
Abstract
Background
Despite vaccination since the 1950s, pertussis has persisted and resurged. It remains a major cause of infant death worldwide and is the most prevalent vaccine-preventable disease in developed countries. The resurgence of pertussis has been associated with the expansion of Bordetella pertussis strains with a novel allele for the pertussis toxin (Ptx) promoter, ptxP3, which have replaced resident ptxP1 strains. Compared to ptxP1 strains, ptxP3 produce more Ptx resulting in increased virulence and immune suppression. To elucidate how B. pertussis has adapted to vaccination, we compared genome sequences of two ptxP3 strains with four strains isolated before and after the introduction vaccination.
Results
The distribution of SNPs in regions involved in transcription and translation suggested that changes in gene regulation play an important role in adaptation. No evidence was found for acquisition of novel genes. Modern strains differed significantly from prevaccination strains, both phylogenetically and with respect to particular alleles. The ptxP3 strains were found to have diverged recently from modern ptxP1 strains. Differences between ptxP3 and modern ptxP1 strains included SNPs in a number of pathogenicity-associated genes. Further, both gene inactivation and reactivation was observed in ptxP3 strains relative to modern ptxP1 strains.
Conclusions
Our work suggests that B. pertussis adapted by successive accumulation of SNPs and by gene (in)activation. In particular changes in gene regulation may have played a role in adaptation.
Similar content being viewed by others


Background
The genus Bordetella comprises nine species, of which four are exclusively respiratory pathogens of mammalian hosts: Bordetella bronchiseptica, Bordetella parapertussis, Bordetella pertussis and Bordetella holmesii [1]. The first three species are closely related, while B. holmesii forms a distinct branch [2]. B. bronchiseptica causes chronic and often asymptomatic respiratory tract infections in a wide variety of mammals and is only sporadically isolated from humans. B. parapertussis consists of two distinct lineages, designated B. parapertussisHU and B. parapertussisOV, which infect humans and sheep respectively [3, 4]. B. parapertussisHU and B. pertussis are exclusive human pathogens and the causative agents of pertussis or whooping cough. Both these species have evolved independently from a B. bronchiseptica-like ancestor, a process which has been accompanied by extensive gene loss [4–6].
By far, most cases of whooping cough are caused by B. pertussis. Despite widespread vaccination, pertussis remains a major cause of infant death worldwide [7]. In the 1990s a resurgence of pertussis was observed in several countries with highly vaccinated populations and pertussis has become the most prevalent vaccine-preventable disease in developed countries [8–10]. In the Netherlands, the estimated rate of infection was 6.6% per year for the 3-79-year age group from 1995 through 1996 [11]. Similar percentages have been found in the United States [12–14]. One of the hallmarks of the pertussis resurgence is a shift in disease prevalence towards older persons who have waning vaccine-induced immunity, while recently vaccinated infants are well protected [15]. The reemergence of pertussis has been attributed to various factors including decreased vaccination coverage due to concerns over side effects, suboptimal vaccines, waning vaccine-induced immunity, and adaptation of B. pertussis [1, 9, 10]. The relative contribution of these factors may differ between countries and is the subject of ongoing debate. Pathogen adaptation is supported by several observations. We and others have shown that antigenic divergence has occurred between vaccine strains and clinical isolates with respect to surface proteins which confer protective immunity; pertussis toxin (Ptx), pertactin and fimbriae [1, 16–18]. Further, in a mouse model, pertussis vaccines were less effective against strains carrying non-vaccine type antigens compared to strains with vaccine-type antigens [19–22]. Recently we found evidence that polymorphism in the promoter for Ptx (ptxP) may also be important in adaptation [23]. In the last twenty years two ptxP alleles, ptxP1 and ptxP3, predominated in the Dutch B. pertussis population. The ptxP3 strains were first observed in 1988, gradually increased in frequency, and nearly completely replaced the resident ptxP1 strains in the late 1990s. In the Netherlands, the increase in frequency of ptxP3 strains was associated with the resurgence of pertussis. The ptxP3 strains are found in Asia, Europe, North and South America, and there is evidence that they have spread worldwide in the 1980s and 1990s [23]. The ptxP3 strains produced more Ptx than the ptxP1 strain and epidemiological data suggest that ptxP3 strains are more virulent. Ptx suppresses both the innate and adaptive immune system [24, 25] and we have proposed that increased Ptx production increases pathogen fitness in vaccinated populations by enhancing transmission by hosts in which vaccine immunity has waned. Thus, both antigenic divergence and increased immune suppression in combination with waning immunity are likely to contribute to the pertussis resurgence [23]. Here we extend our studies on adaptation of B. pertussis using comparative genomics. We determined, annotated and compared genome sequences of six Dutch strains, two of which were isolated before vaccination was introduced in 1953 and four modern strains, isolated approximately 50 years later. The modern strains carried either the ptxP1 allele or the ptxP3 allele, while the pre-vaccination strains carried ptxP1 or ptxP2. We identified novel polymorphisms in specific genes and gene categories which may play a role in the persistence and resurgence of pertussis in the face of intensive vaccination.
