
Abstract
Background
Camptotheca acuminata is a Nyssaceae plant, often called the "happy tree", which is indigenous in Southern China. C. acuminata produces the terpenoid indole alkaloid, camptothecin (CPT), which exhibits clinical effects in various cancer treatments. Despite its importance, little is known about the transcriptome of C. acuminata and the mechanism of CPT biosynthesis, as only few nucleotide sequences are included in the GenBank database.
Results
From a constructed cDNA library of young C. acuminata leaves, a total of 30,358 unigenes, with an average length of 403 bp, were obtained after assembly of 74,858 high quality reads using GS De Novo assembler software. Through functional annotation, a total of 21,213 unigenes were annotated at least once against the NCBI nucleotide (Nt), non-redundant protein (Nr), Uniprot/SwissProt, Kyoto Encyclopedia of Genes and Genomes (KEGG), and Arabidopsis thaliana proteome (TAIR) databases. Further analysis identified 521 ESTs representing 20 enzyme genes that are involved in the backbone of the CPT biosynthetic pathway in the library. Three putative genes in the upstream pathway, including genes for geraniol-10-hydroxylase (CaPG10H), secologanin synthase (CaPSCS), and strictosidine synthase (CaPSTR) were cloned and analyzed. The expression level of the three genes was also detected using qRT-PCR in C. acuminata. With respect to the branch pathway of CPT synthesis, six cytochrome P450s transcripts were selected as candidate transcripts by detection of transcript expression in different tissues using qRT-PCR. In addition, one glucosidase gene was identified that might participate in CPT biosynthesis. For CPT transport, three of 21 transcripts for multidrug resistance protein (MDR) transporters were also screened from the dataset by their annotation result and gene expression analysis.
Conclusion
This study produced a large amount of transcriptome data from C. acuminata by 454 pyrosequencing. According to EST annotation, catalytic features prediction, and expression analysis, novel putative transcripts involved in CPT biosynthesis and transport were discovered in C. acuminata. This study will facilitate further identification of key enzymes and transporter genes in C. acuminata.
Similar content being viewed by others

Background
Camptothecin (CPT) was first extracted from the stems of Camptotheca acuminata in 1966 and subsequently from Nothapodytes foetida, Ophiorrhiza pumila, and Ophiorrhiza japonica[1]. CPT exhibits clinical anti-tumor activity by inhibiting DNA topoisomerase I, an enzyme involved in DNA recombination, repair, replication, and transcription [2]. CPT also inhibits the retroviruses, such as the human immunodeficiency virus [3]. Despite its significant clinical use, the main source of CPT is still from its extraction from C. acuminata. However, the quantity is quite limited and cannot meet worldwide demand. Studies on the molecular mechanism of CPT biosynthesis have long been hindered by the lack of transcriptome and genome information for C. acuminata and other CPT-producing plants. Therefore, it is necessary to obtain transcriptome data and screen candidate transcripts involved in CPT biosynthesis to further understand the CPT biosynthetic pathway.
CPT is synthesized through a modified terpenoid indole alkaloid (TIA) pathway. The upstream biosynthesis pathways for all the TIA products are similar among alkaloid-producing plants, and involve a strictosidine backbone (Figure 1A). Over recent decades, several enzymes in the process of strictosidine biosynthesis in C. acuminata have been isolated and functionally identified. Among them are tryptophan synthase (TSB) [4] and tryptophan decarboxylase (TDC) [5], which are involved in the synthesis of the indole precursor tryptamine, 3-hydroxy-3-methylglutaryl-CoA synthase (HMGR) [6], 1-deoxy-D-xylulose-5-phosphate reductoisomeras (DXR) [7], and 10-hydroxy geraniol oxidoreductase (10HGO) [8] are involved in secologanin synthesis.

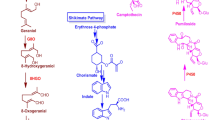
Biosynthetic pathway of CPT from DMAPP to strictosidine and from strictosidine to CPT in C. acuminata. (A) The upstream pathway for the synthesis of backbone strictosidine. (B) The proposed branch pathway of CPT biosynthesis (steps after strictosidine synthesis). TSB: β-subunit of tryptophan synthase; TDC: tryptophan decarboxylase; G10H: geraniol-10-hydroxylase; SCS: secologanin synthase; STR: strictosidine synthase; 10-HGO: 10-hydroxy geraniol oxidoreductase. PGD: putative strictosidine β-D-glucosidase. The arrow with the dotted shaft represents the step that was presumed in the study to be catalyzed by a CYP450.
G10H and SCS, belonging to the CYP76B6 and CYP72A1subfamilies of cytochrome P450 family respectively, were identified in monoterpenoid biosynthesis from Catharanthus roseus[9, 10]. The synthesis of strictosidine is finally catalyzed by STR, a committed enzyme for the CPT backbone biosynthesis, which was isolated and identified in Rauvolfia serpentine, C. roseus, the CPT-producing plant O. japonica, and O. pumila, in previous studies. However, the genes encoding CaG10H, CaSCS and CaSTR, have not been yet cloned and characterized in C. acuminata.
The steps following strictosidine formation (branch pathway) are not very clear and only a proposed biosynthetic pathway based on relative compounds extracted from CPT-producing plants has been reported [11] (Figure 1B). In the proposed pathway, a series of oxidation and hydroxylation reactions are involved in some steps of the pathway which are probably catalyzed by monooxygenases and hydroxylase, belonging to the superfamily of cytochrome P450s [12, 13]. Meanwhile, the branch pathway of CPT biosynthesis is unique among TIA pathways because strictosidine is not immediately deglycosylated as in C. roseus[14]. However, it requires a glucosidase for glycoside hydrolysis, which likely occurs in one of the last steps of CPT biosynthesis in C. acuminata and other CPT-producing plants. At present, the CYP450s and glucosidase involved in CPT biosynthesis have not been studied in C. acuminata.
