrity of RNA was verified in the Agilent Bioanalyzer standard RNA chip. One μg of RNA was used for full length RT-PCR amplification using the Super-SMART PCR cDNA synthesis kit (Clontech) according to the manufacturer’s instructions. The PCR amplified cDNA library was fragmented by nebulization and subjected to library preparation according to the 454 shotgun sequencing protocol. After emulsion PCR titration and amplification, the library was sequenced in a full picotiter plate using the Genome Sequencer FLX platform. A similar approach was used to generate additional midgut libraries from P. vivax infected-mosquitoes at 24 h post-infective blood meal (PIBM) and seven days PIBM, but sequenced as pooled bar-coded libraries in half picotiter plate. A third sequencing 454 run was performed with two cDNA libraries from dissected dorsal vessels, obtained at 18 h post-inoculation (intra-hemocoelic) with 0.25 μl of soluble fraction of zymosan (10 μg glucose-equivalents/ml, Sigma) as described [71] and saline-inoculated mosquitoes respectively. Dorsal vessels were collected in RNAlater (Ambion) and stored at -80°C. After RNAlater removal, total RNA was extracted with the RNAeasy Kit (Qiagen) and amplified with the SMARTER Pico PCR cDNA synthesis Kit (Clontech), and sequenced in a full picotiter plate (one region per library) using the GS FLX Titanium platform. Primer adaptors used for cDNA library generation were trimmed after signal processing using SeqClean. 454 sequence data was submitted to the Sequence Read Archive (SRA) (Accession number: SRA052091).
Illumina sequencing
Total RNA from 50 An. albimanus midguts was extracted using TRIzol, DNased and cleaned with an RNAeasy column (Qiagen) according to the manufacturer’s instructions. Total RNA was then quality controlled for integrity on a Bioanalyzer (Agilent Technologies). mRNA libraries were constructed and sequenced, as previously described [72–74] on a single lane of a Illumina HiSeq 2000, which generated ~210 million 101 bp paired end reads. Illumina sequence data was submitted to the Sequence Read Archive (SRA) (Accession number: SRA051893).
Assembly
The entire Illumina read set was split into eight equal sized read sets. Each one of these Illumina read sets was merged with the 454 and Sanger data and assembled using the Velvet [75] and Oases [76] software packages using three different kmer sizes (43, 45 and 47). The resulting contigs for each assembly were run again through Velvet and Oases to produce a final assembly. We then filtered the final assembly to retain only those loci that contained a single transcript, that were longer than 300 bp, and that had confidence scores of 1.0. To address which contigs contained 454 or Sanger reads, all 454 and Sanger reads were re-mapped to the initial assembly using the GS Reference Mapper v.2.5.3 using default parameters. Unmapped reads were re-assembled using the GS assembler v2.5.3 on cDNA mode to yield an additional set of 935 contigs.
Genome mapping
The An. gambiae (AgamP3) [77] and An. darlingi genomes [18] were softmasked with RepeatMasker [78]. An. albimanus transcripts were aligned to either genome using Exonerate v. 2.2 [79] with the EST2 Genome mode, and a threshold score of 300, and maximum intron length of 20,000 bp.
Transcript annotation
Gene ontology annotations were performed using Blast2Go [34]. For the Initial BLASTX against the NCBI-nr database the command-line option “-e1-e-6” was used. Additionally, transcripts were annotated according to the InterPro databases using InterProScan [35] in six-frame translation mode. Kruskal-Wallis test followed by Dunn’s correction was performed to calculate statistical differences within GO classes and protein percent identity with the Graph Pad PRISM software.
In silico proteome comparison
The entire assembled transcript dataset was used to search for the best hit homologous proteins (BLASTX cut-off e-value 1.0E-5) in the An. gambiae (AgamP3.6), Ae. aegypti (AaegL1.2); Culex quinquefasciatus (CpipJ1.2), P. humanus (PhumU1.2) and Ixodes scapularis (IscaW1.1) predicted proteomes present at VectorBase [77], as well as the An. darlingi[18] and D. melanogaster[80] proteomes. Ortholog prediction was done by performing BLASTX and TBLASTN bidirectional comparisons between An. albimanus An. darlingi and An. gambiae (e value 1.0E-5) to identify the best reciprocal hits within the three species.
