
Abstract
Background
Digitalis purpurea is an important ornamental and medicinal plant. There is considerable interest in exploring its transcriptome.
Results
Through high-throughput 454 sequencing and subsequent assembly, we obtained 23532 genes, of which 15626 encode conserved proteins. We determined 140 unigenes to be candidates involved in cardiac glycoside biosynthesis. It could be grouped into 30 families, of which 29 were identified for the first time in D. purpurea. We identified 2660 mRNA-like npcRNA (mlncRNA) candidates, an emerging class of regulators, using a computational mlncRNA identification pipeline and 13 microRNA-producing unigenes based on sequence conservation and hairpin structure-forming capability. Twenty five protein-coding unigenes were predicted to be targets of these microRNAs. Among the mlncRNA candidates, only 320 could be grouped into 140 families with at least two members in a family. The majority of D. purpurea mlncRNAs were species-specific and many of them showed tissue-specific expression and responded to cold and dehydration stresses. We identified 417 protein-coding genes with regions significantly homologous or complementary to 375 mlncRNAs. It includes five genes involved in secondary metabolism. A positive correlation was found in gene expression between protein-coding genes and the homologous mlncRNAs in response to cold and dehydration stresses, while the correlation was negative when protein-coding genes and mlncRNAs were complementary to each other.
Conclusions
Through comprehensive transcriptome analysis, we not only identified 29 novel gene families potentially involved in the biosynthesis of cardiac glycosides but also characterized a large number of mlncRNAs. Our results suggest the importance of mlncRNAs in secondary metabolism and stress response in D. purpurea.
Similar content being viewed by others

Background
Digitalis purpurea L. (common foxglove, purple foxglove or lady's glove) is an herbaceous biennial flowering plant species. It is native to Europe and has been widely introduced into other parts of the world. Now, this plant becomes naturalized in many countries, such as China, Canada, USA, and New Zealand [1]. D. purpurea is known for its campanulate flowers produced on a tall spike. The flower color varies from purple to pink, yellow or white. Because of the showy flowers, D. purpurea has an ornamental value and is cultivated world widely. However, this plant is highly poisonous to humans and may be fatal if ingested, so it is suggested to be banished from the table [2, 3]. Although D. purpurea is toxic, it has a high medical value in the therapy of congestive heart failure, particularly arrhythmia for over 200 years [1]. Many patients benefited from the use of this plant and its extracts. The main active compounds of D. purpurea are cardiac glycosides (cardenolides), which are a group of cardio-active agents with the ability to inhibit Na+/K+-ATPase and have been proved to be the most effective drugs to treat heart failure during the past two centuries [1, 4, 5]. D. purpurea is also a potential source of anti-tumor drugs because of its high cytotoxicity against human cancer cell lines [6]. The discovery of cardio-active agents in D. purpurea by the 18th century Scottish physician William Withering is thought to help launching the development of modern pharmacology and the pharmaceutical industry, further suggesting the importance of D. purpurea to medicine [7].
D. purpurea tolerates many environmental stresses, such as cold, drought, heat, flooding, full sun and poor soil, although it prefers rich and well-drained acidic soils and half shade and moist environments. This plant can survive in most temperate climates of the world, including the cold temperate in Alaska of the United States and south-western Norway and the hot and drought conditions in some areas of Africa [1, 8]. Being a biennial plant, D. purpurea is able to withstand the winter cold and the summer drought [9]. Significant damage to D. purpurea leaves were happened only at -12°C or below and the threshold for buds and roots was as low as -15°C and -18°C, respectively [9]. Thus, D. purpurea seems to be an ideal species for studying plant responses to drought and cold stresses.
Although D. purpurea exhibits significance in ornament, medicine and biological study, little has been done about genes involved in the growth and development of this plant species. No EST sequences are available and only 77 nucleotide records are found for D. purpurea in the NCBI nucleotide database http://www.ncbi.nlm.nih.gov/nucleotide/. The major efforts to study the molecular aspects of D. purpurea were focused on the purification and characterization of malonyl-coenzyme A: 21-hydroxypregnane 21-O-malonyltransferase (Dp21MaT) and the identification of genes encoding progesterone 5β-reductase (5β-POR) and Δ5-3β-hydroxysteroid dehydrogenase (3βHSD), which are involved in the biosynthesis of cardiac glycosides [5, 10–12].
Non-protein-coding RNAs (npcRNAs), also known as noncoding RNAs (ncRNAs), are transcripts that have neither experimental nor evolutionary evidence for an open reading frame encoding a functional protein [13]. They are produced in both eukaryotes and prokaryotes and represent the vast majority of all transcripts in a cell as demonstrated by genomic tiling arrays, whole transcriptome analysis, reverse transcription-RCR (RT-PCR) and computational prediction [14–26]. In human, more than 90% of the total genome sequence is likely to be transcribed, but only about 1.5% of the genome codes for proteins, suggesting the abundance of human npcRNA species [15, 27]. The high percentage of transcription in non-protein-coding regions was also reported for other eukaryotes, such as mouse, yeast and Drosophila melanogaster [13, 28–32]. Now, npcRNA is a research hotspot of RNA biology and the number of identified npcRNAs is increasing rapidly.
Results from gene expression profiling and functional analysis strongly suggested that most of the npcRNAs could be biologically meaningful rather than merely transcriptional "noise" [13, 32]. Based on the expression profiles and predicted functions, npcRNAs can be classified into two groups, including housekeeping npcRNAs and regulatory npcRNAs [13]. Housekeeping npcRNAs are usually expressed constitutively and include rRNAs, tRNAs, snRNAs and snoRNAs, whereas regulatory npcRNAs are differentially expressed and developmentally regulated and can be subdivided into short regulatory npcRNAs (or small RNAs, sRNAs, < 40 nucleotides), such as microRNAs (miRNAs) and small interfering RNAs (siRNAs), and large regulatory npcRNAs (or long npcRNAs, lncRNAs, > 40 nucleotides) that may or may not be polyadenylated. A subset of lncRNAs, which can undergo splicing and are capped and polyadenylated as conventional mRNAs, are called mRNA-like npcRNAs (mlncRNAs) [33].
Recently, sRNAs, particularly miRNAs, have been intensely studied [34]. The number of identified miRNAs has reached to 16772 according to the most recent release of miRBase (release 17, http://www.mirbase.org/). It includes 3362 miRNAs from plants, 237 from viruses, and more than 13,000 from animals. In plants, miRNAs play crucial roles in organ development and defense responses by targeting other RNA molecules for cleavage, or, in a few cases, for translational repression [35–38]. lncRNAs are much less studied as compared with mRNAs and sRNAs and represent a major unexplored transcript species [13]. So far, only a small number of lncRNAs have been functionally characterized [32]. The known roles of lncRNAs in animals include transcriptional regulation, epigenetic gene regulation, and disease response [13]. Studies of plant lncRNAs were mainly concentrated on systematic searches of mlncRNAs in Arabidopsis thaliana, wheat, and Medicago truncatula [39–46]. Only few were identified in other plant species [47–53]. Several of the identified plant mlncRNAs, including Arabidopsis AtIPS1/At4 and COLDAIR, Cucumis sativus CsM10, Zea mays Zm401 and M. truncatula enod40, were functionally characterized. The results suggested important regulatory roles of plant mlncRNAs in phosphate deprivation response, vernalization response, pollen development, gender-biased expression, and nodulation [47, 49–57]. Functions of the other plant mlncRNAs are largely unknown.
In this study, we performed a comprehensive analysis of D. purpurea transcriptome that had never been explored. We obtained a high-quality unigene set and determined conserved and non-conserved protein-coding genes, including candidates for about 75% of the total cardiac glycoside biosynthesis-associated gene families in D. purpurea. We identified a large number of mlncRNA candidates and 13 microRNA-producing unigenes and predicted 25 microRNA targets. Using a comprehensive approach, we revealed some important characteristics of mlncRNAs and took efforts to identify mlncRNA-regulated protein-coding genes. These results enhance our knowledge of cardiac glycoside biosynthesis and provide useful information for understanding the function of mlncRNAs.
Results
High-throughput sequencing and assembly of D. purpurea transcriptome
In order to explore the transcriptome of D. purpurea, we constructed a cDNA library of the leaves of one-year-old plants and sequenced it using the 454 ultra-high-throughput pyrosequencing platform, the Genome Sequencer FLX System. We obtained a total of 66103 high-quality reads [GenBank SRA database series identifier SRX084241] with an average length of 402 bases. Pre-processing and de novo assembly using the GS De Novo Assembler Software clustered 51056 reads (77.2% of the total high-quality reads) into 8413 contigs [GenBank TSA database accession nos. JO460003-JO467642] and left 15119 singletons, yielding a total of 23532 unigenes. Original reads of contigs ranged from 2 to 268 with an average 6, and lengths ranged from 94 bp to 3927 bp with an average 519 bp. The size distribution showed that most of contigs ranged from 300 bp to 600 bp and the percentage of contigs no less than 300 bp was 76% (Figure 1). These data suggested that most of the pyrosequencing data had been successfully assembled into relatively long contigs.

Size distribution of contigs assembled from 454 pyrosequencing data. The number of contigs with same length (A) and the number of contigs with length less than 300 bp, 300-600 bp and larger than 600 bp (B) are shown.
Functional annotation and gene ontology analysis
Functional annotation of unigenes was carried out by sequence similarity searches against the NCBI non-redundant (Nr) protein database using the Basic Local Alignment Search Tool (BLAST) [58]. We applied a generous e-value cutoff of 10-5 to the BLAST homolog recognition. 15626 (66.4%) of total unigenes were annotated according to the proposed function of the best match for each unigene in the Nr protein database. These annotated unigenes were protein-coding genes conserved in other species. The remaining 7906 unigenes (33.6%), which did not show significant similarity to any sequences in the Nr protein database, could be npcRNAs, UTR regions of known protein-coding genes, or novel protein-coding transcripts.
The D. purpurea unigenes were further analyzed and categorized using the Gene Ontology (GO), which had been widely used to standardize the representation of gene and gene product attributes across species and to describe gene products in terms of their associated biological processes, cellular locations and molecular functions in a species-independent manner [59]. GO analysis annotated a total of 6462 unigenes and assigned one or more GO terms to each of them (Figure 2). The category of molecular function, which included 5750 unigenes, was the largest, followed by the biological process category (4607 unigenes) and the cellular location category (2144 unigenes). GO assignments of 29 subcategories of the three categories were shown in Figure 2. Metabolic process, protein binding, cellular process and catalytic activity were the largest four subcategories.

Gene ontology analysis of D. purpurea unigenes. Percent of unigenes assigned to 29 subcategories of the cellular location, molecular function and biological process categories is shown.
Identification of protein-coding genes involved in cardiac glycoside biosynthesis
D. purpurea is a useful medicinal plant species in heart failure treatment [1]. Isolation and characterization of genes involved in the biosynthesis of secondary metabolites, such as cardiac glycosides, is particularly valuable. However, little research had been done on this area. With the aim of identifying cardiac glycoside biosynthesis-associated genes on a transcriptome-wide scale, we first analyzed the 23532 D. purpurea unigenes by sequence similarity searches against the Kyoto Encyclopedia of Genes and Genomes database (KEGG) and then searched for unigenes associated with metabolic pathways [60]. Using an E-value cutoff of 10-5, we computationally annotated 12921 unigenes (54.9%), of which 1400 were assigned to the KEGG metabolic pathway category, suggesting a number of unigenes associated with metabolic pathways of D. purpurea were obtained. Within the metabolic pathway category, unigenes were separated into 10 subcategories (Figure 3A).