Glandular trichomes in leaves are the main site for CPT accumulation in C. acuminata[15]. However, gene expression involved in CPT synthesis was not detected in glandular trichomes but, instead, in epidermal cells and mesophyll cells in C. acuminata leaves, which implied the translocation of CPT between organs or cells [16]. Multidrug resistance protein (MDR) transporters, belonging to the ATP-binding cassette (ABC) transporter family, were reported to be responsible for uptake or secretion in alkaloid transportation in some plants [17–19]. Therefore, we hypothesized that MDR transporters are responsible for CPT transportation from other cells to glandular trichomes in C. acuminata. At present, no CPT transport mechanism or related genes have been investigated in C. acuminata.
Expressed sequence tags (ESTs) analysis has been a primary tool for the discovery of novel genes, based on the traditional Sanger sequencing principle, which is slow and costly for non-model species with little genomic information. The emergence of high throughput platforms, such as pyrosequencing technology [20], enables comprehensive study of the transcriptome for various purposes, such as development study, miRNA identification, and genetic polymorphisms discovery in plants and animals [21–23]. The Roche/454 GS FLX platform, one of the high throughput sequencing platforms, offers the advantages of longer read length and lower cost which is especially suitable for de novo transcriptome sequencing aimed at gene discovery and analysis in a specific metabolic pathway [24, 25]. Previous studies have indicated that the content of CPT in young leaves is higher than that in old leaves and root [26, 27], suggesting that young leaf is an important tissue for the study of CPT biosynthesis and transport. Therefore, cDNA from C. acuminata young leaves was subjected to de novo transcriptome sequencing to uncover genes involved in CPT biosynthesis and transport, using a Roche/454 GS FLX titanium sequencing platform, a next-generation sequencing system. Based on the sequencing and analysis results, three important genes likely to be involved in the CPT biosynthesis were cloned and analyzed. From data analysis and expression analysis, six cytochrome P450s and one glucosidase gene were found to be candidate genes in the process of CPT biosynthesis. Meanwhile, three MDR transporter genes were also found to be candidate genes involved in CPT transportation.
Results and Discussion
454 sequencing and EST assembly
Through 454 deep pyrosequencing, 74,858 high-quality (HQ, > 99.5% accuracy on single base reads) reads were generated and then submitted to the Sequence Read Archive of NCBI with an accession number SRX033123. The total length of all the reads is 28,746,026 bp, and the average size is 384 bp. After sequence assembly, 30,358 unigenes, with an average length of 403 bp, were generated, including 9,145 contigs and 21,213 singletons. The average coverage was 3.72-fold. The assembled contigs ranged from 96-3848 bp, with a mean length of 525 bp, including 8,485 contigs which were more than 200 bp (about 92.8%). The singletons ranged from 50 bp to 608 bp, with an average length of 351 bp. The length distribution of HQ reads (Figure 2A) and assembled contigs (Figure 2B) are shown for evaluation of the quality of the library. A summary of the sequencing and assembly results is provided in Table 1.

Primary sequencing results for the cDNA library of C. acuminata. (A) Length distribution for ESTs of the 454 dataset. (B) Length distribution of the assembled contigs of the cDNA library.
Annotation and categorization
A total of 21,213 unigenes (69.87%, 21,213/30,358) were functionally characterized against the NCBI nucleotide (Nt), non-redundant protein (Nr), Uniprot/SwissProt, Kyoto Encyclopedia of Genes and Genomes (KEGG), and Arabidopsis thaliana proteome (TAIR) databases [28–32]. An overview of the annotation statistics against public databases (Additional file 1A) and a summary of the most abundant (Additional file 1B) and longest transcripts of the dataset (Additional file 1C) are listed in the supporting information.
To functionally categorize the information in this EST pool, all unigenes were characterized by Gene Ontology (GO) analysis, provided by the TAIR database. A total of 18,172 unigenes were classified into three large categories and forty-five subcategories, based on GO classification [33], accounting for approximately 60% of all the unigenes (Additional file 2).
Transcripts for proteins involved in the backbone biosynthetic pathway of CPT
Putative strictosidine synthesis genes discovered in the dataset
Strictosidine is the precursor and backbone of many TIAs, including CPTs, in plants such as C. acuminata. A proposed biosynthetic pathway of strictosidine is shown in Figure 1, and each of the main enzymes present in the dataset is marked with a bold box (Figure 1-A). From the 454 data pool, 521 ESTs representing 20 enzyme genes involved in strictosidine biosynthesis were discovered. Thirteen of these genes had not been previously reported, including the important enzymes geraniol-10-hydroxylase (G10H), secologanin synthase (SCS), and strictosidine synthase (STR) (Table 2). By searching the annotation information from the Nr, Swissprot, and KEGG databases, we found that transcripts of 1-deoxy-D-xylulose-5-phosphate reductoisomerase (DXR), 10HGO, and SCS were presented many ESTs, indicating that they are highly expressed in the young leaves of C. acuminata. G10H and TDC were both rare transcripts in the dataset, indicating that they are rarely expressed and are possibly rate-limiting genes in the tissue. The specific annotation information of some putative transcripts against the Nr, Swissprot and KEGG databases is shown in the supporting information (Additional file 3).