Results and Discussion
Strain selection
Our long term aim is to identify B. pertussis loci which have contributed to the persistence and resurgence of pertussis in vaccinated populations. We selected two strains isolated in 1949 and 1952 which were characteristic for the Dutch B. pertussis population in the pre-vaccine era [16, 26], and four modern strains isolated in 1999 and 2000, approximately 50 years after the introduction of vaccination. Two of the modern strains carried the ptxP1 allele and two the ptxP3 allele. In our comparisons, we included the Tohama I strain (ptxP1) of which the annotated genome sequence was available [6]. The Tohama I strain was isolated in Japan in the 1950s and has been subcultured in vitro extensively. It is used as model strain in many different laboratories. Strain characteristics are listed in Table 1.
Comparative analysis of Bordetella pertussis genomes
We determined the genome sequences of the six B. pertussis strains through pyrosequencing. As this technology is known to generate errors in homopolymeric nucleotide tracts, SNPs and indels in these regions were filtered out. As a consequence, differences between strains in homopolymeric nucleotide tracts were not identified. However, homopolymeric nucleotide tracts have high mutation rates and may vary during subculturing of a single strain [27]. Thus, genotypes and phenotypes controlled by homopolymeric nucleotide tracts are not stable and changes in these tracts will not represent fixed differences between strains.
We identified 471 SNPs (i.e. bases that were not conserved in one or more of the seven strains), of which 414 and 57 were located in ORFs and intergenic regions, respectively (Additional file 1). Four ORFs were found to contain small insertion or deletions (indels) ranging from eight to 31 bases (Additional file 1). Based on our analyses, the estimated SNP density was 1 SNP per 8,675 bases. Maharjan and coworkers used Microarray-based comparative genome sequencing to detect SNPs in 34% of the Tohama I genome [28]. The Tohama I strain was compared to an Australian isolate from 2006 and a SNP density of 1 SNP per 20,000 bases was found. As we included the whole genome in our comparison and used a larger number of strains, the higher SNP density we found was not unexpected. SNP densities in other monomorphic human pathogens have been found to range from 1 SNP per 2,300 bases in Salmonella enterica serovar Typhi [29] to 1 SNP per 28,400 bases in Mycobacterium leprae [30, 28]. This places B. pertussis among the most monomorphic human pathogens known. In their analyses, Maharjan and coworkers identified 66 SNPs in 1,229 genes and 4 SNPs in 268 intergenic regions. Of these 70 SNPs, 27 (39%) were also detected in one of the six Dutch strains, while 14 (20%) were specific for the Australian strain.
In addition to the SNPs discussed above, we confirmed 13 large regions of difference (RDs) identified in previous studies using microarrays [31–35] and whole genome sequencing [33, 36]. Further, we found a new polymorphism in RD23 (Additional files 2 and 3).