Percentages of D. purpurea unigenes in 10 subcategories of the metabolic pathway category (A), 15 sub-sub-categories of the lipid metabolism subcategory (B), and 14 sub-sub-categories of the biosynthesis of secondary metabolites subcategory (C).
Further analysis of unigene assignment in the subcategories showed that 797 of the 1400 unigenes each were classified to a subcategory, while the other 603 each were assigned to two or more subcategories. Among the 10 subcategories of metabolic pathway, carbohydrate metabolism was assigned the largest amount of unigenes (Figure 3A), followed by amino acid metabolism, energy metabolism, lipid metabolism, biosynthesis of secondary metabolites, and the others (Figure 3A). The biosynthesis of steroids and terpenoids were well-represented by unigenes in the subcategories of lipid metabolism and biosynthesis of secondary metabolites, allowing the effective identification of cardiac glycoside biosynthesis-related genes in D. purpurea (Figure 3B, C).
Digitali s cardiac glycosides are a class of plant secondary metabolites consisting of a steroid nucleus and a sugar side chain varied in length [61]. The putative biosynthetic pathway of plant cardiac glycosides roughly comprises terpenoid backbone biosynthesis, steroid biosynthesis and cardenolide biosynthesis three stages, involving about 40 gene families, of which only a few had been characterized in D. purpurea. Examination of unigenes assigned to the subcategories of lipid metabolism and biosynthesis of secondary metabolites allowed us to identify a total of 140 unigenes that could be associated with the biosynthesis of terpenoid backbone, steroids and cardenolides (see Additional file 1). These unigenes were grouped into 30 gene families, representing about 75% of the total cardiac glycoside biosynthesis-associated gene families.
Among the 140 unigenes, 24 assembled from 103 original 454 reads were involved in the biosynthesis of terpenoid backbone, and represented 13 of the 16 terpenoid backbone biosynthesis-associated gene families (see Additional file 1). Twenty unigenes assembled from 118 original 454 reads were identified to represent 12 of the 13 putative steroid biosynthesis-associated gene families, suggesting genes involved in the steroid biosynthesis were also well-represented in the unigene set (see Additional file 1).
Different from that involved in the biosynthesis of terpenoid backbone and steroids, genes associated with cardenolide biosynthesis were not well-represented. The putative pathway of cardenolide biosynthesis involved at least 11 gene families, of which only 5 were represented by unigenes. It includes those encoding Δ5-3β-hydroxysteroid dehydrogenases (3β-HSD), progesterone 5β-reductases (5β-POR), mono-oxygenases, cardenolide 16'-O-glucohydrolases (CGH), and glycosyltransferases (GT) (see Additional file 1). 3β-HSD and 5β-POR and mono-oxygenase genes are involved in the initial steps of cardenolide biosynthesis toward cardenolide genin formation, GT genes are responsible for addition of sugars to the genins, whereas CGH is involved in the degradation of primary cardiac glycosides. The gene families of mono-oxygenases and GTs are usually super-large in plants and play crucial roles in various cellular and metabolic processes. Consistently, in the 454 EST database, mono-oxygenases were represented by 36 unigenes assembled from 100 original 454 reads, while GTs were represented by 56 unigenes from 133 reads. The exact mono-oxygenase and GT genes involved in cardenolide biosynthesis remained to elucidate. It is worthy to note that the unigene encoding CGH was listed among the 25 most abundant unigenes (see Additional file 2). Low representativeness of unigenes for cardenolide biosynthesis-associated gene families in our 454 EST database and high expression of the gene involved in primary cardiac glycoside hydrolysis were consistent with overall low cardiac glycoside content in D. purpurea leaves, although relative to other organs leaves have the highest content of cardiac glycosides [62]. These data greatly enhanced our knowledge of Digitalis cardiac glycoside biosynthesis and provided candidate genes for improving the production of active medicinal compounds in D. purpurea through genetic engineering.
Transcriptome-wide identification of D. purpurea mlncRNA candidates
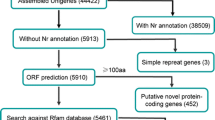
Although npcRNA had been found in various organisms, systematically searches of plant mlncRNAs were carried out only in A. thaliana, wheat and M. truncatula [39–46]. In D. purpurea, among the 23532 unigenes analyzed, 7906 (33.6%) could be novel protein-coding transcripts or npcRNAs because they did not show significant similarity to any sequences in the Nr protein database. Among the 7906 unigenes, 4010, which were not further analyzed, were less than 300 bp in length. The other 3896 with sizes at least 300 bp were subjected to detailed characterization using the pipeline summarized in Figure 4.

Schematic outline of novel protein-coding genes and mlncRNA candidates. The number of unigenes is shown in parentheses. ORF, open reading frame.
The pipeline we used is similar to those reported previously for systematic screens of mlncRNAs from the EST databases of Arabidopsis [39, 40, 42], Medicago [45], Sus scrofa [63], Drosophila [64], and human [65]. A cutoff of 100 amino acids was applied in this study to distinguish npcRNAs from protein-coding transcripts as suggested by Rymarquis et al (2008). The results showed that 1215 of the 3896 unigenes with sizes at least 300 bp had the potential to encode proteins, while the other 2681 unigenes were npcRNA candidates. It includes 2660 mlncRNAs and 21 housekeeping npcRNAs (a tRNA precursor and 20 snoRNA precursors) (see Additional file 3). Low number of non-polyadenylated tRNA and snoRNAs are consistent with our experimental strategy to select polyadenylated RNAs for library construction. Being consistent with the size distribution of contigs, most of the identified mlncRNA candidates have sizes ranged between 300 bp and 600 bp (Figure 5). The others include 22 with 600-699 bp in length and 30 over 700 bp (Table 1). The long ones, such as those with sizes over 600 bp, are very likely to be authentic mlncRNAs, whereas we cannot rule out the possibility that some of the short ones are UTRs of protein coding genes or contain partial ORFs and UTRs of protein coding genes. Further experimental cloning of full-length transcripts may help verify those short mlncRNA candidates. These results suggest the existence of a large number of mlncRNAs in D. purpurea.

Size distribution of mlncRNA candidates.
Identification of 13 D. purpurea microRNAs
microRNAs (miRNAs) are a class of small noncoding RNAs derived from long primary miRNAs and play crucial roles in organ development and defense responses [35–38]. Many miRNAs are deeply conserved across species boundaries. It allows us to identify D. purpurea miRNAs by computational search of our unigene set based on the conservation of miRNA sequences and the secondary structure of unigenes [66]. We, therefore, searched the unigene set for sequences similar to the miRNAs included in the most recent release of miRBase (release 17, http://www.mirbase.org/) [67] using the BLASTN program [58], and then performed mfold analysis of hairpin structures for those unigenes with regions no more than three mismatches to known miRNAs [68]. As a result, we identified a total of 13 miRNA-producing unigenes, which counted for 0.06% of total D. purpurea unigenes (Table 2). Except FXAT9O005F5R10, which is 209 bp in length, the other 12 miRNA-producing unigenes are mlncRNAs with sizes at least 300 bp. It includes JO460086 (mlncR9), one of the mlncRNAs with sizes over 700 bp (Table 1). Based on the predicted miRNA sequences, these unigenes were classified into eight families, of which four, including MIR160, MIR396, MIR397 and MIR408, were each represented by a single member, while the others, including MIR156, MIR166, MIR167 and MIR172, each had two or three members identified (Figure 6 and Table 2). In absence of a small RNA database for D. purpurea, we are not able to reveal those mlncRNAs that produce species-specific miRNAs, but even so, the ratio (~0.06%) of miRNA-producing unigenes to total D. purpurea transcripts is much higher than the ratios (~0.01%) reported previously for other plant species, suggesting the importance of miRNAs in D. purpurea plants [66].

Predicted hairpin structures of D. purpurea miRNA precursors. Mature miRNA sequences are indicated in red. The red, green and blue vertical lines indicate G:C, G:U and A:U pairing, respectively.
Identification of 25 D. purpurea miRNA targets
The regulatory role of plant miRNAs was achieved mainly through targeting other RNA molecules for cleavage [35–38]. Perfect or near-perfect complementarities were found between plant miRNAs and their targets, allowing an effective prediction of target sequences through computation [69]. A useful web server for prediction of plant miRNA targets, known as psRNATarget, has been developed [70]. It searches potential miRNA targets using an improved iterative parallel Smith-Waterman algorithm and a weighted scoring schema [71]. Penalty scores were calculated for mismatched patterns in the miRNA:mRNA duplexes within a 20-base sequence window. Using psRNATarget and setting the penalty score cutoff threshold 0-3, we identified a total of 25 targets from our unigene set for seven D. purpurea miRNA families, which are MIR156, MIR160, MIR166, MIR167, MIR172, MIR396, and MIR 408 (Table 3). The number of targets for each miRNA families range between one and seven. No targets were predicted for MIR397, indicating a very low expression level of miR397 targets in D. purpurea leaves.
As expected, some of the predicted targets are deeply conserved among various plant species. It includes two auxin response factor (ARF) genes regulated by miR160, an APETALA2 (AP2) gene targeted by miR172, and a growth-regulating factor (GRF) gene cleaved by miR396 (Table 3). All of them encode transcription factors that are important to plant development, suggesting the conserved roles of miRNAs in different plant species. Twenty one of the 25 predicted targets appear to be not conserved targets of miRNAs. It is consistent with the previous results showing species-specific targets for many conserved miRNAs in other plant species, such as Populus trichocarpa and Pinus taeda [23, 72, 73]. Among the twenty one targets, twelve are involved in metabolism, RNA process, transcriptional regulation and signal transduction, while the other nine are function-unknown, indicating possible new roles of miRNAs in D. purpure a (Table 3). Computational comparison of gene functions showed that many of these species-specific targets could be important in plant responses to biotic and abiotic stresses. For instance, the glyoxalase I gene targeted by miR160 has 75% identity with a member of Arabidopsis calmodulin-binding proteins, At1g08110, which is involved in plant responses to phytoprostane treatment [74, 75]. The two KH domain-containing proteins regulated by miR408 show at least 75% identity with At5g56140 that is involved in light responses in Arabidopsis plants growing at low temperature [76]. The dead box ATP-dependent RNA helicase gene targted by miR396 is a member of a large gene family involved in defense responses against pathogen infection and various abiotic stresses [77–80]. The heat shock protein encoded by contig05310, a predicted target of miR396, shows 91% identity to Arabidopsis HSP90.1 and HSP81 that are involved in various biotic and abiotic stresses [81, 82]. These results suggested the roles of miRNAs in the development and defense responses of D. purpurea plants. Further experimental validation of the predicted miRNA targets may help add new insights into the regulatory mechanisms of miRNAs in D. purpurea.
Indentification of mlncRNA families based on sequence homology
Among the 2660 D. purpurea mlncRNA candidates with sizes at least 300 bp, 12 produce miRNAs (Table 2), while functions of the other 2648 mlncRNAs are poorly understood. As a first step toward elucidating the roles of these mlncRNAs, we searched sequence homology among them using BLASTN [58]. By applying an e-value cutoff of 10-5 to the homolog recognition, we revealed that 320 of the 2648 mlncRNAs could be grouped into 140 families (see Additional file 4). Number of members for these mlncRNA families is between 2 and 8. The existence of mlncRNA families with multiple members could be a first indication that they actually are important in D. purpurea. No homologs were found for the other 2328 mlncRNAs, indicating that most of the mlncRNAs could be single copy. These results provide a basis for further demonstrating mlncRNA functions.