G10H, SCS, and STR are the most important enzymes in the synthesis of strictosidine in TIA-producing plants, including C. acuminata. CrG10H, the first CYP450 in CPT synthesis, is a rate-limiting enzyme in the process of TIA synthesis in C. roseus. In the 454 dataset, only one read of G10H was found and it had approximately 60% identical to the G10H gene of C. roseus and Swertia mussotii. Based on the EST sequence, a putative G10H gene in C. acuminata (CaPG10H) gene was cloned (GenBank ID: JF508378) and analyzed. Similarity analysis of the amino acid sequence showed that CaPG10H shared 56% identity to CrG10H, which implied it may have catalytic activity in geraniol hydroxylation process as in C. roseus (Additional file 4) . SCS, the second CYP450, is the last enzyme in the biosynthesis of secologanin. Unigenes, assembled from 165 reads in our library, were annotated to the CrSCS. One putative SCS gene in C. acuminata (CaPSCS) was cloned (GenBank ID: HQ605982), according to a contig that had annotated to the CrSCS gene. The molecular weight of the predicted protein was approximately 60 kDa. Protein subcellular localization prediction using the WoLF PSORT program [34] indicated that the presumed protein was likely targeted to the endoplasmic reticulum (ER) membrane. Protein alignment revealed that the predicted protein shared 68% amino acid identity to that in C. roseus, which is involved in a similar terpenoid indole alkaloids biosynthetic pathway (Additional file 5). From the analysis, we inferred that the CaPSCS gene may play a role in secologanin biosynthesis in C. acuminata. STR is the enzyme that catalyzes the reaction of strictosidine synthesis. We cloned the ORF of a putative STR gene (CaPSTR, GenBank ID: JF508375) from C. acuminata. Phylogenetic analysis showed that STR proteins from reported alkaloid-producing plants were clustered together, which implied that the candidate gene possibly plays a role in CPT biosynthesis (Figure 3A).

Phylogenetic tree analysis of strictosidine synthases (STR) and the expression profile detection of CaG10H, CaSCS , and CaSTR. (A) Protein sequences for 22 STRs were aligned using the ClustalW module and phylogenetic tree was constructed using MEGA 4.0. (B) Relative expression of three genes after induction by MeJA. Expression levels in young leaves without treatment served as controls (M0). M1, M3, M6, M12 and M24 indicate that the treatment times of 1 h, 3 h, 6 h, 12 h and 24 h, respectively. (C) The quantification of three genes involved in CPT biosynthesis in different tissues. Expression levels in young leaves served as controls. Y: young leaves; O: old leaves; P: petioles; S: stems; H: root bark; R: root.
Expression analysis of transcripts for proteins involved in strictosidine synthesis
Methyl jasmonate (MeJA)-induced accumulation of secondary metabolites and related gene expression has been reported in medicinal plants such as Panax ginseng and C. roseus[35–37]. A previous report determined that the CPT content responded to MeJA and jasmonic acid, and that the response curve for jasmonic acid treatment was a waveform, with two time-specific CPT accumulation peaks in C. acuminata suspension cells [38]. However, there are few reports of the effect of MeJA treatment on the expression of genes of CPT biosynthesis in C. acuminata. In response to MeJA treatment, transcripts of G10H, SCS and STR were regulated in a waveform manner, including two expression peaks during 24 hours of induction (Figure 3B). The trend of the curve was consistent with the result of a previous report for genes in anthocyanin biosynthesis [39]. In this study, all the detected genes responded to MeJA immediately, with a common peak within one hour of induction, and then decreased rapidly to even lower levels than the control. The expression levels increased again to the second peak, whose timing was gene-specific. Therefore, we speculate that transcripts of CaG10H, CaSCS, and CaSTR were most likely to be involved in CPT biosynthesis. It has been reported that TDC genes, which are responsible for the production of tryptamine for auxin and CPTs, do not respond to MeJA [40]. The expression of HMGR genes is even inhibited by MeJA in C. acuminata[41]. This is likely to be because the substrates of TDCs and HMGRs link primary and secondary metabolism, and their expression profiles are complicated.
Previous reports had shown that young and actively growing tissues, showed the highest level of CPT [26]. In this study, the mRNA levels of CaG10H, CaSCS and CaSTR were detected using real-time PCR. The results demonstrated that the expression levels of the three genes were all higher in young leaves and petioles than in old leaves (Figure 3C). Therefore, young leaves and young petioles are the possible sites of active CPT synthesis, as well as sites of accumulation, in C. acuminata compared with the mature tissues. This difference served as a standard for real-time PCR detection for downstream candidate gene selection [42]. Meanwhile, the expression levels of these genes were also relatively lower in the root and root bark which implied that root may not be a main synthetic tissue. This was consistent with the expression pattern of the TDC1 and 10HGO genes reported in a previous study [16].