Genetic relationship based on whole genome sequencing
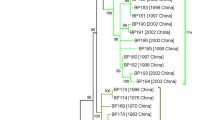
A maximum likelihood phylogenetic tree based on 471 SNPs was derived (Figure 1). The genome sequence of B. bronchiseptica was used as outgroup to root the tree [6]. The tree suggested that B1193, which harbors the ptxP2 allele, represented a distinct lineage which diverged from the six other strains relatively early in the history of B. pertussis. Although the Tohama I strain was more closely related to the four remaining Dutch strains, it also formed a distinct, relatively deep branch. This may be due to the fact that the Tohama I strain has been subcultured in vitro since the 1950s resulting in a relaxation of selective forces, or it could reflect geographical isolation, as the Tohama I strains was isolated in Japan, while all other strains were from the Netherlands. The four recently isolated Dutch strains were closely related. The tree indicated that the ptxP3 strains comprise a monophyletic lineage which recently diverged from modern ptxP1 strains.

Phylogenetic tree of strains used in this study based on 471 SNPs. Colored strain designations indicate ptxP alleles: black, ptxP1; blue, ptxP2; red, ptxP3. The location of the root was based on the sequence of B. bronchiseptica RB50 strain [6].
Evidence for selection in cis regulatory regions
Although it is often assumed that mutations located outside protein coding sequences (CDSs) are neutral, such mutations can affect fitness if they are located in cis-regulatory elements (i.e. in regions involved in binding of transcriptional factors or in regions that affect translation). To explore the role of cis regulatory elements in adaptation, we determined the distribution of all SNPs in intergenic regions relative to the start codon of predicted CDSs in segments of 25 bases (Figure 2). Assuming random mutation (the null hypothesis), one expects a SNP distribution which reflects the frequency of the 25 base intergenic segments in the chromosome. This frequency is highest for the proximate 25 base segment and decreases with distance to the start codon (Figure 2). The actual frequency distribution was compared with the frequency based on null hypothesis and found to be significantly different (P 0.01). In view of the low number of SNPs observed, the region 200 to > 500 will be ignored. In the regions 50-99 and 175-199 more SNPs were observed than in the random mutation model, suggesting diversifying selection. These regions may contain binding sites for regulatory proteins such as BvgA, as shown for Ptx [37]. The regions 125-174 and 0-24 contained less SNPs compared to the random model, possibly reflecting purifying selection. The region 0-24 contains the binding site for RNA-polymerase and regions involved in initiation of translation and it seems that such regions tend to be conserved in B. pertussis. Thus our results provide evidence that regions involved in binding of regulatory proteins are subject to diversifying selection suggesting a role in adaptation. The low degree of polymorphism, the limited number of strains sequenced and the absence of silent mutations in some CDSs did not allow calculation of dN/dS ratios to identify purifying or diversifying selection in ORFs [38–40].

Distribution of SNPs in intergenic regions relative to the start codons of predicted protein coding genes. The number of SNPs was plotted as a function of the distance to the initiation codon of genes using a window of 25 bases. Blue columns show the expected distribution based on the frequency distribution of 25 bases segments in intergenic regions. Red columns show the actual distribution. The two distributions are significantly different (Chi-square test, P 0.01).
Polymorphisms in pathogenicity-associated genes
The 89 pathogenicity-associated genes (Additional file 4) contained 34 polymorphisms (SNPs and small indels) distributed over 24 genes and three promoter regions (Figure 3 and Additional file 5). To the best of our knowledge, 16 of the polymorphisms in pathogenicity-associated genes were not described before. Nineteen of the 34 polymorphisms were specific for the ptxP2 strain, again underlining the distinct nature of this strain. It was noteworthy that for Ptx, Prn (unpublished data), the type III secretion toxin (BteA) and tracheal colonization factor (TcfA), polymorphisms were found in both the protein coding sequences [41, 42] and the (putative) promoter regions, suggesting both structural and regulatory adaptations for these virulence factors.