Conservation of D. purpurea mlncRNAs
The conservation of mlncRNAs was analyzed by searching D. purpurea mlncRNA candidates against the NONCODE database of known npcRNAs using the BLASTN program [58, 83]. An e-value cutoff of 10-5 was applied. Among the 2648 non-microRNA-producing mlncRNA candidates, only eight were found to be conserved (see Additional file 5). It suggests the majority of D. purpurea mlncRNAs are species-specific. The low degree of evolutionary constraint of mlncRNAs was also found in other plant species including M. truncatula, Arabidopsis, wheat, and animals such as Drosophila and mouse, indicating low conservation of mlncRNAs is a common phenomenon in organisms [42, 45, 46, 64, 84]. Among the eight conserved mlncRNAs, two are homologues of GUT15, an npcRNA probably involved in hormone response in Nicotiana tabacum and Arabidopsis [85, 86]. Functions of the other six conserved mlncRNAs are currently unknown.
Tissue-specific expression of mlncRNAs in D. purpurea
Except miR408-producing npcR9 (JO460086), expression patterns of the other 29 mlncRNAs with sizes over 700 bp were analyzed in one-year-old, greenhouse grown D. purpurea plants by quantitative real-time RT-PCR (qPCR). Ubiquitin gene was selected as a reference since it showed stable expression in the D. purpurea tissues analyzed compared with actin and 18S rRNA (see Additional file 6) [87]. Among the 29 mlncRNAs analyzed, two, including mlncR8 and mlncR11, were undetected, indicating low levels in the tissues analyzed. The other 27 were found to be expressed in at least one tissue (Figure 7). mlncR1, mlncR4, mlncR10, mlncR23, mlncR29 and mlncR30 are abundant in leaves, while mlncR17 and mlncR24 are expressed mainly in roots. The levels of mlncR2, mlncR3, mlncR6, mlncR14 and mlncR26 are higher in leaves and roots than stems and flowers, while mlncR15, mlncR18 and mlncR28 exhibit high expression in leaves and stems compared with that in flowers and roots. Differential expression was also found for the other mlncRNAs (Figure 7). It suggests the level of mlncRNAs is developmentally regulated and indicates the importance of mlncRNAs in D. purpurea growth and development.

Expression of mlncRNAs in leaves (L), stems (S), flowers (F) and roots (R) of one-year-old D. purpurea plants. Fold changes of mlncRNA levels were shown. Expression levels were quantified by qPCR. Ubiquitin was used as a reference gene. The mlncRNA levels in leaves were arbitrarily set to 1. Error bars represent the standard deviations of three PCR replicates.
Identification of 24 cold- and 27 dehydration-responsive mlncRNAs
D. purpurea plants tolerate various environmental stresses [1, 8, 9]. To address whether mlncRNAs are involved in plant response to cold and dehydration stresses, we analyzed the levels of 29 non-miRNA-producing mlncRNAs with sizes over 700 bp in D. purpurea plantlets treated with cold and dehydration for 1, 5, 10, 24 hours and compared them with the levels in untreated plantlets. We were able to detect 27 mlncRNAs in plantlets, except mlncR7, an mlncRNA detected in leaves, stems and flowers of one-year old mature plants, and mlncR11, which was also undetected in the analyzed tissues of mature plants (Figure 7, 8 and 9). D. purpurea mlncR7 exhibited opposite expression patterns with mlncR8, which expressed in plantlets but not in the analyzed tissues of one-year-old plants. It suggests that the expression of both mlncR7 and mlncR8 is developmentally regulated, while their biological functions are distinct. D. purpurea mlncR11 appears to play a temporary role in D. purpurea because it is undetected in all of the tissues analyzed.

Expression of mlncRNAs in D. purpurea plantlets treated with cold stress for 0, 1, 5, 10 and 24 hours. Fold changes of mlncRNA levels were shown. Expression levels were quantified by qPCR. Ubiquitin was used as a reference gene. The mlncRNA levels in leaves were arbitrarily set to 1. Error bars represent the standard deviations of three PCR replicates.

Expression of mlncRNAs in D. purpurea plantlets treated with dehydration stress for 0, 1, 5, 10 and 24 hours. Fold changes of mlncRNA levels were shown. Expression levels were quantified by qPCR. Ubiquitin was used as a reference gene. The mlncRNA levels in leaves were arbitrarily set to 1. Error bars represent the standard deviations of three PCR replicates.
Among the 27 mlncRNAs expressed in plantlets, 24 and 27 showed more than 2-fold changes between at least two time-points in plantlets treated with cold and dehydration, respectively, and thus was considered as cold- or dehydration-responsive mlncRNAs (Figure 8 and 9). It includes 3 responsive to dehydration only and 24 responsive to cold and dehydration, suggesting all of the mlncRNAs analyzed are stress-responsive and the majority is not only cold-responsive but also dehydration-responsive. It demonstrates the importance of mlncRNAs in stress responses and indicates the existence of a crosstalk among mlncRNAs in response to cold and dehydration stresses. The results of almost all analyzed mlncRNAs to be cold- and/or dehydration-responsive indicate that the plant materials were probably exposed to environmental stresses before being used for EST library construction.
Based on the expression patterns in response to cold stress, mlncRNAs can be roughly categorized into three major types (Figure 8). Type A mlncRNAs, including mlncR1, mlncR2 and mlncR15, showed less than 2-fold changes between any two time-points. These mlncRNAs appears to be not responsive to cold. Type B, such as mlncR17 and mlncR21, exhibited an immediate decrease after treatment for 1 hour. The decrease continued for at least 10 hours and then some of them, such as mlncR21, showed a trend of recovering to the level in untreated tissues. By contrast, type C mlncRNAs, accounting for more than 80% or 22 of the total 27 mlncRNAs, showed a rapid increase after treatment. The highest level was reached, for most type C mlncRNAs, at 1 h after stress, or in a few cases, at 5 (mlncR30) or 10 hours (mlncR10). After reaching to a maximum, the mlncRNA levels quickly declined to near, or in most cases, far below the levels in untreated tissues. Rapid changes of expression level indicate the importance of type B and type C mlncRNAs in cold-stress response, although the underlying biological functions can be distinct between type B and type C mlncRNAs.
Similarly, based on the expression patterns in response to dehydration stress, mlncRNAs can also be roughly classified into 3 groups (Figure 9). The level of group I mlncRNAs, such as mlncR22, kept constant within the first 10 hours of dehydration stress and then increased at 24 hours, showing a relatively slow response. Group II and group III mlncRNAs showed much quicker responses to dehydration stress. The expression pattern of group II mlncRNAs in response to dehydration is similar to type B mlncRNAs in response to cold stress, exhibiting a rapid decrease at 1 hour of stress (Figure 8 and 9). Except mlncR15, which showed an increase at 10 hours, down-regulation of most group II mlncRNAs lasted for at least 24 hours. Group II, consisting of 20 mlncRNAs, is much larger than type B which includes only two, although group II and type B mlncRNAs exhibited similar expression patterns in response to dehydration and cold stress, respectively. Group III consists of 6 mlncRNAs, including mlncR6, mlncR8, mlncR23, mlncR24, mlncR28 and mlncR30. Expression of mlncRNAs in this group was induced at 1 hour of stress and then immediately down-regulated to far below the level in untreated tissues. It is similar to the pattern of type C mlncRNAs in response to cold stress (Figure 8 and 9). The similarity of mlncRNA expression patterns in response to cold and dehydration suggests that the cold signaling network and the dehydration signaling network are probably overlapped.
Identification of 417 protein-coding genes with regions significantly homologous or complementary to 375 mlncRNAs
Genes with conserved sequences usually show structural, functional or evolutionary relationships. In order to elucidate the relationship between mlncRNAs and protein-coding genes in D. purpurea, we searched our unigene set for protein-coding genes with regions significantly homologous or complementary to mlncRNAs using BLASTN and applying a generous e-value cutoff of 10-5 to the BLAST homolog recognition [58]. A total of 417 protein-coding genes were identified (see Additional file 7). They showed sequence similarity to 375 mlncRNAs in coding sequences (CDS), untranslated regions (UTR) or CDS-UTR junctions. Among the 417 protein-coding genes, 373 each had only one corresponding mlncRNAs, while the others each had 2-9 hits, implying protein-coding genes can be related to one or several mlncRNAs. Similarly, among the 375 mlncRNAs, 51 each showed sequence similarity to 2-10 protein-coding genes, indicating some mlncRNAs are related to multiple protein-coding genes. The results demonstrate the complex relationship between mlncRNAs and protein-coding genes in D. purpurea. Among the 417 protein-coding genes, many encode known proteins involved in metabolism, signal transduction, gene regulation, stress response, protein folding and degradation, or nucleic acid and chromatin modification, indicating the importance of mlncRNAs in the growth and development of D. purpurea plants (Table 4 see Additional file 7).
Expression analysis of 4 protein-coding genes homologous or complementary to mlncRNAs
Four mlncRNA/protein-coding gene pairs were selected for further analysis (Figure 10). It includes mlncR1 (JO460015)/vacuolar H+-ATP synthase subunit E gene (VHA-E, FXAT9O005FSUW), mlncR6 (JO460006)/SNF1-related protein kinase gene (SnRK, JO461197), mlncR8 (JO463019)/4-hydroxy-3-methylbut-2-en-1-yl diphosphate synthase gene (HDS, JO463639), and mlncR31 (FXAT9O005F8J63)/solanesyl diphosphate synthase gene (SPS, JO463507). We first determined the transcriptional direction of protein-coding genes based on the direction of annotated proteins, and then experimentally identified the transcriptional direction of mlncRNAs using 3' RACE. Results show that mlncR1 and mlncR6 and mlncR8 have the same transcriptional direction as their corresponding protein-coding genes in the conserved regions, suggesting these mlncRNAs are homologous to the corresponding protein-coding genes. Among them, mlncR1 and mlncR8 share homologous sequences with the 5' UTR of VHA-E and the 3' UTR of HDS, respectively, while mlncR6 has a region highly similar to the CDS of SnRK. In contrast, mlncR31 appears to be complementary to the 5' UTR of SPS gene (Figure 10).

Protein-coding genes homologous or complementary to mlncRNAs. Protein-coding genes are presented by heavy black lines (coding sequences) and thin lines (nontranslated regions). mlncRNAs are represented by thin lines. The transcriptional direction of protein-coding genes was determined based on the direction of annotated proteins, while that of mlncRNAs was analyzed by 3' RACE. VHA-E, SnRK and HDS are homologous to mlncR1, mlncR6 and mlncR8, respectively (A, B and C). SPS shares an antisense homology with mlncR31 (D). Homologous or complementary regions are shaded in grey and the nucleotide positions are shown. Sequence identities and Watson-Crick pairing (vertical dashes) between protein-coding genes and the corresponding mlncRNAs are indicated.