To identify the proportion of the core eukaryotic genome covered by the An. albimanus transcriptome, we used HMM profiles corresponding to the 458 core eukaryotic proteins as provided by the CEGMA algorithm [32]. Local HMMER3 searches [81] were calibrated using the An. gambiae core eukaryotic protein validated dataset consisting of 453 sequences [82]. HMMER3 was performed using hmmscan command and the “-T 40” and “--domT 40” filters against the An. albimanus predicted proteome, as well as the predicted proteomes of An. darlingi An. gambiae Ae. aegypti C. quinquefasciatus and D. melanogaster.
Web interface
Many web-based genome browsers [83–85] are available as open source software and are well suited to displaying transcript annotations. However they are heavily dependent on the availability of a genome sequence to act as a coordinate system. It is possible to adapt genome browsers to work without genomes [86] but it is not easy to keep them synchronized with developments in the “parent” software. In this work, we chose to develop a standalone “genome-free” web application, called GDAV (Genome-Delinked Annotation Viewer). It was designed with flexibility in mind; it can handle any kind of sequence annotations and integrates with genome browsers of closely related species via the DAS protocol [87]. The free open source code is available at https://github.com/VectorBase/GDAV and the software was developed within the auspices of VectorBase [77].
The key to GDAV’s flexibility is its use of three simple, open text file formats for loading data: FASTA for sequences, tab-delimited files for annotations and GFF3 for genome alignments. Only the loading of one or more FASTA files is mandatory, thereafter zero or more annotation and alignment files may be loaded into GDAV’s small MySQL schema using the supplied Perl scripts. The annotation file consists of rows of data identified by the sequence ID in the first column, and subsequent named columns providing arbitrary text annotations. The Java-based web interface is simple to deploy within a Java web server such as Apache Tomcat. The web interface, with its integrated search facility, treats all annotations as plain text—no special treatment of numeric data (e.g. range queries or unit conversions) is provided. Link-outs to third party databases from specific columns containing suitable IDs are possible through the configuration file. A Java-based DAS server based on Dazzle [88] is bundled with GDAV. It can be used to display GFF3 file-derived gapped alignments (e.g. exon-intron structure) of the sequences stored in GDAV with respect to the genomes of one or more closely related species. Any alignment features shown via DAS in genome browsers link back to the sequence report page in GDAV.
In this study, the annotation files loaded into the system include the InterPro domain assignments and the BLASTX results providing “best hits” to several other proteomes. The GFF3-format exonerate alignments described above were also loaded into the system.
Author contributions
JMB conceived the study, participated in the generation of some cDNA libraries, performed data analysis, helped in designing web application specification and drafted the manuscript. MHR, RRD, AR and CAM contributed to study conception and design and helped drafting the manuscript. JGG performed transcriptome assembly. RMM performed InterPro annotation and in conjunction with SNR designed the web application specification and developed the web application, database and curation tools. CU, CAM and DEG participated in data analysis and interpretation. REG, JTS, MOM, LGC, SHM and FCR participated in mosquito experiments design, cDNA library generation, 454 and Sanger sequencing and data analysis. All authors read and approved the final manuscript.
Abbreviations
- BGBP:
-
β-glucan Binding Proteins
- CEG:
-
Core Eukaryotic Gene
- CTL’s:
-
C-Type Lectin
- DAS:
-
Distributed Annotation System
- FREP:
-
Fibrinogen Related Protein
- GDAV:
-
Genome De-linked Annotation Viewer
- GO:
-
Gene Ontology
- GST:
-
Glutathion-S-Transferase
- HSP:
-
High Scoring Pair
- IRG’s:
-
Immune Response Genes
- LRIM:
-
Leucine Rich Repeat Immune Protein
- LRR:
-
Leucine Rich Repeat
- My:
-
Million Years
- NGS:
-
Next Generation Sequencing
- PGRP:
-
Peptidoglycan Recognition Protein
- UTR:
-
Untranslated Region.
References
Collins FH, Paskewitz SM: Malaria: Current and future prospects for control. Ann rev entomol. 1995, 40 (1): 195-219. 10.1146/annurev.en.40.010195.001211.