Transcripts for proteins likely to be involved in the branch pathway of CPT synthesis
Strictosidine rapidly forms the intermediate product strictosamide in C. acuminata. The steps after strictosamide synthesis remain somewhat unclear. Based on the proposed branch steps, an intermediate step between strictosamide and 3(S)-pumiloside in the CPT biosynthetic pathway was presumed to be catalyzed by a cytochrome P450, with another P450 possibly in the last steps of CPT biosynthesis (Figure 1B). Cytochrome P450s, are a large and complex superfamily, which play important roles through catalysis of oxidation and hydroxylation reactions. In C. acuminata, no cytochrome P450 involved in the downstream CPT biosynthetic pathway had been cloned and identified. After EST annotation against the Swissprot database, 99 putative cytochrome P450 transcripts were identified in the 454 ESTs pool (Additional file 6), belonging to 28 cytochrome P450 subfamilies, according to the standard CYP family categories (Additional file 7). According to clan classification, transcripts of CYP71 clan and CYP72 subfamilies are likely to be involved in secondary metabolism [43]. A total of 27 cytochrome P450 transcripts belonging to these two subfamilies were discovered as candidate genes for further screening. Glucosidases, which is a superfamily involved in various biological process including cell wall assembly, polysaccharides, plant defense and secondary metabolism, catalyze the action of deglycosylation [44]. It had been reported that β-D-glucosidase plays a role in glycoside hydrolysis in TIA biosynthesis in plants such as C. roseus[11], Psychotria ipecacuanha[45] and R. serpentine[46, 47]. In the 454 dataset, one transcript (contig 00133) annotated as strictosidine β-D-glucosidase in C. roseus (CrSGD) was identified with a predicted homologous peptide of 178 amino acids. The peptide was found to share 70% similarity to amino acids 47-224 of CrSGD and 62% similarity to amino acids 18-195 of the β-D-glucosidase IpeGlu1 of Psychotria ipecacuanha (PiIpeGlu1), which is involved in ipecac alkaloid synthesis. When compared with raucaffricine-O-beta-D-glucosidase (RsRD) and SGD (RsSGD) of another TIA-producing plant, R. serpentine, these proteins showed 69% and 74.8% (Additional file 8) similarity, respectively. CrSGD, RsRD, RsSGD and IpeGlu1 all belong to the glycosyl hydrolase (GH) family, which catalyzes the deglycosylation reaction in the TIA pathway, and their substrates are strictosidine, raucaffricine and N-deacetyli(so)pecoside, respectively. The predicted glucosidase peptide demonstrated high amino acid similarity with the glucosidases identified above as being involved in the alkaloid biosynthetic pathway. Therefore, CaPGD is likely to be a key enzyme in CPT synthesis through removal of a glucose moiety. Analysis of the amino acids of the predicted peptide demonstrated that three key amino acids 161-His, 207-Glu and 210-Thr, which were key amino acids for catalytic activity [47], are found in the corresponding residues of the predicted peptide of the PGD transcript in C. acuminata. The 388-Trp was not included in the peptide.
After initial screening, relative expression analysis in young/old leaves of the 27 cytochrome P450s and the CaPGD was performed for C. acuminata. Consequently, six cytochrome P450 transcripts and one CaPGD transcript that were shown to be expressed three-fold higher than the control were identified as candidate genes for participating in the branch pathway of CPT biosynthesis (Figure 4).

Expression analysis of the cytochrome P450s and CaPGD transcripts in different tissues of the dataset. The expression in old leaves was set to be the control. O: old leaves; Y: young leaves. 1-27 represent 27 cytochrome P450 transcripts in this dataset.
Transcripts for proteins likely to participate in CPT transport
MDR is a subfamily of the ABC transporter family that has been reported to be related to the transport of alkaloids metabolites [18]. From the annotated databases, 21 MDR transporters were found in the 132 ABC transporter transcripts in the library. Some of the transcripts were possibly responsible for CPT transport from synthesis site to the glandular trichomes in leaves through the plasma membrane [48]. Previous studies showed that the CPT content was four to five-fold higher in young C. acuminata leaves compared with mature leaves [49]. It is possible that CPT transporters were more abundant in the young leaves than in mature ones [50]. Subsequently, the 21 annotated MDR transporter transcripts were subjected to expression analysis in young leaves and old leaves of C. acuminata by real-time PCR. The results showed that the expression level of three transcripts (FXAT9O006HB5TT, FXAT9O006HKTK5, and contig05927) among the annotated MDR transporters were three-fold higher in young leaves than in the mature leaves. Thus, they represent candidate genes for CPT transportation in leaves (Figure 5).

Expression analysis of MDR transcripts in the dataset. O: old leaves; Y: young leaves. The gene expression in old leaves was served as the control. 1-21 were 21 MDR transcripts in the annotated dataset.
Probable site of CPT biosynthesis
Young leaves are the main site for CPT accumulation; therefore, this tissue was used to identify new genes in CPT biosynthetic pathway by high throughput sequencing. After assembly and annotation, 20 enzyme genes that act before the step of strictosidine synthesis were found in the dataset, including the key genes encoding G10H, SCS and STR. This result indicated that many putative genes in CPT synthesis are expressed in young leaves, which demonstrates that young leaves are likely to be active tissues for CPT biosynthesis as well as accumulation. Expression profile analysis indicated that the biosynthesis of strictosidine may be more active in young leaves and petioles than in mature leaves and roots. This result indicates that CPT is likely to be synthesized in young leaves, which is consistent with a recent study [16]. Young leaves and petioles are likely to be the main sites for CPT biosynthesis. The lower expression in roots implies that roots may not be a main tissue for CPT biosynthesis; however, CPT does accumulate in roots. Therefore, our results support the hypothesis that in C. acuminata, the main CPT synthesis site is the young leaf.
The subcellular site for CPT synthesis in C. acuminata, has not been reported previously. In this study, we predicted that the CaSCS gene was localized in the ER. The results indicated that secologanin in C. acuminata is possibly biosynthesized in the ER, which was consistent with the studies of CPT location in hairy roots of O. pumila[48]. Therefore, we hypothesize that in C. acuminata CPT is likely to be biosynthesized in the ER and then transported to a vacuole [15] or excreted outside the cytoplasmic membrane, as reported in O. pumila[51].
Conclusion
In this study, a high quality cDNA library was established to mine effective transcriptome information in CPT biosynthesis and transport in C. acuminata. A method was adopted for gene discovery using a combination of sequence annotation, chemical catalytic features prediction and transcripts expression profiling for deep mining of target genes of the CPT metabolism pathway. Consequently, a number of putative transcripts, including genes encoding G10H, SCS, STR, cytochrome P450s, glucosidase, and MDR transporter genes, were identified as possibly being involved in CPT biosynthesis and transport. Meanwhile, three important genes encoding proteins involved in CPT backbone biosynthesis were cloned and analyzed. The transcriptome data represents a valuable genetic resource for further identification of genes involved in CPT biosynthesis and transport. This dataset could be beneficial for further research of the CPT metabolism pathway and molecular genetic breeding.