We next analyzed the expression patterns of VHA-E, SnRK, HDS and SPS in response to cold and dehydration stresses and compared them with the corresponding mlncRNAs (Figure 11). In general, the expression of VHA-E, SnRK and HDS showed a positive correlation with the homologous mlncRNAs, mlncR1, mlncR6 and mlncR8, respectively, although variation was found at very early stage after treatment, such as 1 hour. The most conspicuous positive correlation of expression was found at 5, 10 and 24 hours of stress. It suggests the existence of a close relationship between protein-coding genes and the homologous mlncRNAs and indicates that some protein-coding genes can be regulated by mlncRNAs based on the sense:sense interaction through a currently unknown mechanism or the protein-coding genes and mlncRNAs may be regulated by the same cis/trans factors. In contrast, the expression of SPS gene showed a negative correlation with mlncR31 after 5 hours of stress, being most noticeable in plantlets treated with cold stress (Figure 11). It indicates the presence of sense:antisense interaction between protein-coding genes and the complementary mlncRNAs.

Expression of mlncR1, mlncR6, mlncR8 and mlncR31 and the corresponding protein-coding genes, VHA-E, SnRK, HDS and SPS , in D. purpurea plantlets treated with cold (left panel) and dehydration (right panel) stresses for 0, 1, 5, 10 and 24 hours. Fold changes of expression levels were shown. The levels of mlncRNAs and protein-coding genes were quantified by qPCR. Ubiquitin was used as a reference gene. The levels of mlncRNAs and protein-coding genes in untreated tissues were arbitrarily set to 1. Error bars represent the standard deviations of three PCR replicates.
Discussion
Some mlncRNAs shown sense or antisense homology with protein-coding genes involved in secondary metabolism in D. purpurea
Transcriptomic analysis is an effective approach to discover genes particularly for those organisms without whole genome information, such as D. purpurea, an economically and ecologically important plant species. Through high-throughput 454 sequencing and subsequent assembly, we obtained a high-quality D. purpurea unigene set comprising a total of 23532 genes, which include 140 putatively involved in the biosynthesis of terpenoids/cardiac glycosides, the most effective drugs to treat heart failure during the past two centuries [1, 4, 5]. The 140 genes were grouped into 30 families, representing about 75% of total cardiac glycoside biosynthesis-associated gene families in D. purpurea. These sequence data greatly enhance our knowledge of cardiac glycoside biosynthesis and provides useful information for manipulating cardiac glycoside biosynthesis in D. purpurea through biotechnology. More importantly, we found that 5 mlncRNAs showed sense or antisense homology with 5 protein-coding genes involved in secondary metabolism, including one putatively involved in terpenoid biosynthesis (Table 4).
HDS is the penultimate enzyme of the seven-step DXP pathway, one of the two pathways generating the C5 backbone of all terpenoids including the hormone GA and CGs (see Additional file 8). It catalyzes the conversion of 2-C-methyl-D-erythritol 2,4-cyclodiphosphate into 1-hydroxy-2-methyl-2-butenyl 4-diphosphate and is encoded by a single gene in various plants, including Arabidopsis, Hevea brasiliensis and Ginkgo biloba [88–91]. Through sequence similarity search and subsequent transcriptional direction analysis, we found that the 3' UTR of D. purpurea HDS gene contained a 90 bp-region with 87% identities to an mlncRNA, mlncR8 (Figure 10). The expression of HDS and mlncR8 was positively correlated at 5, 10 and 24 hours of cold and dehydration stresses (Figure 11). It indicates the existence of close relationship between HDS and mlncR8 through a currently unknown mechanism.
SPS gene involved in the biosynthesis of ubiquinone and plastoquinone is related to mlncR31. It utilizes farnesyl diphosphate (FPP) and geranylgeranyl diphosphate (GGPP) as substrates and is responsible for the biosynthesis of isoprenoid side chain of ubiquinone and plastoquinone in Arabidopsis [92]. The 5' UTR of D. purpurea SPS gene harbors a 102 bp-region highly complementary to mlncR31 (Figure 10). The expression of SPS gene is negatively correlated with mlncR31, which is most noticeable in plantlets treated with cold stress for 5, 10 and 24 hours (Figure 11), indicating SPS gene is regulated by mlncR31 through RNA-mediated gene silencing in D. purpurea [93–95]. Both ubiquinone and plastoquinone are electron carriers. Plastoquinone is a member of the photosynthetic electron transport chain in chloroplast, while ubiquinone is a component of respiratory electron transport chain in mitochondria. Thus, mlncR31 may be important in maintaining the activities of plant cells under environmental stress conditions.
In addition, mlncRNAs are probably involved in the biosynthesis of flavonoid, carotenoid and alkaloid (Table 4). The mlncRNA, FXAT9O005FZSI2, share homology with a unigene (FXAT9O005FZ5UJ) encoding dihydroflavonal-4-reductase (DFR), which catalyze the reduction of dihydroflavonol into flavan-3,4-diol (leucoanthocyanin), a key step in the biosynthesis of anthocyanins, proanthocyanidins and other flavonoids important for plant survival and human nutrition [96]. FXAT9O005GD1W7 appears the other mlncRNA involved in secondary metabolism. It shows sequence similarity to a unigene (FXAT9O005GCP3D) encoding phytoene dehydrogenase (PDS). PDS, which catalyzes the desaturation of phytoene into phytofluene, is a rate-limiting enzyme in carotenoid biosynthesis in plants [97]. The mlncRNA probably involved in alkaloid biosynthesis is FXAT9O005FYD4C, which shares homology with the coding region of aromatic amino acid decarboxylase (AADC) gene (FXAT9O005FK4AG). AADC catalyzes the decarboxylation of tyrosine into tyramine, a key step of plant alkaloid biosynthesis [98, 99].
Taken together, these results strongly suggest the importance of mlncRNAs in secondary metabolism in D. purpurea.
D. purpurea mlncRNAs exhibit species-specific characteristics and are important in plant development and stress responses
Using a computational mlncRNA identification pipeline, we identified 2660 D. purpurea mlncRNA candidates, of which only about 12% can be grouped into families with at least two members in a family, while the others are single gene family members. Furthermore, the conservation of mlncRNAs is very low. Searching the NONCODE database of known npcRNAs, we found that only about 0.3% of the mlncRNAs analyzed was conserved, suggesting the vast majority of D. purpurea mlncRNAs are species-specific. These results are consistent with those obtained from other plant species including M. truncatula, Arabidopsis, wheat, and animals such as Drosophila and mouse [42, 45, 46, 64, 84], indicating mlncRNAs may be evolved with a low degree of constraint and many of them are probably undergoing frequent birth and death. It is particularly true for the mlncRNAs which produce non-conserved microRNA families represented by single genes [100].
In this study, we identified a total of 13 microRNA-producing mlncRNAs based on the conservation of miRNA sequences and the secondary structure of unigenes [66–68]. The mature microRNAs were classified into eight families, including MIR156, MIR160, MIR166, MIR167, MIR172, MIR396, MIR397 and MIR408 (Figure 6, Table 2). They were predicted to regulate at least 25 protein-coding genes, of which 4 encode conserved transcriptional factors, including two ARFs, an AP2 and a GRF (Table 3). ARFs play regulatory roles in the development of plant organs, such as seed, gynoecium, embryo, hypocotyl, root and petal in Arabidopsis [101–107]. AP2 is involved in floral transition and floral development [108]. GRF control cell proliferation in Arabidopsis leaves [109, 110]. It suggests the importance of microRNAs in the development of D. purpurea plants. Additionally, among the 417 protein-coding genes shown sense or antisense homology with mlncRNAs, several, such as JO464993, FXAT9O005FR21D, FXAT9O005GE5AF, FXAT9O005F8DZ7, FXAT9O005F4WF9 and FXAT9O005GCSS5, encode transcription factors and other development-related proteins, indicating some non-microRNA-producing mlncRNAs are also probably involved in the development of D. purpurea plants (see Additional file 7). It is consistent with the findings showing the involvement of mlncRNA (Zm401) in pollen development in Zea mays and the regulatory role of mlncRNA (enod40) in symbiotic nitrogen-fixing nodule formation in legumes [49, 111, 112].
Expressional analysis of mlncRNAs allowed us to identify 24 cold- and 27 dehydration-responsive mlncRNAs, suggesting the importance of mlncRNAs in stress responses in D. purpurea (Figure 8 and 9). In addition, the majority of mlncRNAs analyzed were responsive to both cold and dehydration and many of them exhibited similar expression patterns under cold and dehydration conditions, indicating the cold signaling network and the dehydration signaling network are probably overlapped. Stress responses of mlncRNAs have been reported previously in various plants, including Arabidopsis [39, 44, 113] and wheat [46]. For example, Arabidopsis IPS1 and M. truncatula Mt4 mlncRNAs are involved in phosphate starvation through fine-tuning the expression of miR399/PHO2 gene [54, 56, 113]. Arabidopsis COOLAIR (cold induced long antisense intragenic RNA) and COLDAIR (COLD ASSISTED INTRONIC NONCODING RNA) play significant roles in cold response and are required for the vernalization-mediated epigenetic repression of FLC [44, 55]. Several wheat mlncRNAs are responsive to powdery mildew infection and/or heat stress [46]. Thus, mlncRNAs seem to be very important for the whole plant kingdom to survive in the stressful environments.
Functional mechanisms of mlncRNAs in D. purpurea
mlncRNAs have been found to interact with RNA, DNA and protein molecules [13, 114, 115]. A small portion of the identified D. purpurea mlncRNAs produce conserved microRNAs. Since no small RNA sequences are available and experimental setup does not allow us to detect the processed miRNAs, we are not able to identify those producing non-conserved microRNAs from the set of D. purpurea mlncRNA candidates. However, it is rational to believe the existence of non-conserved microRNA-producing D. purpurea mlncRNAs based on the increasing number of non-conserved microRNAs in the miRBase [67]. These mlncRNAs play crucial roles in organ development and defense responses through producing conserved or non-conserved microRNAs, which target other RNA molecules for cleavage, or, in a few cases, for translational repression [35–38]. Additionally, some mlncRNAs play regulatory roles in plants through generating siRNAs. These mlncRNAs can be antisense transcripts of protein-coding genes, ta-siRNA precursors, repeat-associated transcripts, or transcripts sharing antisense homology with protein-coding genes [116–118]. It is also possible that some mlncRNAs may function through inhibiting the action of microRNAs. For example, the Arabidopsis mlncRNA, IPS1, contains a 24 nt region complementary to miR399, a microRNA involved in phosphate starvation by targeting PHO2 gene for cleavage. IPS1 mimics PHO2, except that the base-pairing is interrupted by a mismatched loop at the miR399 cleavage. Therefore, IPS1 is not cleaved but instead sequesters miR399, resulting in inhibition of miR399 activity on PHO2 [113]. The siRNA-generating mlncRNAs and the mimics of microRNA targets in our mlncRNA set remain to be identified.
In this study, we predicted a total of 2660 D. purpurea mlncRNA candidates using a computational mlncRNA identification pipeline. According to the sequence similarity between mlncRNAs and protein-coding genes and the location of homologous regions in mlncRNAs and protein-coding genes, the majority of mlncRNAs candidates appear to be not the fragments of UTRs of protein-coding genes (Figure 11; see Additional file 7). Among the 2660 mlncRNA candidates, 375 mlncRNAs share sense or antisense homology with 417 protein-coding genes, indicating that some mlncRNAs and the corresponding protein-coding gene are probably homologous genes derived from a common ancestral gene, or these mlncRNAs are new born genes originated from the corresponding protein-coding genes, as the case of long non-coding RNA, Xist, and the protein-coding gene, Lnx3, in mammals [119, 120]. The mlncRNAs sharing antisense homology with protein-coding genes, such as D. purpurea mlncR31, seems to play a role in post-transcriptional regulation of the corresponding protein-coding genes. It is evidenced by the negative correlation of expression between mlncR31 and the SPS gene after 5 hours of stress (Figure 11). The roles in post-transcriptional regulation have been previously reported for some long non-coding RNAs identified from various animal species, such as human and mouse [13, 93, 95]. It is not well-known for the mechanisms of mlncRNAs acting on protein-coding genes with sense homology, such as mlncR1, mlncR6 and mlncR8 and their homologous protein-coding genes, VHA-E, SnRK and HDS, respectively. One of the possible mechanisms is co-suppression, a phenomenon in plants in which a sense transgene sometimes cause suppression of both the endogenous gene and the transgene [121–123]. Further identification of the interaction mechanism between mlncRNAs and protein-coding genes will give us a much clearer picture of how mlncRNAs function in D. purpurea.