Besansky NJ:http://www.vectorbase.org/Help/Anopheles_species_cluster_white_paper#tab=Document,
Cohuet A, Harris C, Robert V, Fontenille D: Evolutionary forces on Anopheles: what makes a malaria vector?. Trends Parasitol. 2010, 26 (3): 130-136. 10.1016/j.pt.2009.12.001.
Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan M-S, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DMA: Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002, 419 (6906): 498-511. 10.1038/nature01097.
Holt RA, Subramanian GM, Halpern A, Sutton GG, Charlab R, Nusskern DR, Wincker P, Clark AG, Ribeiro JC, Wides R, Salzberg SL, Loftus B, Yandell M, Majoros WH, Rusch DB, Lai Z, Kraft CL, Abril JF, Anthouard V, Arensburger P, Atkinson PW, Baden H, de Berardinis V, Baldwin D, Benes V, Biedler J, Blass C, Bolanos R, Boscus D, Barnstead M: The genome sequence of the malaria mosquito Anopheles gambiae. Science. 2002, 298 (5591): 129-149. 10.1126/science.1076181.
Cirimotich CM, Dong Y, Garver LS, Sim S, Dimopoulos G: Mosquito immune defenses against Plasmodium infection. Dev Comp Immunol. 2010, 34 (4): 387-395. 10.1016/j.dci.2009.12.005.
Marois E: The multifaceted mosquito anti-Plasmodium response. Curr Opin Microbiol. 2011, 14 (4): 429-435. 10.1016/j.mib.2011.07.016.
White BJ, Lawniczak MKN, Cheng C, Coulibaly MB, Wilson MD, Sagnon NF, Costantini C, Simard F, Christophides GK, Besansky NJ: Adaptive divergence between incipient species of Anopheles gambiae increases resistance to Plasmodium. Proc Natl Acad Sci. 2011, 108 (1): 244-249. 10.1073/pnas.1013648108.
The malERA Consultative Group on Basic Science Enabling Technologies: A research agenda for malaria eradication: Basic science and enabling technologies. PLoS Med. 2011, 8 (1): e1000399-
Dinglasan RR, Jacobs-Lorena M: Flipping the paradigm on malaria transmission-blocking vaccines. Trends Parasitol. 2008, 24 (8): 364-370. 10.1016/j.pt.2008.05.002.
Parish LA, Colquhoun DR, Ubaida Mohien C, Lyashkov AE, Graham DR, Dinglasan RR: Ookinete-interacting proteins on the microvillar surface are partitioned into detergent resistant membranes of Anopheles gambiae midguts. J prot res. 2011, 10 (11): 5150-5162. 10.1021/pr2006268.
An C, Budd A, Kanost MR, Michel K: Characterization of a regulatory unit that controls melanization and affects longevity of mosquitoes. Cell mol life sci CMLS. 2011, 68 (11): 1929-1939. 10.1007/s00018-010-0543-z.
Carlton JM, Sina BJ, Adams JH: Why is Plasmodium vivax a neglected tropical disease?. PLoS Negl Trop Dis. 2011, 5 (6):
Mueller I, Galinski MR, Baird JK, Carlton JM, Kochar DK, Alonso PL, del Portillo HA: Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet Infect Dis. 2009, 9 (9): 555-566. 10.1016/S1473-3099(09)70177-X.
Sinka ME, Rubio-Palis Y, Manguin S, Patil AP, Temperley WH, Gething PW, Van Boeckel T, Kabaria CW, Harbach RE, Hay SI: The dominant Anopheles vectors of human malaria in the Americas: occurrence data, distribution maps and bionomic precis. Parasites & vectors. 2010, 3: 72-10.1186/1756-3305-3-72.
Zimmerman RH: Ecology of malaria vectors in the Americas and future direction. Memórias do Instituto Oswaldo Cruz. 1992, 87: 371-383.
Krzywinski J, Besansky NJ: Molecular systematics of Anopheles: from subgenera to subpopulations. Ann rev entomol. 2003, 48: 111-139. 10.1146/annurev.ento.48.091801.112647.