Methods
Materials preparation and treatment
Young leaves (the first leaf from the apex of side branches, including the apex) for library construction and gene cloning [52] were collected from a C. acuminata tree with a diameter of 14 cm cultivated in the greenhouse of the IMPLAD (Institute of Medicinal Plant Development), Beijing, China. Root, root bark, stem, petiole (the petiole of the first young leaf), young leaves (the first leaf from the apex of the side branches) and old leaves (the fifth leaf from the apex of the side branches) were prepared from the same tree for expression analysis as described previously [52]. The second young leaves from the apex of each branch (about 4 cm long) were cut off for treatment. For the MeJA induction experiment, young leaves were soaked in 100 μM MeJA, with unsoaked leaves serving as a control. The experimental materials were then immediately frozen in liquid nitrogen and stored at -80°C for further processing. All the real-time experiments were repeated three times.
RNA preparation
Total RNA was isolated using the Universal Plant RNA Isolation Mini Kit (BioTeke, Beijing, China), according to the manufacturer's recommendation. Total RNA quantity and quality were determined with a GeneQuant100 spectrophotometer (GE Healthcare, UK) and 1% agarose gels.
cDNA library construction
Total RNA was extracted from young leaves of C. acuminata. RNA samples were digested with RNase-free DNase I (TURBO DNase; Ambion, TX, USA) immediately after RNA extraction. The digested RNA was converted to cDNA using a SMART cDNA synthesis kit (Clontech, CA, USA) and then amplified by applying the Advantage II polymerase (Clontech, USA) to increase the total quantity of the sample for sequencing. Purification of the amplified products was carried out with the PureLink™ PCR purification kit (Invitrogen, USA). Sequences shorter than 300 bp were removed, and approximately 5 μg purified cDNA was sent for a 1/8 run using the 454 GS FLX platform shotgun sequencing (454 Life Sciences, Roche).
EST assembly
GS FLX De Novo Assembly Software v2.0.01 (454 Life Sciences, Roche) was used for EST processing and assembly. ESTs with weak signals and low quality were filtered through the software analysis (using default parameters). Sequencing adaptors were trimmed using the software, and then high-quality (> 99.5% accuracy on single base reads) reads were generated (using default parameters). The SMART PCR primers (Clontech) were then screened, and HQ reads that were shorter than 50 bp were removed for data cleaning of the cDNA library. The remaining HQ ESTs were used for de novo assembly using the GS FLX De Novo Assembly Software v2.0.01 (using default parameters), with a quality score threshold set at 40. After assembly, all the sequences, including contigs (obtained from one cluster) and singletons (appeared only once), were named as "unigenes" for subsequent annotation.
Functional annotation and classification
Similarity searches were carried out against a series of nucleotide and protein databases, such as the Nt, Nr, SwissProt, Kegg, and TAIR databases [28–32], with a common significance threshold cutoff of E-value ≤ 1e-5. For the database annotation, the top five results based on BLAST scores were retained for transcriptome analysis. Gene Ontology classification of TAIR was used to assign the functional roles of C. acuminata through similarity searches. All unigenes were classified into forty-five subcategories belonging to three major categories: cellular component, molecular function and biological process.
In this study, the transcripts were identified and screened by searching the annotation for scores over 100 and were checked manually.
ORF cloning of putative genes encoding proteins from the backbone of CPT biosynthesis
RNA samples of young leaves for gene cloning were converted to first-strand cDNA of the 5' and 3' ends according to the SMART™ RACE cDNA Amplification Kit User Manual (Clontech, USA). RACE PCR Primers for G10H and STR cloning were designed based on the sequence of FXAT9O006GXSI6 and contig03632 respectively in the dataset (Table S1). Primers for G10H and STR genes cloning were designed according to the entire assembled sequence of RACE PCR. Gene cloning of SCS was performed using the annotated unigene contig00661 in the cDNA library, which had integrated ORF sequences. Primers for SCS cloning were designed from the 3' end and 5' untranslated region of contig 00661, which contained an entire ORF. Advantage 2 Polymerase Mix (Clontech, USA) was used for PCR amplification of 3' ends, 5' ends and ORFs of the three genes. All three genes were amplified at 95°C for 3 min; followed by 25 cycles of 95°C for 30 sec, 57°C for 30 sec and 72°C for 1 min 30 sec; and a final step at 72°C for 10 min. The recycled products were integrated into a pMD® 18-T vector (Takara, Dalian, China) and transferred into E. coli DH5α competent cells (Transgene, Beijing, China). The isolated clones were sequenced on a 3730XL (ABI, USA). Sequence alignment with CrG10H and CrSCS in C. roseus was carried out using the DNAMAN software (Lynnon Biosoft, USA). A phylogenetic tree of CaPSTR was constructed according to the amino acid sequences of selected plants. The evolutionary analysis was generated using the software of MEGA 4.0.
Expression analysis
To determine the expression profile of the transcripts involved in CPT biosynthesis, mRNA levels of the transcripts at different tissues and under different treatments were analyzed using Quantitative Real-time PCR. The PrimeScript™ 1st Strand cDNA Synthesis Kit (TaKaRa, Dalian, China) was used for single-strand cDNA synthesis using 1 μg RNase-free DNase I-treated (TaKaRa, Dalian, China) total RNA. Quantitative PCR (Q-PCR) was carried out at least three times each with SYBR® Premix Ex TaqTM (Perfect Real Time) (TaKaRa, Dalian, China) on an IQ5 Multicolor Real-Time PCR Detection System (Bio-Rad, USA). Each qRT-PCR system contained 10 μL 2 × SYBR® Premix Ex Taq™, 0.2 μM forward and reverse primers and 1 μL cDNA template. The PCR amplification program was as follows: 50°C for 2 min; 95°C for 30 sec; 40 cycles of 95°C for 3 sec and 62°C for 40 sec; followed by a melting-curve program of 55°C to 85°C, with a 5-sec hold at each temperature. The gene expression patterns of all genes were normalized to an internal reference (18S rRNA) [53]. The relative gene expression analysis was performed using BIO-RAD IQ™5 optical system software version 2.0 with the 2-ΔΔCt method. All the real-time PCR primers were designed using OMIGA software (Accelrys, USA) with suitable parameters (length: 100-300 bp; Tm: approximately 62°C). The sequences of all primers are listed in the supporting information (Additional file 9).