Conclusions
The comprehensive analysis of transcriptome data from high-throughput sequencing allow us to discover many novel unigenes involved in the biosynthesis of secondary metabolites, such as cardiac glycosides, ubiquinone, plastoquinone, flavonoid, carotenoid and alkaloid, and to identify a large number of mlncRNA candidates in D. purpurea. Detailed analysis suggests that the majority of D. purpurea mlncRNAs are species-specific and most mlncRNA families are single gene families. These mlncRNAs exhibited tissue-specific expression and responded to cold and dehydration stresses. Since at least 24 mlncRNAs were found to be not only cold-responsive but also dehydration-responsive, a crosstalk could exist among mlncRNAs in response to cold and dehydration stresses. The identified mlncRNAs appears to be involved in many aspects, such as plant development, stress response and secondary metabolism. The regulatory role is associated with protein-coding genes sharing sequence similarity with the mlncRNAs. These results provide novel and significant information for understanding the biosynthesis of secondary metabolites and the function of mlncRNAs. In addition, a large number of RNAseq data of D. purpurea have been recently released in the NCBI SRA database under the series identifier SRP006029, which provides an additional source for the discovery of genes and mlncRNAs in D. purpurea.
Methods
Plant materials
Leaves, stems, flowers and roots were collected from one-year-old, greenhouse grown D. purpurea 'GIANT SHIRLY' plants and then used for EST library construction and tissue-specific expression analysis of mlncRNAs. For cold and dehydration treatments, D. purpurea seeds were sterilized in 70% ethanol for 1 min and then in 0.1% mercuric chloride for 8 min. After washing three times in sterile water, the seeds were grown on MS medium supplemented with 3% sucrose and 0.9% agar for 3 weeks at 25°C under 14 hours light:10 hours dark. The plantlets were treated at 4°C, or removed from the agar and then dehydrated in glass dishes at 25°C under dim light, for 0, 1, 5, 10, 24 hours. Six seedlings were collected in liquid nitrogen for each example.
EST library construction and 454 sequencing
Total RNA was extracted from D. purpurea leaves using the Universal Plant Total RNA Extraction kit (Bioteke). mRNA was isolated from total RNA using the Oligotex mRNA kit (Qiagen). Single-stranded cDNA was prepared from mRNA using the SMART cDNA Synthesis kit (Clontech). Double-stranded cDNA was amplified by PCR polymerase Advantage II (Clontech). PCR products less than 300 bp in length were removed using the PureLink™ PCR Purification kit (Invitrogen). Pyrosequencing was performed by 454 Life Sciences using the Genome Sequencer FLX System.
EST assembly, annotation and classification
The GS-FLX Software (454 Life Sciences, Roche) was used for trimming adapters and filtering low-quantity sequences. High-quantity reads were assembled into unigenes by the GS De Novo Assembler Software, an application of the GS FLX Software. The unigenes obtained were annotated by sequence similarity searches against the NCBI non-redundant (Nr) protein database using the Basic Local Alignment Search Tool (BLAST) [58]. A generous e-value cutoff of 10-5 was applied to the BLAST homolog recognition. Gene Ontology (GO) terms were assigned using InterProScan [59].
mlncRNA identification
The procedure for computational mlncRNA identification is outlined in Figure 4. Unigenes without Nr annotation and with sizes at least 300 bp were selected for open-reading frame (ORF) prediction using ESTScan 2 [124]. Computational prediction was performed on a local server through batch analysis using the default parameters. A cutoff of 100 amino acids was applied to distinguish mlncRNAs from protein-coding transcripts as described [43]. Housekeeping npcRNAs were searched against Rfam 10.0 (January 2010) using BLASTN [58, 125]. Sequence homology among the D. purpurea mlncRNA candidates was analyzed using BLASTN [58]. The conservation of mlncRNAs was analyzed by searching D. purpurea mlncRNA candidates against the NONCODE database of known npcRNAs using the BLASTN program [58, 83]. An e-value cutoff of 10-5 was applied to the homolog recognition.
Analysis of conserved miRNAs
Plant microRNA sequences were downloaded from miRBase (Released 17, http://www.mirbase.org/) [67]. Unigenes with regions no more than three mismatches to known microRNAs were searched against the D. purpurea unigene set using BLASTN [58]. A word size of 4 was used as described [126]. The secondary structures were predicted using mfold [68]. Criteria described by Meyers et al (2008) were applied to annotate D. purpurea microRNAs. The minimal folding free energy index (MFEI) was calculated as described previously [127].
Prediction of microRNA targets
Prediction of microRNA targets was performed on the psRNATarget Server using the default parameters [70].
qPCR
Total RNA was extracted from plant tissues using the Concert™ Plant RNA Reagent (Invitrogen) and then digested with RNase-free DNase I (Promega) to remove the genomic DNA contamination. Reverse transcription was performed on 2 μg total RNA for each example by 200 U SuperScript III Reverse Transcriptase (Invitrogen) in a 20 μl volume. The reaction was carried out at 65°C for 5 minutes, 50°C for 60 minutes and 70°C for 15 minutes. The resulting cDNA was diluted to 1,600 μl with sterile water. qPCR was carried out in triplicate reactions using the BIO-RAD CFX system (BIO-RAD). Gene-specific primers were listed in (see Additional file 9). Ubiquitin gene was selected as a reference. PCR was carried out in a 20 μl volume containing 2 μl diluted cDNA, 250 nM forward primer, 250 nM reverse primer, and 1 × SYBR Premix Ex Taq II (TaKaRa) using the following conditions: 95°C for 3 min, 40 cycles of 95°C for 15 sec, 60°C for 15 sec and 72°C for 15 sec. Specificity of the amplification was verified by the Bio-Rad CFX Manage software based on dissociation curves. The results from gene-specific amplification were analyzed using the comparative Cq method which uses an arithmetic formula, 2-ΔΔCq, to achieve results for relative quantification [128]. Cq represents the threshold cycle.
3' RACE for determining the transcriptional direction of mlncRNAs
3' RACE was performed on total RNA isolated from D. purpurea plantlets treated with cold stress for 1 hour using the GeneRacer kit (Invitrogen). PCR amplifications were carried out using the GeneRacer 3' primer and the nesting gene-specific primers. Nested PCR amplifications were performed using the GeneRacer 3' nested primer and the nested gene-specific primers (see Additional file 10). PCR products were purified, cloned and sequenced.
Abbreviations
- 3βHSD :
-
Δ5-3β-hydroxysteroid dehydrogenase
- 5β-POR :
-
progesterone 5β-reductase
- AACT :
-
acetyl-CoA acetyltransferases
- AADC :
-
aromatic amino acid decarboxylase
- AP2 :
-
APETALA2
- ARF :
-
auxin response factor
- BLAST :
-
Basic Local Alignment Search Tool
- BLASTN :
-
nucleotide BLAST
- CDS :
-
coding sequences
- CGH :
-
16'-O-glucohydrolases
- DFR :
-
dihydroflavonal-4-reductase: Dp21MaT:malonyl-coenzyme A: 21-hydroxypregnane 21-O-malonyltransferas: EST:expressed sequence tag
- FLC :
-
FLOWERING LOCUS C
- FPP :
-
farnesyl diphosphate
- GGPP :
-
geranylgeranyl diphosphate
- GO :
-
Gene Ontology
- GRF :
-
growth-regulating factor
- GT :
-
glycosyltransferases
- HDS :
-
4-hydroxy-3-methylbut-2-en-1-yl diphosphate synthase
- HMGS :
-
3-hydroxyl-3-methyl-glutaryl-CoA synthases
- KEGG :
-
Kyoto Encyclopedia of Genes and Genomes database
- MEP :
-
2-C-methyl-D-erythritol 4-phosphate
- miRNA :
-
microRNA
- mlncRNA :
-
mRNA-like npcRNA
- MVA :
-
mevalonate
- ncRNAs :
-
noncoding RNAs
- npcRNAs :
-
non-protein-coding RNAs
- Nr :
-
non-redundant
- ORF :
-
open-reading frame
- PDS :
-
phytoene dehydrogenase
- psRNATarget :
-
A Plant Small RNA Target Analysis Server
- RACE :
-
rapid amplification of cDNA ends
- RT-PCR :
-
reverse transcription-RCR
- siRNAs :
-
small interfering RNAs
- snoRNAs :
-
Small nucleolar RNAs
- SnRK :
-
SNF1-related protein kinase
- snRNAs :
-
small nuclear ribonucleic acid
- SPS :
-
solanesyl diphosphate synthase
- sRNAs :
-
small RNAs
- ta-siRNA :
-
trans small interfering RNAs
- UTR :
-
untranslated regions
- VHA-E :
-
vacuolar H+-ATP synthase subunit E.
References
Warren B: Digitalis purpurea. Am J Cardiol. 2005, 95: 544-
Lin CC, Yang CC, Phua DH, Deng JF, Lu LH: An outbreak of foxglove leaf poisoning. J Chin Med Assoc. 2010, 73: 97-100.
Maffe S, Cucchi L, Zenone F, Bertoncelli C, Beldi F, Colombo ML, Bielli M, Paino AM, Parravicini U, Paffoni P, Dellavesa P, Perucca A, Pardo NF, Signorotti F, Didino C, Zanetta M: Digitalis must be banished from the table: a rare case of acute accidental Digitalis intoxication of a whole family. J Cardiovasc Med (Hagerstown). 2009, 10: 727-732.
Hauptman PJ, Garg R, Kelly RA: Cardiac glycosides in the next millennium. Prog Cardiovasc Dis. 1999, 41: 247-254.
Kuate SP, Padua RM, Eisenbeiss WF, Kreis W: Purification and characterization of malonyl-coenzyme A: 21-hydroxypregnane 21-O-malonyltransferase (Dp21MaT) from leaves of Digitalis purpurea L. Phytochemistry. 2008, 69: 619-626.
Lopez-Lazaro M, Palma De La Pena N, Pastor N, Martin-Cordero C, Navarro E, Cortes F, Ayuso MJ, Toro MV: Anti-tumour activity of Digitalis purpurea L. subsp. heywoodii. Planta Med. 2003, 69: 701-704.
Lesney MS: Flowers for the heart. Mod Drug Discov. 2002, 5: 46-48.
Sletvold N, Rydgren K: Population dynamics in Digitalis purpurea: the interaction of disturbance and seed bank dynamics. J Ecol. 2007, 95: 1346-1359.
Bruelheide H, Heinemeyer A: Climatic factors controlling the eastern and altitudinal Bruelheide boundary of Digitalis purpurea L. in Germany. Flora. 2002, 197: 475-490.
Gavidia I, Tarrio R, Rodriguez-Trelles F, Perez-Bermudez P, Seitz HU: Plant progesterone 5beta-reductase is not homologous to the animal enzyme. Molecular evolutionary characterization of P5betaR from Digitalis purpurea. Phytochemistry. 2007, 68: 853-864.