Anopheles darlingi genome:http://www.ncbi.nlm.nih.gov/genome/2624,
Ghosh AK, Jacobs-Lorena M: Plasmodium sporozoite invasion of the mosquito salivary gland. Curr Opin Microbiol. 2009, 12 (4): 394-400. 10.1016/j.mib.2009.06.010.
Kumar S, Christophides GK, Cantera R, Charles B, Han YS, Meister S, Dimopoulos G, Kafatos FC, Barillas-Mury C: The role of reactive oxygen species on Plasmodium melanotic encapsulation in Anopheles gambiae. Proc Natl Acad Sci U S A. 2003, 100 (24): 14139-14144. 10.1073/pnas.2036262100.
Crawford JE, Guelbeogo WM, Sanou A, Traore A, Vernick KD, Sagnon N, Lazzaro BP: De novo transcriptome sequencing in Anopheles funestus using Illumina RNA-seq technology. PLoS One. 2010, 5 (12): e14202-10.1371/journal.pone.0014202.
Sandmann T, Vogg MC, Owlarn S, Boutros M, Bartscherer K: The head-regeneration transcriptome of the planarian Schmidtea mediterranea. Genome Biol. 2011, 12 (8): R76-10.1186/gb-2011-12-8-r76.
Adamidi C, Wang Y, Gruen D, Mastrobuoni G, You X, Tolle D, Dodt M, Mackowiak SD, Gogol-Doering A, Oenal P, Rybak A, Ross E, Sanchez Alvarado A, Kempa S, Dieterich C, Rajewsky N, Chen W: De novo assembly and validation of planaria transcriptome by massive parallel sequencing and shotgun proteomics. Genome Res. 2011, 21 (7): 1193-1200. 10.1101/gr.113779.110.
Poelchau MF, Reynolds JA, Denlinger DL, Elsik CG, Armbruster PA: A de novo transcriptome of the Asian tiger mosquito, Aedes albopictus, to identify candidate transcripts for diapause preparation. BMC genomics. 2011, 12: 619-10.1186/1471-2164-12-619.
Li J, Riehle MM, Zhang Y, Xu J, Oduol F, Gomez SM, Eiglmeier K, Ueberheide BM, Shabanowitz J, Hunt DF, Ribeiro JM, Vernick KD: Anopheles gambiae genome reannotation through synthesis of ab initio and comparative gene prediction algorithms. Genome Biol. 2006, 7 (3): R24-10.1186/gb-2006-7-3-r24.
Sharakhova MV, Hammond MP, Lobo NF, Krzywinski J, Unger MF, Hillenmeyer ME, Bruggner RV, Birney E, Collins FH: Update of the Anopheles gambiae PEST genome assembly. Genome Biol. 2007, 8 (1): R5-10.1186/gb-2007-8-1-r5.
Vectorbase Anopheles gambiae genome project description:http://www.vectorbase.org/Anopheles_gambiae/Info/Index,
Zdobnov EM, von Mering C, Letunic I, Torrents D, Suyama M, Copley RR, Christophides GK, Thomasova D, Holt RA, Subramanian GM, Mueller H-M, Dimopoulos G, Law JH, Wells MA, Birney E, Charlab R, Halpern AL, Kokoza E, Kraft CL, Lai Z, Lewis S, Louis C, Barillas-Mury C, Nusskern D, Rubin GM, Salzberg SL, Sutton GG, Topalis P, Wides R, Wincker P: Comparative genome and proteome analysis of Anopheles gambiae and Drosophila melanogaster. Science. 2002, 298 (5591): 149-159. 10.1126/science.1077061.
Keren H, Lev-Maor G, Ast G: Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet. 2010, 11 (5): 345-355. 10.1038/nrg2776.
Malko DB, Makeev VJ, Mironov AA, Gelfand MS: Evolution of exon-intron structure and alternative splicing in fruit flies and malarial mosquito genomes. Genome Res. 2006, 16 (4): 505-509. 10.1101/gr.4236606.
Nagano T, Fraser P: No-nonsense functions for long noncoding RNAs. Cell. 2011, 145 (2): 178-181. 10.1016/j.cell.2011.03.014.