Acknowledgements
This study was supported by the Program for the National Key Technology R&D Foundation of China (No.2006BAI09B05-3, 81130069) and the National Natural Science Foundation of China (30970307 and 30900113). We also thank Professor Yu-Lin Lin (Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, China) for his kind help in the identification of the plant of C. acuminata.
References
Lorence A, Nessler CL: Camptothecin, over four decades of surprising findings. Phytochemistry. 2004, 65 (20): 2735-49. 10.1016/j.phytochem.2004.09.001.
Pommier Y: DNA Topoisomerase I Inhibitors: Chemistry, Biology, and Interfacial Inhibition. Chem Rev. 2009, 109 (7): 2894-2902. 10.1021/cr900097c.
Oberlies NH, Kroll DJ: Camptothecin and taxol: historic achievements in natural products research. J Nat Prod. 2004, 67: 129-135. 10.1021/np030498t.
Lu H, McKnight TD: Tissue-Specific Expression of the β-Subunit of Tryptophan Synthase in Camptotheca acuminata, an Indole Alkaloid-Producing Plant. Plant Physiol. 1999, 120: 43-52. 10.1104/pp.120.1.43.
López-Meyer M, Nessler CL: Tryptophan decarboxylase is encoded by two autonomously regulated genes in Camptotheca acuminata which are differentially expressed during development and stress. Plant J. 1997, 11 (6): 1167-1175. 10.1046/j.1365-313X.1997.11061167.x.
Burnett RJ, Maldonado-Mendoza IE, McKnight TD, Nessler CL: Expression of a 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Gene from Camptotheca acuminata Is Differentially Regulated by Wounding and Methyl Jasmonate. Plant Physiol. 1994, 103: 41-48.
Yao H, Gong Y, Zuo K, Ling H, Qiu C, Zhang F, Wang Y, Pi Y, Liu X, Sun X, et al: Molecular cloning, expression profiling and functional analysis of a DXR gene encoding 1-deoxy-d-xylulose 5-phosphate reductoisomerase from Camptotheca acuminata. J Plant Physiol. 2008, 165 (2): 203-213. 10.1016/j.jplph.2006.12.001.
Keat HT, Elizabeth G, McKnight TD: Characterization and cloning of 10-hydroxygeraniol oxidoreductase. Plant Biology. 2000
Collua G, Unvera N, Peltenburg-Loomana AMG, van der Heijdena R, Verpoortea R, Memelink J: Geraniol 10-hydroxylase, a cytochrome P450 enzyme involved in terpenoid indole alkaloid biosynthesis. FEBS Lett. 2001, 508: 215-220. 10.1016/S0014-5793(01)03045-9.
Irmler S, SchroÈ der G, St-Pierre B, Crouch NP, Hotze M, Schmidt J, Strack D, Matern U, SchroÈ der J: Indole alkaloid biosynthesis in Catharanthus roseus: new enzyme activities and identification of cytochrome P450 CYP72A1 as secologanin synthase. Plant J. 2000, 24 (6): 797-804. 10.1046/j.1365-313x.2000.00922.x.
Connor SEO, Maresh JJ: Chemistry and biology of monoterpene indole alkaloid biosynthesis. Nat Prod Rep. 2006, 23: 532-547. 10.1039/b512615k.
Coon MJ: Cytochrome P450: nature's most versatile biological catalyst. Annu Rev Pharmacol Toxicol. 2005, 45: 1-25. 10.1146/annurev.pharmtox.45.120403.100030.
Morant M, Bak S, Moller BL, Werck-Reichhart D: Plant cytochromes P450: tools for pharmacology, plant protection and phytoremediation. Curr Opin biotech. 2003, 14 (2): 151-162. 10.1016/S0958-1669(03)00024-7.
Hutchinson CR, Heckendorf AH, Straughn JL, Daddona PE, Cane DE: Biosynthesis of camptothecin: III. Definition of strictosamide as the penultimate biosynthetic precursor assisted by carbon-13 and deuterium NMR spectroscopy. J Am Chem Soc. 1979, 101: 3358-3369. 10.1021/ja00506a037.
Pasqua G, Monacelli B, Valletta A: Cellular localisation of the anti-cancer drug camptothecin in Camptotheca acuminata Decne (Nyssaceae). Eur J Histochem. 2004, 48: 321-328.
Valletta A, Trainotti L, Santamaria AR, Psaqua G: Cell-specific expression of tryptophan decarboxylase and 10-hydroxygeraniol oxidoreductase, key genes involved in camptothecin biosynthesis in Camptotheca acuminata Decne (Nyssaceae). BMC Plant Biol. 2010, 10: 1-27. 10.1186/1471-2229-10-1.
Gaertner LS, Murray CL, Morris CE: Transepithelial transport of nicotine and vinblastine in isolated malpighian tubules of the tobacco hornworm (Manduca sexta) suggests a P-glycoprotein-like mechanism. J Exp Biol. 1998, 201: 2637-2645.
Sakai K, Shitan N, Sato F, Ueda K, Yazaki K: Characterization of berberine transport into Coptis japonica cells and the involvement of ABC protein. J Exp Bot. 2002, 53: 1879-1886. 10.1093/jxb/erf052.