Herl V, Frankenstein J, Meitinger N, Muller-Uri F, Kreis W: Delta 5-3beta-hydroxysteroid dehydrogenase (3 beta HSD) from Digitalis lanata. Heterologous expression and characterisation of the recombinant enzyme. Planta Med. 2007, 73: 704-710.
Perez-Bermudez P, Garcia AA, Tunon I, Gavidia I: Digitalis purpurea P5 beta R2, encoding steroid 5 beta-reductase, is a novel defense-related gene involved in cardenolide biosynthesis. New Phytol. 2010, 185: 687-700.
Ponting CP, Oliver PL, Reik W: Evolution and functions of long noncoding RNAs. Cell. 2009, 136: 629-641.
Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, Zhu X, Rinn JL, Tongprasit W, Samanta M, Weissman S, Gerstein M, Snyder M: Global identification of human transcribed sequences with genome tiling arrays. Science. 2004, 306: 2242-2246.
Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, Boyle PJ, Cao H, Carter NP, Clelland GK, Davis S, Day N, Dhami P, Dillon SC, Dorschner MO, Fiegler H, Giresi PG, Goldy J, Hawrylycz M, Haydock A, Humbert R, James KD, Johnson BE, Johnson EM, Frum TT, Rosenzweig ER, Karnani N, Lee K, Lefebvre GC, Navas PA, Neri F, Parker SC, Sabo PJ, Sandstrom R, Shafer A, Vetrie D, Weaver M, Wilcox S, Yu M, Collins FS, Dekker J, Lieb JD, Tullius TD, Crawford GE, Sunyaev S, Noble WS, Dunham I, Denoeud F, Reymond A, Kapranov P, Rozowsky J, Zheng D, Castelo R, Frankish A, Harrow J, Ghosh S, Sandelin A, Hofacker IL, Baertsch R, Keefe D, Dike S, Cheng J, Hirsch HA, Sekinger EA, Lagarde J, Abril JF, Shahab A, Flamm C, Fried C, Hackermuller J, Hertel J, Lindemeyer M, Missal K, Tanzer A, Washietl S, Korbel J, Emanuelsson O, Pedersen JS, Holroyd N, Taylor R, Swarbreck D, Matthews N, Dickson MC, Thomas DJ, Weirauch MT, Gilbert J, Drenkow J, Bell I, Zhao X, Srinivasan KG, Sung WK, Ooi HS, Chiu KP, Foissac S, Alioto T, Brent M, Pachter L, Tress ML, Valencia A, Choo SW, Choo CY, Ucla C, Manzano C, Wyss C, Cheung E, Clark TG, Brown JB, Ganesh M, Patel S, Tammana H, Chrast J, Henrichsen CN, Kai C, Kawai J, Nagalakshmi U, Wu J, Lian Z, Lian J, Newburger P, Zhang X, Bickel P, Mattick JS, Carninci P, Hayashizaki Y, Weissman S, Hubbard T, Myers RM, Rogers J, Stadler PF, Lowe TM, Wei CL, Ruan Y, Struhl K, Gerstein M, Antonarakis SE, Fu Y, Green ED, Karaoz U, Siepel A, Taylor J, Liefer LA, Wetterstrand KA, Good PJ, Feingold EA, Guyer MS, Cooper GM, Asimenos G, Dewey CN, Hou M, Nikolaev S, Montoya-Burgos JI, Loytynoja A, Whelan S, Pardi F, Massingham T, Huang H, Zhang NR, Holmes I, Mullikin JC, Ureta-Vidal A, Paten B, Seringhaus M, Church D, Rosenbloom K, Kent WJ, Stone EA, Batzoglou S, Goldman N, Hardison RC, Haussler D, Miller W, Sidow A, Trinklein ND, Zhang ZD, Barrera L, Stuart R, King DC, Ameur A, Enroth S, Bieda MC, Kim J, Bhinge AA, Jiang N, Liu J, Yao F, Vega VB, Lee CW, Ng P, Yang A, Moqtaderi Z, Zhu Z, Xu X, Squazzo S, Oberley MJ, Inman D, Singer MA, Richmond TA, Munn KJ, Rada-Iglesias A, Wallerman O, Komorowski J, Fowler JC, Couttet P, Bruce AW, Dovey OM, Ellis PD, Langford CF, Nix DA, Euskirchen G, Hartman S, Urban AE, Kraus P, Van Calcar S, Heintzman N, Kim TH, Wang K, Qu C, Hon G, Luna R, Glass CK, Rosenfeld MG, Aldred SF, Cooper SJ, Halees A, Lin JM, Shulha HP, Xu M, Haidar JN, Yu Y, Iyer VR, Green RD, Wadelius C, Farnham PJ, Ren B, Harte RA, Hinrichs AS, Trumbower H, Clawson H, Hillman-Jackson J, Zweig AS, Smith K, Thakkapallayil A, Barber G, Kuhn RM, Karolchik D, Armengol L, Bird CP, de Bakker PI, Kern AD, Lopez-Bigas N, Martin JD, Stranger BE, Woodroffe A, Davydov E, Dimas A, Eyras E, Hallgrimsdottir IB, Huppert J, Zody MC, Abecasis GR, Estivill X, Bouffard GG, Guan X, Hansen NF, Idol JR, Maduro VV, Maskeri B, McDowell JC, Park M, Thomas PJ, Young AC, Blakesley RW, Muzny DM, Sodergren E, Wheeler DA, Worley KC, Jiang H, Weinstock GM, Gibbs RA, Graves T, Fulton R, Mardis ER, Wilson RK, Clamp M, Cuff J, Gnerre S, Jaffe DB, Chang JL, Lindblad-Toh K, Lander ES, Koriabine M, Nefedov M, Osoegawa K, Yoshinaga Y, Zhu B, de Jong PJ: Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007, 447: 799-816.
Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, Brenner SE, Batalov S, Forrest AR, Zavolan M, Davis MJ, Wilming LG, Aidinis V, Allen JE, Ambesi-Impiombato A, Apweiler R, Aturaliya RN, Bailey TL, Bansal M, Baxter L, Beisel KW, Bersano T, Bono H, Chalk AM, Chiu KP, Choudhary V, Christoffels A, Clutterbuck DR, Crowe ML, Dalla E, Dalrymple BP, de Bono B, Della Gatta G, di Bernardo D, Down T, Engstrom P, Fagiolini M, Faulkner G, Fletcher CF, Fukushima T, Furuno M, Futaki S, Gariboldi M, Georgii-Hemming P, Gingeras TR, Gojobori T, Green RE, Gustincich S, Harbers M, Hayashi Y, Hensch TK, Hirokawa N, Hill D, Huminiecki L, Iacono M, Ikeo K, Iwama A, Ishikawa T, Jakt M, Kanapin A, Katoh M, Kawasawa Y, Kelso J, Kitamura H, Kitano H, Kollias G, Krishnan SP, Kruger A, Kummerfeld SK, Kurochkin IV, Lareau LF, Lazarevic D, Lipovich L, Liu J, Liuni S, McWilliam S, Madan Babu M, Madera M, Marchionni L, Matsuda H, Matsuzawa S, Miki H, Mignone F, Miyake S, Morris K, Mottagui-Tabar S, Mulder N, Nakano N, Nakauchi H, Ng P, Nilsson R, Nishiguchi S, Nishikawa S, Nori F, Ohara O, Okazaki Y, Orlando V, Pang KC, Pavan WJ, Pavesi G, Pesole G, Petrovsky N, Piazza S, Reed J, Reid JF, Ring BZ, Ringwald M, Rost B, Ruan Y, Salzberg SL, Sandelin A, Schneider C, Schonbach C, Sekiguchi K, Semple CA, Seno S, Sessa L, Sheng Y, Shibata Y, Shimada H, Shimada K, Silva D, Sinclair B, Sperling S, Stupka E, Sugiura K, Sultana R, Takenaka Y, Taki K, Tammoja K, Tan SL, Tang S, Taylor MS, Tegner J, Teichmann SA, Ueda HR, van Nimwegen E, Verardo R, Wei CL, Yagi K, Yamanishi H, Zabarovsky E, Zhu S, Zimmer A, Hide W, Bult C, Grimmond SM, Teasdale RD, Liu ET, Brusic V, Quackenbush J, Wahlestedt C, Mattick JS, Hume DA, Kai C, Sasaki D, Tomaru Y, Fukuda S, Kanamori-Katayama M, Suzuki M, Aoki J, Arakawa T, Iida J, Imamura K, Itoh M, Kato T, Kawaji H, Kawagashira N, Kawashima T, Kojima M, Kondo S, Konno H, Nakano K, Ninomiya N, Nishio T, Okada M, Plessy C, Shibata K, Shiraki T, Suzuki S, Tagami M, Waki K, Watahiki A, Okamura-Oho Y, Suzuki H, Kawai J, Hayashizaki Y: The transcriptional landscape of the mammalian genome. Science. 2005, 309: 1559-1563.
Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, Long J, Stern D, Tammana H, Helt G, Sementchenko V, Piccolboni A, Bekiranov S, Bailey DK, Ganesh M, Ghosh S, Bell I, Gerhard DS, Gingeras TR: Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005, 308: 1149-1154.
Geissmann T, Chevalier C, Cros MJ, Boisset S, Fechter P, Noirot C, Schrenzel J, Francois P, Vandenesch F, Gaspin C, Romby P: A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nucleic Acids Res. 2009, 37: 7239-7257.
He H, Wang J, Liu T, Liu XS, Li T, Wang Y, Qian Z, Zheng H, Zhu X, Wu T, Shi B, Deng W, Zhou W, Skogerbo G, Chen R: Mapping the C. elegans noncoding transcriptome with a whole-genome tiling microarray. Genome Res. 2007, 17: 1471-1477.
Jia H, Osak M, Bogu GK, Stanton LW, Johnson R, Lipovich L: Genome-wide computational identification and manual annotation of human long noncoding RNA genes. RNA. 2010, 16: 1478-1487.
Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermuller J, Hofacker IL, Bell I, Cheung E, Drenkow J, Dumais E, Patel S, Helt G, Ganesh M, Ghosh S, Piccolboni A, Sementchenko V, Tammana H, Gingeras TR: RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007, 316: 1484-1488.
Li Z, Liu M, Zhang L, Zhang W, Gao G, Zhu Z, Wei L, Fan Q, Long M: Detection of intergenic non-coding RNAs expressed in the main developmental stages in Drosophila melanogaster. Nucleic Acids Res. 2009, 37: 4308-4314.
Lu S, Sun YH, Amerson H, Chiang VL: MicroRNAs in loblolly pine (Pinus taeda L.) and their association with fusiform rust gall development. Plant J. 2007, 51: 1077-1098.
Tran TT, Zhou F, Marshburn S, Stead M, Kushner SR, Xu Y: De novo computational prediction of non-coding RNA genes in prokaryotic genomes. Bioinformatics. 2009, 25: 2897-2905.
Vercruysse M, Fauvart M, Cloots L, Engelen K, Thijs IM, Marchal K, Michiels J: Genome-wide detection of predicted non-coding RNAs in Rhizobium etli expressed during free-living and host-associated growth using a high-resolution tiling array. BMC Genomics. 2010, 11: 53-
Voss B, Georg J, Schon V, Ude S, Hess WR: Biocomputational prediction of non-coding RNAs in model cyanobacteria. BMC Genomics. 2009, 10: 123-
International Human Genome Sequencing Consortium: Finishing the euchromatic sequence of the human genome. Nature. 2004, 431: 931-945.
Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, Schroder K, Cloonan N, Steptoe AL, Lassmann T, Waki K, Hornig N, Arakawa T, Takahashi H, Kawai J, Forrest AR, Suzuki H, Hayashizaki Y, Hume DA, Orlando V, Grimmond SM, Carninci P: The regulated retrotransposon transcriptome of mammalian cells. Nat Genet. 2009, 41: 563-571.
Kapranov P, Willingham AT, Gingeras TR: Genome-wide transcription and the implications for genomic organization. Nat Rev Genet. 2007, 8: 413-423.
Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M: The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008, 320: 1344-1349.
Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, Goodhead I, Penkett CJ, Rogers J, Bahler J: Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008, 453: 1239-1243.
Wilusz JE, Sunwoo H, Spector DL: Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009, 23: 1494-1504.
Griffiths-Jones S: Annotating noncoding RNA genes. Annu Rev Genomics Hum Genet. 2007, 8: 279-298.
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ: miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008, 36: D154-158.
Bartel DP: MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004, 116: 281-297.
Chen X: Small RNAs and their roles in plant development. Annu Rev Cell Dev Biol. 2009, 25: 21-44.
Jones-Rhoades MW, Bartel DP, Bartel B: MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol. 2006, 57: 19-53.
Voinnet O: Origin, biogenesis, and activity of plant microRNAs. Cell. 2009, 136: 669-687.
Ben Amor B, Wirth S, Merchan F, Laporte P, d'Aubenton-Carafa Y, Hirsch J, Maizel A, Mallory A, Lucas A, Deragon JM, Vaucheret H, Thermes C, Crespi M: Novel long non-protein coding RNAs involved in Arabidopsis differentiation and stress responses. Genome Res. 2009, 19: 57-69.
Hirsch J, Lefort V, Vankersschaver M, Boualem A, Lucas A, Thermes C, d'Aubenton-Carafa Y, Crespi M: Characterization of 43 non-protein-coding mRNA genes in Arabidopsis, including the MIR162a-derived transcripts. Plant Physiol. 2006, 140: 1192-1204.
Kurihara Y, Matsui A, Hanada K, Kawashima M, Ishida J, Morosawa T, Tanaka M, Kaminuma E, Mochizuki Y, Matsushima A, Toyoda T, Shinozaki K, Seki M: Genome-wide suppression of aberrant mRNA-like noncoding RNAs by NMD in Arabidopsis. Proc Natl Acad Sci USA. 2009, 106: 2453-2458.
MacIntosh GC, Wilkerson C, Green PJ: Identification and analysis of Arabidopsis expressed sequence tags characteristic of non-coding RNAs. Plant Physiol. 2001, 127: 765-776.
Rymarquis LA, Kastenmayer JP, Huttenhofer AG, Green PJ: Diamonds in the rough: mRNA-like non-coding RNAs. Trends Plant Sci. 2008, 13: 329-334.
Swiezewski S, Liu F, Magusin A, Dean C: Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature. 2009, 462: 799-802.
Wen J, Parker BJ, Weiller GF: In silico identification and characterization of mRNA-like noncoding transcripts in Medicago truncatula. In Silico Biol. 2007, 7: 485-505.
Xin M, Wang Y, Yao Y, Song N, Hu Z, Qin D, Xie C, Peng H, Ni Z, Sun Q: Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC Plant Biol. 2011, 11: 61-
Cho J, Koo DH, Nam YH, Han CK, Lim HT, Bang JW, Hur Y: Isolation and characterization of cDNA clones expressed under male sex expression conditions in a monoecious cucumber plant (Cucumis sativus L.cv. Winter Long). Euphytica. 2005, 146: 271-281.
Kouchi H, Takane K, So RB, Ladha JK, Reddy PM: Rice ENOD40: isolation and expression analysis in rice and transgenic soybean root nodules. Plant J. 1999, 18: 121-129.
Ma J, Yan B, Qu Y, Qin F, Yang Y, Hao X, Yu J, Zhao Q, Zhu D, Ao G: Zm401, a short-open reading-frame mRNA or noncoding RNA, is essential for tapetum and microspore development and can regulate the floret formation in maize. J Cell Biochem. 2008, 105: 136-146.
Rohrig H, John M, Schmidt J: Modification of soybean sucrose synthase by S-thiolation with ENOD40 peptide A. Biochem Biophys Res Commun. 2004, 325: 864-870.
Rohrig H, Schmidt J, Miklashevichs E, Schell J, John M: Soybean ENOD40 encodes two peptides that bind to sucrose synthase. Proc Natl Acad Sci USA. 2002, 99: 1915-1920.
Sugiyama R, Kazama Y, Miyazawa Y, Matsunaga S, Kawano S: CCLS96.1, a member of a multicopy gene family, may encode a non-coding RNA preferentially transcribed in reproductive organs of Silene latifolia. DNA Res. 2003, 10: 213-220.
Vleghels I, Hontelez J, Ribeiro A, Fransz P, Bisseling T, Franssen H: Expression of ENOD40 during tomato plant development. Planta. 2003, 218: 42-49.
Burleigh SH, Harrison MJ: The down-regulation of Mt4-like genes by phosphate fertilization occurs systemically and involves phosphate translocation to the shoots. Plant Physiol. 1999, 119: 241-248.
Heo JB, Sung S: Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science. 2011, 331: 76-79.
Liu C, Muchhal US, Raghothama KG: Differential expression of TPS11, a phosphate starvation-induced gene in tomato. Plant Mol Biol. 1997, 33: 867-874.
Shin H, Shin HS, Chen R, Harrison MJ: Loss of At4 function impacts phosphate distribution between the roots and the shoots during phosphate starvation. Plant J. 2006, 45: 712-726.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000, 25: 25-29.
Kanehisa M, Goto S: KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28: 27-30.
Kreis W: Biochemistry of sterols, cardiac glycosides, brassinosteroids, phytoecdysteroids and steroid saponins. Annual plant reviews. Edited by: Wink M. 2010, Oxford: Garsington Road, 40: 304-363. 2
Usai M, Atzei AD, Marchetti M: Cardenolides content in wild Sardinian Digitalis purpurea L. populations. Nat Prod Res. 2007, 21: 798-804.
Xiao B, Zhang X, Li Y, Tang Z, Yang S, Mu Y, Cui W, Ao H, Li K: Identification, bioinformatic analysis and expression profiling of candidate mRNA-like non-coding RNAs in Sus scrofa. J Genet Genomics. 2009, 36: 695-702.
Inagaki S, Numata K, Kondo T, Tomita M, Yasuda K, Kanai A, Kageyama Y: Identification and expression analysis of putative mRNA-like non-coding RNA in Drosophila. Genes Cells. 2005, 10: 1163-1173.
Szell M, Bata-Csorgo Z, Kemeny L: The enigmatic world of mRNA-like ncRNAs: their role in human evolution and in human diseases. Semin Cancer Biol. 2008, 18: 141-148.
Zhang BH, Pan XP, Wang QL, Cobb GP, Anderson TA: Identification and characterization of new plant microRNAs using EST analysis. Cell Res. 2005, 15: 336-360.
Kozomara A, Griffiths-Jones S: miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39: D152-157.
Zuker M: Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31: 3406-3415.
Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP: Prediction of plant microRNA targets. Cell. 2002, 110: 513-520.
Dai X, Zhuang Z, Zhao PX: Computational analysis of miRNA targets in plants: current status and challenges. Briefings in bioinformatics. 2011, 12: 115-121.
Zhang Y: miRU: an automated plant miRNA target prediction server. Nucleic Acids Res. 2005, 33: W701-704.
Lu S, Sun YH, Chiang VL: Stress-responsive microRNAs in Populus. Plant J. 2008, 55: 131-151.
Lu S, Sun YH, Shi R, Clark C, Li L, Chiang VL: Novel and mechanical stress-responsive microRNAs in Populus trichocarpa that are absent from Arabidopsis. Plant Cell. 2005, 17: 2186-2203.
Loeffler C, Berger S, Guy A, Durand T, Bringmann G, Dreyer M, von Rad U, Durner J, Mueller MJ: B1-phytoprostanes trigger plant defense and detoxification responses. Plant Physiol. 2005, 137: 328-340.
Reddy VS, Ali GS, Reddy AS: Genes encoding calmodulin-binding proteins in the Arabidopsis genome. J Biol Chem. 2002, 277: 9840-9852.
Soitamo AJ, Piippo M, Allahverdiyeva Y, Battchikova N, Aro EM: Light has a specific role in modulating Arabidopsis gene expression at low temperature. BMC Plant Biol. 2008, 8: 13-
Gong Z, Dong CH, Lee H, Zhu J, Xiong L, Gong D, Stevenson B, Zhu JK: A DEAD box RNA helicase is essential for mRNA export and important for development and stress responses in Arabidopsis. Plant Cell. 2005, 17: 256-267.
Kant P, Kant S, Gordon M, Shaked R, Barak S: STRESS RESPONSE SUPPRESSOR1 and STRESS RESPONSE SUPPRESSOR2, two DEAD-box RNA helicases that attenuate Arabidopsis responses to multiple abiotic stresses. Plant Physiol. 2007, 145: 814-830.
Kim JS, Kim KA, Oh TR, Park CM, Kang H: Functional characterization of DEAD-box RNA helicases in Arabidopsis thaliana under abiotic stress conditions. Plant Cell Physiol. 2008, 49: 1563-1571.
Li D, Liu H, Zhang H, Wang X, Song F: OsBIRH1, a DEAD-box RNA helicase with functions in modulating defence responses against pathogen infection and oxidative stress. J Exp Bot. 2008, 59: 2133-2146.
Ascencio-Ibanez JT, Sozzani R, Lee TJ, Chu TM, Wolfinger RD, Cella R, Hanley-Bowdoin L: Global analysis of Arabidopsis gene expression uncovers a complex array of changes impacting pathogen response and cell cycle during geminivirus infection. Plant Physiol. 2008, 148: 436-454.
Yabe N, Takahashi T, Komeda Y: Analysis of tissue-specific expression of Arabidopsis thaliana HSP90-family gene HSP81. Plant Cell Physiol. 1994, 35: 1207-1219.
He S, Liu C, Skogerbo G, Zhao H, Wang J, Liu T, Bai B, Zhao Y, Chen R: NONCODE v2.0: decoding the non-coding. Nucleic Acids Res. 2008, 36: D170-172.
Numata K, Kanai A, Saito R, Kondo S, Adachi J, Wilming LG, Hume DA, Hayashizaki Y, Tomita M: Identification of putative noncoding RNAs among the RIKEN mouse full-length cDNA collection. Genome Res. 2003, 13: 1301-1306.
Taylor CB, Green PJ: Identification and characterization of genes with unstable transcripts (GUTs) in tobacco. Plant Mol Biol. 1995, 28: 27-38.
van Hoof A, Kastenmayer JP, Taylor CB, Green PJ: GUT15 cDNAs from tobacco (Accession No. U84972) and Arabidopsis (Accession No. U84973) correspond to transcripts with unusual metabolism and a short conserved ORF (PGR97-048). Plant Physiol. 1997, 113: 1004-
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F: Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3: RESEARCH0034-
Gil MJ, Coego A, Mauch-Mani B, Jorda L, Vera P: The Arabidopsis csb3 mutant reveals a regulatory link between salicylic acid-mediated disease resistance and the methyl-erythritol 4-phosphate pathway. Plant J. 2005, 44: 155-166.