Parra G, Bradnam K, Korf I: CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007, 23 (9): 1061-1067. 10.1093/bioinformatics/btm071.
Heger A, Ponting CP: Evolutionary rate analyses of orthologs and paralogs from 12 Drosophila genomes. Genome Res. 2007, 17 (12): 1837-1849. 10.1101/gr.6249707.
Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A: High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36 (10): 3420-3435. 10.1093/nar/gkn176.
Zdobnov EM, Apweiler R: InterProScan – an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001, 17 (9): 847-848. 10.1093/bioinformatics/17.9.847.
Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA, Kaufman TC, Kellis M, Gelbart W, Iyer VN, Pollard DA, Sackton TB, Larracuente AM, Singh ND, Abad JP, Abt DN, Adryan B, Aguade M, Akashi H, Anderson WW, Aquadro CF, Ardell DH, Arguello R, Artieri CG, Barbash DA, Barker D, Barsanti P, Batterham P, Batzoglou S, Begun D: Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007, 450 (7167): 203-218. 10.1038/nature06341.
Sackton TB, Lazzaro BP, Schlenke TA, Evans JD, Hultmark D, Clark AG: Dynamic evolution of the innate immune system in Drosophila. Nat Genet. 2007, 39 (12): 1461-1468. 10.1038/ng.2007.60.
Waterhouse RM, Kriventseva EV, Meister S, Xi Z, Alvarez KS, Bartholomay LC, Barillas-Mury C, Bian G, Blandin S, Christensen BM, Dong Y, Jiang H, Kanost MR, Koutsos AC, Levashina EA, Li J, Ligoxygakis P, Maccallum RM, Mayhew GF, Mendes A, Michel K, Osta MA, Paskewitz S, Shin SW, Vlachou D, Wang L, Wei W, Zheng L, Zou Z, Severson DW: Evolutionary dynamics of immune-related genes and pathways in disease-vector mosquitoes. Science. 2007, 316 (5832): 1738-1743. 10.1126/science.1139862.
Wu DD, Wang GD, Irwin DM, Zhang YP: A profound role for the expansion of trypsin-like serine protease family in the evolution of hematophagy in mosquito. Mol Biol Evol. 2009, 26 (10): 2333-2341. 10.1093/molbev/msp139.
Ranson H, Claudianos C, Ortelli F, Abgrall C, Hemingway J, Sharakhova MV, Unger MF, Collins FH, Feyereisen R: Evolution of supergene families associated with insecticide resistance. Science. 2002, 298 (5591): 179-181. 10.1126/science.1076781.
VectorBase release VB-2011-12: Anopheles gambiae (AgamP3) Top 500 InterPro hits.http://www.vectorbase.org/Anopheles_gambiae/Info/IPtop500?db=core,
Marinotti O, Nguyen QK, Calvo E, James AA, Ribeiro JM: Microarray analysis of genes showing variable expression following a blood meal in Anopheles gambiae. Insect mol biol. 2005, 14 (4): 365-373. 10.1111/j.1365-2583.2005.00567.x.
Neira Oviedo M, VanEkeris L, Corena-Mcleod MDP, Linser PJ: A microarray-based analysis of transcriptional compartmentalization in the alimentary canal of Anopheles gambiae (Diptera: Culicidae) larvae. Insect mol biol. 2008, 17 (1): 61-72. 10.1111/j.1365-2583.2008.00779.x.
van Wijk SJ, Timmers HT: The family of ubiquitin-conjugating enzymes (E2s): deciding between life and death of proteins. FASEB j official publ Federation Am Soc Experiment Biol. 2010, 24 (4): 981-993. 10.1096/fj.09-136259.
Li HM, Margam V, Muir WM, Murdock LL, Pittendrigh BR: Changes in Drosophila melanogaster midgut proteins in response to dietary Bowman-Birk inhibitor. Insect mol biol. 2007, 16 (5): 539-549.