Terasaka K, Sakai K, Sato F, Yamamoto H, Yazaki K: Thalictrum minus cell cultures and ABC-transporter. Phytochemistry. 2003, 62: 483-489. 10.1016/S0031-9422(02)00548-4.
Emrich SJ, Barbazuk WB, Li L, Schnable PS: Gene discovery and annotation using LCM-454 transcriptome sequencing. Genome Res. 2007, 17: 69-73.
Li P, Ponnala L, Gandotra N, Wang L, Si Y, Tausta SL, Kebrom TH, Provart N, Patel R, Myers CR, et al: The developmental dynamics of the maize leaf transcriptome. Nat Genet. 2010, 42: 1060-1069. 10.1038/ng.703.
Liang CW, Zhang XW, Zou J, Xu D, Su F, Ye NH: Identification of miRNA from Porphyra yezoensis by High-Throughput Sequencing and Bioinformatics Analysis. PLoS One. 2010, 5 (5): e10698-10.1371/journal.pone.0010698.
Crawford JE, Guelbeogo WM, Vernick KD, Sagnon NF, Lazzaro BP: De Novo Transcriptome Sequencing in Anopheles funestus Using Illumina RNA-Seq Technology. PLoS One. 2010, 5 (12): e14202-10.1371/journal.pone.0014202.
Chen S, Luo H, Li Y, Sun Y, Wu Q, Niu Y, Song J, Lv A, Zhu Y, Sun C, et al: 454 EST analysis detects genes putatively involving in ginsenoside Biosynthesis in Panaxginseng. Plant cell Rep. 2011, 30: 1593-1601. 10.1007/s00299-011-1070-6.
Luo HM, Li Y, Sun C, Wu Q, Song JY, Sun YZ, Steinmetz A, Chen SL: Comparison of 454-ESTs from Huperzia serrata and Phlegmariurus carinatus reveals putative genes involved in lycopodium alkaloid biosynthesis and developmental regulation. BMC Plant Biology. 2010, 10: 209-10.1186/1471-2229-10-209.
Lopez-Meyer M, Nessler CL, McKnight TD: Sites of accumulation of the antitumor alkaloid camptothecin in Camptotheca acuminata. Planta Med. 1994, 60: 558-560. 10.1055/s-2006-959571.
Li SY, Yi YJ, Wang YJ, Zhang ZZ, Beasley RS: Camptothecin accumulation and variations in Camptotheca. Plant Med. 2002, 68: 1010-1016. 10.1055/s-2002-35652.
KEGG Database. [http://www.genome.jp/kegg/]
Nr Database. [ftp://ftp.ncbi.nih.gov/blast/db/FASTA/nr.gz]
Nt Database. [ftp://ftp.ncbi.nih.gov/blast/db/FASTA/nt.gz]
The TAIR Database. [ftp://ftp.arabidopsis.org/home/tair/Sequences/blast_datasets/TAIR9_blastsets/]
The UniProt-SwissProt Database. [http://www.uniprot.org/downloads]
Berardini TZ, Mundodi S, Reiser L, Huala E, Garcia-Hernandez M, Zhang P, Mueller LA, Yoon J, Doyle A, Lander G: Functional annotation of the Arabidopsis genome using controlled vocabularies. Plant physiol. 2004, 135 (2): 745-755. 10.1104/pp.104.040071.
The WoLF PSORT Protein Subcellular Localization Prediction website. [http://wolfpsort.org/]
Yendo ACA, Gosmann FCG, Fett-Neto AG: Production of Plant Bioactive Triterpenoid Saponins: Elicitation Strategies and Target Genes to Improve Yields. Mol Biotechnol. 2010, 46: 94-104. 10.1007/s12033-010-9257-6.
Aerts RJ, Gisi D, Carolis ED, Luca VD, Baumann TW: Methyl jasmonate vapor increases the developmentally controlled synthesis of alkaloids in Catharanthus and Cinchona seedlings. Plant J. 1994, 5: 635-643. 10.1111/j.1365-313X.1994.00635.x.
Rischer H, Orešič M, Seppa" nen-Laakso T, Katajamaa M, Lammertyn F, Ardiles-Diaz W, Van Montagu MCE, Inze D, Oksman-Caldentey KM, Goossens A: Gene-to-metabolite networks for terpenoid indole alkaloid biosynthesis in Catharanthus roseus cells. Proc Natl Acad Sci. 2006, 103 (14): 5614-5619. 10.1073/pnas.0601027103.
Song SH, Byun SY: Elicitation of Camptothecin Production in Cell Cultures of Camptotheca acuminata. Biotechnol Bioprocess Eng. 1998, 3: 91-95. 10.1007/BF02932509.
Belhadj A, Telef N, Saigne C, Cluzet S, Barrieu F, Said Hamdi, Me'rillon JM: Effect of methyl jasmonate in combination with carbohydrates on gene expression of PR proteins, stilbene and anthocyanin accumulation in grapevine cell cultures. Plant Physiol Bioch. 2008, 46: 493-499. 10.1016/j.plaphy.2007.12.001.
López-Meyer M, Nessler CL: Tryptophan decarboxylase is encoded by two autonomously regulated genes in Camptotheca acuminata which are differentially expressed during development and stress. Plant J. 1997, 11: 1167-1175. 10.1046/j.1365-313X.1997.11061167.x.
Maldonado-Mendoza IE, Vincent RM, Nessler CL: Molecular characterization of three differentially expressed members of the Camptotheca acuminata 3-hydroxy-3-methylglutaryl CoA reductase (HMGR) gene family. Plant Mol Bio. 1997, 34: 781-790. 10.1023/A:1005866813347.
Seki H, Ohyama K, Sawai S, Mizutani M, Ohnishi T, Sudo H, Akashi T, Aoki T, Saito K, Muranaka T: Licorice β-amyrin 11-oxidase, a cytochrome P450 with a key role in the biosynthesis of the triterpene sweetener glycyrrhizin. Proc Natl Acad Sci. 2008, 105: 14204-14209. 10.1073/pnas.0803876105.