Polymorphisms found in virulence-associated genes. The distribution of polymorphisms over the analyzed strains is shown. Within rows, polymorphisms are distinguished by color. BP1962 is part of RD10 which is deleted from ptxP3 strains (See Fig. 6). Deleted genes are indicated by "Δ". An overview of all known alleles is given in Additional file 5. Abbreviation: indel, insertion/deletion. References [16, 23, 28, 41, 42, 54–58].
Polymorphisms which distinguish strains isolated before and after the introduction of vaccination
The comparison of polymorphisms which distinguish strains isolated before and after the introduction of vaccination (for convenience designated as pre- and post-strains) may be useful to identify mutations which increase fitness in vaccinated populations and hence reduce vaccine efficacy. We identified 14 non-silent, 11 silent SNPs, three intergenic SNPs, one nonsense mutation and two indels which distinguished pre- and post-strains (pseudo-genes were excluded). The distribution of alleles and SNPs are shown in Additional file 6 and Figure 4, respectively. Here, only polymorphisms believed to be particularly interesting based on our current knowledge are discussed. The post-strains were distinguished by a large deletion (RD3) comprising 24 CDSs. King et al. [34] analyzed a larger number of post-strains (N = 43), all of which were found to miss RD3. RD3 contains four ORFs coding for putative exported proteins and its removal may reduce the antigenic profile of B. pertussis. Alternatively, the deletion of this region may be unrelated to vaccination and reflect the ongoing genome reduction [32, 35]. One gene, pitA coding for a putative phosphate transporter, was inactivated in post-strains by a nonsense mutation. The nonsense mutation was also observed in an Australian strain [28]. Post-strain-specific, non-silent, polymorphisms were found in two genes for exported virulence factors, prn and ptxA, in agreement with previous studies [18]. There is evidence that polymorphism in these two genes affects vaccine efficacy in a mouse model [19–22]. The locus for another (putative) exported protein (BP2028) also contained a non-silent polymorphism specific for post-strains. Blast searches revealed 100% identity at the amino acid level between the prevaccination variant and homologues found in B. bronchiseptica and B. parapertussis, suggesting that the prevaccination strains harbor the ancestral allele. No significant similarity was found with CDSs from other bacteria, suggesting a highly specific function. Although the function of the protein is not known, the fact that it is exported suggests a role in host-pathogen interaction. Also of interest is a SNP in the promoter region for the type III secretion toxin and chaperone genes, bteA and btcA respectively. For B. bronchiseptica it has been shown that BteA is translocated into the host and is necessary and sufficient for rapid cytotoxicity in a wide range of mammalian cells [43, 44]. The SNP, which involves an A/G transition, is located in a putative TATA box of btcA and therefore could influence its expression. We investigated the temporal trends of the two alleles, designated allele A and allele G (Figure 5). Allele A was found in all strains from 1949 to 1970 (N = 12). Allele G was first observed in 1977 and gradually increased in frequency to ~90% in 2000-2008. The allele A was found in B. bronchiseptica suggesting that it is the ancestral type, consistent with the trend observed in the Netherlands.

Polymorphisms which distinguish strains isolated before (pre) and after (post) the introduction of vaccination. Within rows, polymorphisms are distinguished by color. Genes truncated in all strains were omitted. An overview of alleles is given in Additional file 6. Abbreviation: indel, insertion/deletion. Ps, pseudogene. References [16, 28, 55–57, 59].

Temporal trend in the frequency of a SNP located in the promoter region for the type III secretion system. The SNP involved an A/G transition and was located between the type III secretion toxin gene bteA and its chaperone btcA. The two genes are oriented back to back. The period 1949-2008 was investigated using 44 strains.