Guberman JM, Ai J, Arnaiz O, Baran J, Blake A, Baldock R, Chelala C, Croft D, Cros A, Cutts RJ, Di Génova A, Forbes S, Fujisawa T, Gadaleta E, Goodstein DM, Gundem G, Haggarty B, Haider S, Hall M, Harris T, Haw R, Hu S, Hubbard S, Hsu J, Iyer V, Jones P, Katayama T, Kinsella R, Kong L, Lawson D: BioMart Central Portal: an open database network for the biological community. Database (Oxford). 2011, 18: bar041-
Baker DA, Nolan T, Fischer B, Pinder A, Crisanti A, Russell S: A comprehensive gene expression atlas of sex- and tissue-specificity in the malaria vector. Anopheles gambiae. BMC genomics. 2011, 12: 296-10.1186/1471-2164-12-296.
Pinto SB, Lombardo F, Koutsos AC, Waterhouse RM, McKay K, An C, Ramakrishnan C, Kafatos FC, Michel K: Discovery of Plasmodium modulators by genome-wide analysis of circulating hemocytes in Anopheles gambiae. Proc Natl Acad Sci U S A. 2009, 106 (50): 21270-21275. 10.1073/pnas.0909463106.
Vlachou D, Schlegelmilch T, Christophides GK, Kafatos FC: Functional genomic analysis of midgut epithelial responses in Anopheles during Plasmodium Invasion. Curr Biol. 2005, 15 (13): 1185-1195. 10.1016/j.cub.2005.06.044.
Koutsos AC, Blass C, Meister S, Schmidt S, MacCallum RM, Soares MB, Collins FH, Benes V, Zdobnov E, Kafatos FC, Christophides GK: Life cycle transcriptome of the malaria mosquito Anopheles gambiae and comparison with the fruitfly Drosophila melanogaster. Proc Natl Acad Sci U S A. 2007, 104 (27): 11304-11309. 10.1073/pnas.0703988104.
Immuno DB: Insect immune-related genes and gene families.http://cegg.unige.ch/Insecta/immunodb,
Obbard DJ, Welch JJ, Kim K-W, Jiggins FM: Quantifying adaptive evolution in the Drosophila immune system. PLoS Genet. 2009, 10: e1000698-
Warr E, Das S, Dong Y, Dimopoulos G: The Gram-negative bacteria-binding protein gene family: Its role in the innate immune system of Anopheles gambiae and in anti-Plasmodium defence. Insect mol biol. 2008, 17 (1): 39-51. 10.1111/j.1365-2583.2008.00778.x.
Schnitger AK, Yassine H, Kafatos FC, Osta MA: Two C-type lectins cooperate to defend Anopheles gambiae against Gram-negative bacteria. J Biol Chem. 2009, 284 (26): 17616-17624. 10.1074/jbc.M808298200.
Dong Y, Dimopoulos G: Anopheles fibrinogen-related proteins provide expanded pattern recognition capacity against bacteria and malaria parasites. J Biol Chem. 2009, 284 (15): 9835-9844. 10.1074/jbc.M807084200.
Blandin SA, Marois E, Levashina EA: Antimalarial responses in Anopheles gambiae: from a complement-like protein to a complement-like pathway. Cell host & microbe. 2008, 3 (6): 364-374. 10.1016/j.chom.2008.05.007.
Waterhouse RM, Povelones M, Christophides GK: Sequence-structure-function relations of the mosquito leucine-rich repeat immune proteins. BMC genomics. 2010, 11: 531-10.1186/1471-2164-11-531.
Christophides GK, Zdobnov E, Barillas-Mury C, Birney E, Blandin S, Blass C, Brey PT, Collins FH, Danielli A, Dimopoulos G, Hetru C, Hoa NT, Hoffmann JA, Kanzok SM, Letunic I, Levashina EA, Loukeris TG, Lycett G, Meister S, Michel K, Moita LF, Müller H-M, Osta MA, Paskewitz SM, Reichhart J-M, Rzhetsky A, Troxler L, Vernick KD, Vlachou D, Volz J: Immunity-related genes and gene families in Anopheles gambiae. Science. 2002, 298 (5591): 159-165. 10.1126/science.1077136.
Gupta L, Molina-Cruz A, Kumar S, Rodrigues J, Dixit R, Zamora RE, Barillas-Mury C: The STAT pathway mediates late-phase immunity against Plasmodium in the mosquito Anopheles gambiae. Cell host & microbe. 2009, 5 (5): 498-507. 10.1016/j.chom.2009.04.003.