Nelson DR, Schuler MA, Paquette SM, Werck-Reichhart D, Bak Søren: Comparative Genomics of Rice and Arabidopsis. Analysis of 727 Cytochrome P450 Genes and Pseudogenes from a Monocot and a Dicot. Plant Physiol. 2004, 135: 756-772. 10.1104/pp.104.039826.
Ketudat Cairns JR, Esen A: β-Glucosidases. Cell Mol Life Sci. 2010, 67: 3389-3405. 10.1007/s00018-010-0399-2.
Nomura T, Quesada AL, Kutchan TM: The New β-D-Glucosidase in Terpenoid-Isoquinoline Alkaloid Biosynthesis in Psychotria ipecacuanha. J Biol Chem. 2008, 283: 34650-34659. 10.1074/jbc.M806953200.
Warzechaa H, Gerasimenkoa I, Kutchan TM, StoÈ ckigt J: Molecular cloning and functional bacterial expression of a plant glucosidase specically involved in alkaloid biosynthesis. Phytochemistry. 2000, 54: 657-666. 10.1016/S0031-9422(00)00175-8.
Barleben L, Panjikar S, Ruppert M, Koepke J, Stöckigt J: Molecular Architecture of Strictosidine Glucosidase: The Gateway to the Biosynthesis of the Monoterpenoid Indole Alkaloid Family. Plant Cell. 2007, 19: 2886-2897. 10.1105/tpc.106.045682.
Sirikantaramas S, Sudo H, Asano T, Yamazaki M, Saito K: Transport of camptothecin in hairy roots of Ophiorrhiza pumila. Phytochemistry. 2007, 68: 2881-2886. 10.1016/j.phytochem.2007.08.028.
Li SY, Yi YJ, Wang YJ, Zhang ZZ, Beasley RS: Camptothecin accumulation and variations in Camptotheca. Planta Med. 2002, 68: 1010-1016. 10.1055/s-2002-35652.
Shitan N, Bazin I, Dan K, Obata K, Kigawa K, Ueda K, Sato F, Forestier C, Yazaki K: Involvement of CjMDR1, a plant MDR-type ABC protein, in alkaloid transport in Coptis japonica. Proc Natl Acad Sci. 2003, 100: 751-756. 10.1073/pnas.0134257100.
Lorence A, Medina-Bolivar F, Nessler CL: Camptothecin and 10-hydroxycamptothecin from Camptotheca acuminata hairy roots. Plant Cell Rep. 2004, 22: 437-441. 10.1007/s00299-003-0708-4.
Liu ZJ, Carpenter SB, Bourgeois WJ, Yu Y, Constantin RJ, Falcon MJ, Adams JC: Variations in the secondary metabolite camptothecin in relation to tissue age and season in Camptotheca acuminata. Tree Physiology. 1998, 18: 265-270.
Pi Y, Liao Z, Jiang K, Huang B, Deng Z, Zhao D, Zeng H, Sun X, Tang K: Molecular cloning, characterization and expression of a jasmonate biosynthetic pathway gene encoding allene oxide cyclase from Camptotheca acuminata. Biosci Rep. 2008, 28: 349-355. 10.1042/BSR20060001.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
YZS contributed to collect experiment samples, designed and carried on the experiment of post sequencing and drafted the manuscript. HML prepared the 454-library and helped to modify the draft. YL, YJZ analyzed the data. YYN, CS, JYS and TE helped to modify the manuscript. LD helped to make the chemical draw. APL and SLC initiated the project, helped to conceive the study and participated in the design and coordination. All authors had read and approved the final manuscript.
Electronic supplementary material
12864_2011_3726_MOESM1_ESM.DOC
Additional file 1:Annotation statistics against public databases. Word document for the summary of the annotation result. (DOC 62 KB)
12864_2011_3726_MOESM2_ESM.TIFF
Additional file 2:Gene Ontology analysis of the 454 sequencing library. TIFF document for the function categorization of the library against the Arabidopsis database. (TIFF 531 KB)
12864_2011_3726_MOESM3_ESM.XLS
Additional file 3:Gene discoveries for CPT biosynthesis against the Nr, Swissprot and Kegg databases. Excel document of specific information for mining genes in CPT biosynthesis. (XLS 50 KB)
12864_2011_3726_MOESM4_ESM.TIFF
Additional file 4:Amino acid alignment between CaG10H and CrG10H. TIFF document of protein sequence alignment of CaG10H and CrG10H. (TIFF 1 MB)
12864_2011_3726_MOESM5_ESM.TIFF
Additional file 5:Peptide alignment between CaSCS and CrSCS. TIFF document of protein sequence alignment of CaSCS and CrSCS. (TIFF 1 MB)
12864_2011_3726_MOESM6_ESM.XLS
Additional file 6:Transcripts of CYP450s discovered in this dataset. Excel document of all the discovered transcripts of cytochrome P450. (XLS 22 KB)
12864_2011_3726_MOESM7_ESM.DOC
Additional file 7:Classification of transcripts annotated to cytochrome P450s in this library. Word document of the classification of cytochrome P450s transcripts. (DOC 70 KB)
12864_2011_3726_MOESM8_ESM.TIFF
Additional file 8:Amino acid alignment between the predicted CaPGD and RsSGD. TIFF document of the comparison of CaPGD and RsSGD. (TIFF 696 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sun, Y., Luo, H., Li, Y. et al. Pyrosequencing of the Camptotheca acuminata transcriptome reveals putative genes involved in camptothecin biosynthesis and transport. BMC Genomics 12, 533 (2011). https://doi.org/10.1186/1471-2164-12-533
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-12-533