Kim SM, Kim SU: Characterization of 1-hydroxy-2-methyl-2-(E)-butenyl-4-diphosphate synthase (HDS) gene from Ginkgo biloba. Mol Biol Rep. 2010, 37: 973-979.
Querol J, Campos N, Imperial S, Boronat A, Rodriguez-Concepcion M: Functional analysis of the Arabidopsis thalian a GCPE protein involved in plastid isoprenoid biosynthesis. FEBS Lett. 2002, 514: 343-346.
Sando T, Takeno S, Watanabe N, Okumoto H, Kuzuyama T, Yamashita A, Hattori M, Ogasawara N, Fukusaki E, Kobayashi A: Cloning and characterization of the 2-C-methyl-D-erythritol 4-phosphate (MEP) pathway genes of a natural-rubber producing plant, Hevea brasiliensis. Biosci Biotechnol Biochem. 2008, 72: 2903-2917.
Hirooka K, Bamba T, Fukusaki E, Kobayashi A: Cloning and kinetic characterization of Arabidopsis thaliana solanesyl diphosphate synthase. Biochem J. 2003, 370: 679-686.
Khachane AN, Harrison PM: Mining mammalian transcript data for functional long non-coding RNAs. PLoS One. 2010, 5: e10316-
Pagano A, Castelnuovo M, Tortelli F, Ferrari R, Dieci G, Cancedda R: New small nuclear RNA gene-like transcriptional units as sources of regulatory transcripts. PLoS Genet. 2007, 3: e1-
Royo H, Cavaille J: Non-coding RNAs in imprinted gene clusters. Biol Cell. 2008, 100: 149-166.
Xie DY, Jackson LA, Cooper JD, Ferreira D, Paiva NL: Molecular and biochemical analysis of two cDNA clones encoding dihydroflavonol-4-reductase from Medicago truncatula. Plant Physiol. 2004, 134: 979-994.
Bartley GE, Scolnik PA: Plant carotenoids: pigments for photoprotection, visual attraction, and human health. Plant Cell. 1995, 7: 1027-1038.
Facchini PJ, Huber-Allanach KL, Tari LW: Plant aromatic L-amino acid decarboxylases: evolution, biochemistry, regulation, and metabolic engineering applications. Phytochemistry. 2000, 54: 121-138.
Lehmann T, Pollmann S: Gene expression and characterization of a stress-induced tyrosine decarboxylase from Arabidopsis thaliana. FEBS Lett. 2009, 583: 1895-1900.
Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL, Carrington JC: High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One. 2007, 2: e219-
Gutierrez L, Bussell JD, Pacurar DI, Schwambach J, Pacurar M, Bellini C: Phenotypic plasticity of adventitious rooting in Arabidopsis is controlled by complex regulation of AUXIN RESPONSE FACTOR transcripts and microRNA abundance. Plant Cell. 2009, 21: 3119-3132.
Hardtke CS, Berleth T: The Arabidopsis gene MONOPTEROS encodes a transcription factor mediating embryo axis formation and vascular development. EMBO J. 1998, 17: 1405-1411.
Harper RM, Stowe-Evans EL, Luesse DR, Muto H, Tatematsu K, Watahiki MK, Yamamoto K, Liscum E: The NPH4 locus encodes the auxin response factor ARF7, a conditional regulator of differential growth in aerial Arabidopsis tissue. Plant Cell. 2000, 12: 757-770.
Schruff MC, Spielman M, Tiwari S, Adams S, Fenby N, Scott RJ: The AUXIN RESPONSE FACTOR 2 gene of Arabidopsis links auxin signalling, cell division, and the size of seeds and other organs. Development. 2006, 133: 251-261.
Sessions A, Nemhauser JL, McColl A, Roe JL, Feldmann KA, Zambryski PC: ETTIN patterns the Arabidopsis floral meristem and reproductive organs. Development. 1997, 124: 4481-4491.
Tatematsu K, Kumagai S, Muto H, Sato A, Watahiki MK, Harper RM, Liscum E, Yamamoto KT: MASSUGU2 encodes Aux/IAA19, an auxin-regulated protein that functions together with the transcriptional activator NPH4/ARF7 to regulate differential growth responses of hypocotyl and formation of lateral roots in Arabidopsis thaliana. Plant Cell. 2004, 16: 379-393.
Varaud E, Brioudes F, Szecsi J, Leroux J, Brown S, Perrot-Rechenmann C, Bendahmane M: AUXIN RESPONSE FACTOR8 regulates Arabidopsis petal growth by interacting with the bHLH transcription factor BIGPETALp. Plant Cell. 2011, 23: 973-983.
Yant L, Mathieu J, Dinh TT, Ott F, Lanz C, Wollmann H, Chen X, Schmid M: Orchestration of the floral transition and floral development in Arabidopsis by the bifunctional transcription factor APETALA2. Plant Cell. 2010, 22: 2156-2170.
Liu D, Song Y, Chen Z, Yu D: Ectopic expression of miR396 suppresses GRF target gene expression and alters leaf growth in Arabidopsis. Physiol Plant. 2009, 136: 223-236.
Rodriguez RE, Mecchia MA, Debernardi JM, Schommer C, Weigel D, Palatnik JF: Control of cell proliferation in Arabidopsis thaliana by microRNA miR396. Development. 2010, 137: 103-112.
Campalans A, Kondorosi A, Crespi M: Enod40, a short open reading frame-containing mRNA, induces cytoplasmic localization of a nuclear RNA binding protein in Medicago truncatula. Plant Cell. 2004, 16: 1047-1059.
Dai X, Yu J, Ma J, Ao G, Zhao Q: Overexpression of Zm401, an mRNA-like RNA, has distinct effects on pollen development in maize. Plant Growth Regul. 2007, 52: 229-139.
Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio-Somoza I, Leyva A, Weigel D, Garcia JA, Paz-Ares J: Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet. 2007, 39: 1033-1037.
Kim DH, Zografos BR, Sung S: Mechanisms underlying vernalization-mediated VIN3 induction in Arabidopsis. Plant Signal Behav. 2010, 5:
Szymanski M, Barciszewska MZ, Zywicki M, Barciszewski J: Noncoding RNA transcripts. J Appl Genet. 2003, 44: 1-19.
Carthew RW, Sontheimer EJ: Origins and mechanisms of miRNAs and siRNAs. Cell. 2009, 136: 642-655.
Chen X: Small RNAs - secrets and surprises of the genome. Plant J. 2010, 61: 941-958.
Vazquez F, Legrand S, Windels D: The biosynthetic pathways and biological scopes of plant small RNAs. Trends Plant Sci. 2010, 15: 337-345.
Duret L, Chureau C, Samain S, Weissenbach J, Avner P: The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science. 2006, 312: 1653-1655.
Elisaphenko EA, Kolesnikov NN, Shevchenko AI, Rogozin IB, Nesterova TB, Brockdorff N, Zakian SM: A dual origin of the Xist gene from a protein-coding gene and a set of transposable elements. PLoS One. 2008, 3: e2521-
Napoli C, Lemieux C, Jorgensen R: Introduction of a chimeric chalcone synthase gene into Petunia results in reversible co-suppression of homologous genes in trans. Plant Cell. 1990, 2: 279-289.
Smith CJ, Watson CF, Bird CR, Ray J, Schuch W, Grierson D: Expression of a truncated tomato polygalacturonase gene inhibits expression of the endogenous gene in transgenic plants. Mol Gen Genet. 1990, 224: 477-481.
van der Krol AR, Mur LA, Beld M, Mol JN, Stuitje AR: Flavonoid genes in petunia: addition of a limited number of gene copies may lead to a suppression of gene expression. The Plant cell. 1990, 2: 291-299.
Lottaz C, Iseli C, Jongeneel CV, Bucher P: Modeling sequencing errors by combining Hidden Markov models. Bioinformatics. 2003, 19 (Suppl 2): ii103-112.
Gardner PP, Daub J, Tate J, Moore BL, Osuch IH, Griffiths-Jones S, Finn RD, Nawrocki EP, Kolbe DL, Eddy SR, Bateman A: Rfam: Wikipedia, clans and the "decimal" release. Nucleic Acids Res. 2011, 39: D141-145.
Lelandais-Briere C, Naya L, Sallet E, Calenge F, Frugier F, Hartmann C, Gouzy J, Crespi M: Genome-wide Medicago truncatula small RNA analysis revealed novel microRNAs and isoforms differentially regulated in roots and nodules. Plant Cell. 2009, 21: 2780-2796.
Zhang BH, Pan XP, Cox SB, Cobb GP, Anderson TA: Evidence that miRNAs are different from other RNAs. Cell Mol Life Sci. 2006, 63: 246-254.
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001, 25: 402-408.
Acknowledgements
We thank Professor Yulin Lin for identifying the plants of D. purpurea. This work was supported by Natural Science Foundation of China (81072993), Beijing Natural Science Foundation (5112026), and Program for Xiehe Scholars in Chinese Academy of Medical Sciences & Peking Union Medical College.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
BW analyzed the data, performed qRT-PCR and RACE, and participated in writing the manuscript. YL and HL constructed the EST library. YL, HY, YM and LY participated in data analysis. SC participated in designing the experiment. SL designed the experiment and wrote the manuscript. All authors have read and approved the version of manuscript.
Electronic supplementary material
12864_2011_3832_MOESM1_ESM.PDF
Additional file 1: Unigenes probably involved in cardiac glycoside biosynthesis. Complete set of unigenes probably involved in cardiac glycoside biosynthesis. (PDF 86 KB)
12864_2011_3832_MOESM2_ESM.PDF
Additional file 2: The 25 most abundant unigenes in D. purpurea. Complete set of the 25 most abundant unigenes in D. purpurea. (PDF 10 KB)
12864_2011_3832_MOESM6_ESM.PDF
Additional file 6: Expression of housekeeping genes. (A) Cq value of ubiquitin, actin and 18s rRNA in leaves (L), stems (S), flowers (F) and roots (R) of one-year-old D. purpurea plants and plantlets treated with cold (C0, C1, C5, C10 and C24) and dehydration (D0, D1, D5, D10 and D24) stresses for 0, 1, 5, 10 and 24 hours. Expression levels were quantified by qPCR. The mean Cq value of three PCR replicates is shown. (B) The expression stability value (M) was calculated by geNorm software [87]. (PDF 59 KB)
12864_2011_3832_MOESM7_ESM.PDF
Additional file 7: Protein-coding genes significantly homologous or complementary to mlncRNAs. Complete set of the protein-coding genes significantly homologous or complementary to mlncRNAs. (PDF 69 KB)
12864_2011_3832_MOESM8_ESM.PDF
Additional file 8: The putative biosynthetic pathway of cardiac glycosides in Digitalis purpurea. The pathway roughly comprises terpenoid backbone biosynthesis, steroid biosynthesis and cardenolide biosynthesis three stages. The enzymes with corresponding unigenes are framed. (PDF 150 KB)
12864_2011_3832_MOESM10_ESM.PDF
Additional file 10: Primers used for indentifying the transcriptional direction of mlncRNAs. Complete set of the primers used for indentifying the transcriptional direction of mlncRNAs. (PDF 50 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wu, B., Li, Y., Yan, H. et al. Comprehensive transcriptome analysis reveals novel genes involved in cardiac glycoside biosynthesis and mlncRNAs associated with secondary metabolism and stress response in Digitalis purpurea. BMC Genomics 13, 15 (2012). https://doi.org/10.1186/1471-2164-13-15
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-13-15