Stec WJ, Zeidler MP: Drosophila SOCS proteins. J signal trans. 2011, 2011: 894510-
Meister S, Kanzok SM, Zheng XL, Luna C, Li TR, Hoa NT, Clayton JR, White KP, Kafatos FC, Christophides GK, Zheng L: Immune signaling pathways regulating bacterial and malaria parasite infection of the mosquito Anopheles gambiae. Proc Natl Acad Sci U S A. 2005, 102 (32): 11420-11425. 10.1073/pnas.0504950102.
Chang YY, Neufeld TP: Autophagy takes flight in Drosophila. FEBS Lett. 2010, 584 (7): 1342-1349. 10.1016/j.febslet.2010.01.006.
Hou Y-CC, Chittaranjan S, Barbosa SG, McCall K, Gorski SM: Effector caspase Dcp-1 and IAP protein Bruce regulate starvation-induced autophagy during Drosophila melanogaster oogenesis. J Cell Biol. 2008, 182 (6): 1127-1139. 10.1083/jcb.200712091.
Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, Kumar S: Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009, 19 (20): 1741-1746. 10.1016/j.cub.2009.08.042.
Chan AS RM, Torres JA, Rodríguez MC, Villarreal C: Susceptibility of three laboratory strains of Anopheles albimanus (Diptera: Culicidae) to coindigenous Plasmodium vivax in southern México. J Med Entomol. 1994, 31 (3): 4-
Gonzalez-Ceron L, Rodriguez MH, Santillan F, Chavez B, Nettel JA, Hernandez-Avila JE, Kain KC: Plasmodium vivax: Ookinete destruction and oocyst development arrest are responsible for Anopheles albimanus resistance to circumsporozoite phenotype VK247 parasites. Exp Parasitol. 2001, 98 (3): 152-161. 10.1006/expr.2001.4626.
Gonzalez-Ceron L, Santillan F, Rodriguez MH, Mendez D, Hernandez-Avila JE: Bacteria in midguts of field-collected Anopheles albimanus block Plasmodium vivax sporogonic development. J Med Entomol. 2003, 40 (3): 371-374. 10.1603/0022-2585-40.3.371.
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using Phred.I. Accuracy Assessment. Genome Res. 1998, 8 (3): 175-185.
Phred, Phrap, Consed:http://www.phrap.org/phredphrapconsed.html,
DFCI Gene Indices Software Tools:http://compbio.dfci.harvard.edu/tgi/software/,
Hernández-Martínez S, Román-Martínez U, Martínez-Barnetche J, Garrido E, Rodríguez MH, Lanz-Mendoza H: Induction of DNA synthesis in Anopheles albimanus tissue cultures in response to a Saccharomyces cerevisiae challenge. Arch Insect Biochem Physiol. 2006, 63 (4): 12-
Gibbons JG, Beauvais A, Beau R, McGary KL, Latgé J-P, Rokas A: Global transcriptome changes underlying colony growth in the opportunistic human pathogen Aspergillus fumigatus. Eukaryotic Cell. 2012, 11 (1): 68-78. 10.1128/EC.05102-11.
Gibbons JG, Janson EM, Hittinger CT, Johnston M, Abbot P, Rokas A: Benchmarking next-generation transcriptome sequencing for functional and evolutionary genomics. Mol Biol Evol. 2009, 26 (12): 2731-2744. 10.1093/molbev/msp188.
Hittinger CT, Johnston M, Tossberg JT, Rokas A: Leveraging skewed transcript abundance by RNA-Seq to increase the genomic depth of the tree of life. Proc Natl Acad Sci U S A. 2010, 107 (4): 1476-1481. 10.1073/pnas.0910449107.
Zerbino DR, Birney E: Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18 (5): 821-829. 10.1101/gr.074492.107.