Polymorphisms which distinguish ptxP3 strains
Strains carrying the ptxP3 allele have spread worldwide and have contributed to the pertussis epidemic in The Netherlands [23]. It seems likely that the ptxP3 allele confers increased fitness compared to the ptxP1 allele it replaced in current human populations. Alternatively, it may be a marker for other selective changes in the same haplotype. To identify other genes that may have contributed to the increased fitness of the ptxP3 lineage, we sought polymorphisms which were unique for the ptxP3 lineage. We identified 26 polymorphisms specific for one or both ptxP3 strains, 11 non-silent SNPs, eight silent SNPs, four intergenic SNPs, two small indels (eight and 31 bases) and one large deletion (RD10, comprising 2.2 kbases). The distribution of alleles and polymorphisms are shown in Additional file 7 and Figure 6, respectively. Only polymorphisms we believe to be particularly interesting based on our current knowledge are discussed. The association of the RD10 deletion with ptxP3 strains was noted before using a larger collection of strains (N = 15) [34]. One of the intergenic SNPs was located in the ptx promoter and defines the ptxP3 lineage. The remaining three intergenic SNPs were located proximal to the genes for glycyl tRNA synthetase alpha chain gene (glyQ), a transporter, an integral membrane transport protein and a dihydrodipicolinate synthetase. It is not clear whether these SNPs affect transcription. We have shown previously that the SNP in the Ptx promoter region affects production of Ptx, however [23]. Two ptxP3-specific SNPs were in known virulence genes, fim3 and bscI, coding for the serotype 3 fimbriae and a component of the type III secretion system, respectively [43, 45]. Two indels, located in BP0880 and BP2946, were ptxP3- specific. BP0880 codes for a putative exported protein which shows similarity with metal dependent phosphohydrolases and signal transduction proteins, while the BP2496 product belongs to the family of arsS transcriptional regulators. BP0880 was a pseudogene in all strains, except the two ptxP3 strains in which the reading frame was restored by deletion of an eight base repeat. Conversely, the reading frame of arsS was intact in all strains except the two ptxP3 strains, due to the deletion of a 31 base repeat. Both BP0880 and BP2496 are intact in B. bronchiseptica and B. parapertussis. The BP0880 and BP2496 genes from three ptxP3 strains were sequenced completely and the ORFs were found to be intact and truncated, respectively. It seems likely that BP0880 and BP2946 are subject to phase variation, i.e. the reversible on/off switching of genes by varying DNA sequence repeat units [46]. Phase variation has been observed for a number of other B. pertussis genes [27, 45, 47]. Based on the phylogenetic relationships, it seems likely that BP0880 and BP2949 were, respectively, reactivated and inactivated in the ptxP3 lineage. This qualifies these loci for further analyses as to their role in the emergence of ptxP3 strains.

Polymorphisms associated with ptxP3 strains. The distribution of polymorphisms over the analyzed strains is shown. Within rows, polymorphisms are distinguished by color. Genes truncated in all strains were omitted. An overview of all known alleles is given in Additional file 7. Abbreviation: indel, insertion/deletion; ps, pseudogene.
Conclusions
We provide the first comprehensive genomic comparison of a bacterial pathogen circulating in a highly vaccinated population. In this study, we included two strains from the prevaccination era and four strains isolated ~50 years later. We confirmed and extended the observation that modern B. pertussis strains differed significantly from prevaccination strains, both phylogenetically and with respect to particular alleles [1]. Further, we identified one highly divergent, possibly ancient, B. pertussis lineage, characterized by the ptxP2 allele. Our work confirmed that B. pertussis strains differ significantly in gene content due to gene loss, a process which may still be ongoing [32, 35]. Further, we found no evidence that acquisition of novel genes has played a role in adaptation, as has been suggested for B. holmesii [2]. In contrast, B. pertussis seems to adapt mainly by the successive accumulation of SNPs. Our work shows that, based on SNP density, B. pertussis is one of the most monomorphic human pathogens. This suggests a recent origin of this species or, more likely, a recent population bottle neck [1, 4].