Zerbino D, Oases : De novo transcriptome assembler for very short reads.http://www.ebi.ac.uk/~zerbino/oases/,
Megy K, Emrich SJ, Lawson D, Campbell D, Dialynas E, Hughes DST, Koscielny G, Louis C, MacCallum RM, Redmond SN, Sheehan A, Topalis P, Wilson D, VectorBase Consortium: VectorBase: improvements to a bioinformatics resource for invertebrate vector genomics. Nucleic Acids Res. 2011, 40: D729-
RepeatMasker Web Server:http://www.repeatmasker.org/cgi-bin/WEBRepeatMasker,
Slater GS, Birney E: Automated generation of heuristics for biological sequence comparison. BMC Bioinforma. 2005, 6:
FlyBase: A database of Drosophila genes and genomes:http://flybase.org/,
HMMER: biosequence analysis using profile hidden Markov models.http://hmmer.janelia.org/,
CEGMA: Ian Korf Lab. Genome Center UCDavis.http://korflab.ucdavis.edu/Datasets/cegma/index.html,
Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, Gil L, Gordon L, Hendrix M, Hourlier T, Johnson N, Kähäri AK, Keefe D, Keenan S, Kinsella R, Komorowska M, Koscielny G, Kulesha E, Larsson P, Longden I, McLaren W, Muffato M, Overduin B, Pignatelli M, Pritchard B, Riat HS: Ensembl 2012. Nucleic Acids Res. 2011, 40: D84-
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler AD: The Human Genome Browser at UCSC. Genome Res. 2002, 12 (6): 996-1006.
Podicheti R, Gollapudi R, Dong Q: WebGBrowse–a web server for GBrowse. Bioinformatics. 2009, 25 (12): 1550-1551. 10.1093/bioinformatics/btp239.
Ueno S, Le Provost G, Leger V, Klopp C, Noirot C, Frigerio JM, Salin F, Salse J, Abrouk M, Murat F, Brendel O, Derory J, Abadie P, Leger P, Cabane C, Barre A, de Daruvar A, Couloux A, Wincker P, Reviron MP, Kremer A, Plomion C: Bioinformatic analysis of ESTs collected by Sanger and pyrosequencing methods for a keystone forest tree species: oak. BMC genomics. 2010, 11: 650-10.1186/1471-2164-11-650.
Prlic A, Down TA, Kulesha E, Finn RD, Kahari A, Hubbard TJ: Integrating sequence and structural biology with DAS. BMC Bioinforma. 2007, 8: 333-10.1186/1471-2105-8-333.
Dazzle:http://biojava.org/wiki/Dazzle,
Acknowledgements
We thank Gabriel Hernandez of ROCHE technical support for help with 454 sequencing; Travis Clark and Chelsea Baker of the Vanderbilt Genome Technology Core for help with Illumina sequencing; Bruno Arce for SQL database support, Bernardo Cortina and Kelly Scoggins for manuscript assistance; and Fidel de la Cruz Hernandez for critically reviewing the manuscript. The authors are very grateful to Imperial College London for high performance computing facilities. We also thank the anonymous reviewers for their suggestions improving the manuscript. This investigation received financial support from the UNICEF/UNDP/WORLD BANK/WHO Special Programme for Research and Training in Tropical Diseases (TDR), project ID: A50256 (for JMB) and CONACyT-SEP-2007SEP-62389 (for MHR); from the Bloomberg Family Foundation and the Johns Hopkins Malaria Research Institute (JHMRI) (for RRD) and grant HHSN268201000032C (N01-HV-00240) from NHLBI, NIH (CU); the Advanced Computing Center for Research and Education at Vanderbilt University. JGG is funded by the Graduate Program in Biological Sciences at Vanderbilt University and the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH, NIAID: F31AI091343-01). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or the NIH. Research in AR’s lab is supported by the Searle Scholars Program and the National Science Foundation (DEB-0844968). VectorBase is supported by National Institutes of Health/National Institute for Allergy and Infectious Diseases contracts HHSN266200400039C and HSN272200900039C). Experimental mosquito infections received support from the CONACYT SALUD-2004-C01-119 grant (for LGC).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Martínez-Barnetche, J., Gómez-Barreto, R.E., Ovilla-Muñoz, M. et al. Transcriptome of the adult female malaria mosquito vector Anopheles albimanus. BMC Genomics 13, 207 (2012). https://doi.org/10.1186/1471-2164-13-207
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-13-207