Our results provide evidence that regions involved in binding of regulatory proteins are subject to diversifying selection suggesting a role in adaptation. Indeed, this is exemplified by the rapid emergence of ptxP3 strains with increased pertussis toxin production [23]. A number of recent studies have highlighted the importance of changes in gene regulation in adaptation of pathogens [48–51]. It is noteworthy that for a number of virulence factors (Ptx, Prn and TcfA), SNPs were found in both the protein-encoding genes and the (putative) promoter regions, suggesting adaptation at both the structural and regulatory level for the same phenotype. Of interest is a SNP in the promoter region for the type III secretion toxin (BteA) and chaperone genes (BtcA). For B. bronchiseptica it has been shown that BteA is necessary and sufficient for rapid cytotoxicity in a wide range of mammalian cells [43, 44]. The ancestral allele was found in all strains from before 1977, but subsequently replaced by a novel allele which increased in frequency to ~90% in 2000-2008. This suggests that the novel allele may significantly affect strain fitness, although it is also possible that its increase in frequency is due to hitchhiking with other loci which affect fitness. In any case, this allele may be an important marker for successful lineages. The ptxP3 lineage, associated with the resurgence of pertussis in the Netherlands, has emerged recently and spread worldwide [23]. We found that the ptxP3 strains comprised a young branch which diverged recently from modern ptxP1 strains. Several alleles were identified, which were uniquely associated with the ptxP3 lineage and may thus have contributed to its success. Two ptxP3-specific SNPs were in known virulence genes, fim3 and bscI, coding for the serotype 3 fimbriae and a component of the type III toxin secretion system, respectively. We also observed both reactivation and inactivation of genes in the ptxP3 lineage. In conclusion, this work has identified a number of genetic loci which are associated with highly successful strains. Further analyses of these loci can contribute to our understanding of the evolution of bacterial pathogens.
Methods
Strain, culture conditions and DNA isolation
The six clinical isolates used in this study are described in Table 1. Strains were grown on Bordet Gengou (BG) agar supplemented with 15% sheep blood and incubated for 3 days at 35°C. DNA was isolated using QIAGEN Genomic-tip 100/G kit, according to the manufacturer's instructions.
Genome sequencing and detection of polymorphisms
B1831 was sequenced using the 454 GS-G20 sequencer (Roche) and the other five isolates were sequenced using the 454 GS-FLX sequencer (Roche), according to the manufacturer's instructions. The generated reads were assembled de novo into contigs using the Newbler assembler (Roche).
SNPs, insertions and deletions were detected by mapping the contigs to the previously sequenced and annotated B. pertussis Tohama I genome [6] using BLAST. We filtered SNP calls as described in [29]. Briefly, SNPs with low base quality, SNPs within 15 base of the end of a contig, SNPs in repetitive sequences, such as insertion sequence elements, and SNPs in homopolymeric tracts were removed. Single base insertions or deletions in homopolymeric tracts were ignored as these are often a result of 454 sequencing errors [52]. To determine the accuracy of these SNPs we verified 88 SNPs in the sequenced strains by combining PCR with mass spectrometry (Sequenom). All 88 SNPs were correct. Only for a subset of small indels (≥ 6 b), which distinguished sets of strains, were checked by resequencing and included in the analyses. Genome sequences have been submitted to the NCBI Nucleotide Sequence data base (accessions numbers; strain B0558, ADKR00000000; strain B1193, ADKS00000000; strain B1831, ADKT00000000; strain B1834, ADKU00000000; strain B1917, ADKV00000000; strain B1920, ADKW00000000).
Phylogenetic analysis
Only SNPs for which the allele at the orthologous nucleotide was determined in all strains were included in the phylogenetic analysis. A Maximum Likelihood tree was derived using PhyML [53] with the following parameters: model, HKY; transition/transversion ratio; estimated; proportion of invariable sites, estimated; number of relative substitution rate categories, 4; gamma distribution parameter, estimated; starting tree, BIONJ; optimize tree topology, yes; optimize branch lengths and rate parameters, yes.
Statistical analyses
To determine if there is evidence for selection in cis-regulatory loci, we compared the expected distribution of the distance of SNPs to the start of the gene with the actual distribution of these distances using the Chi square test. This was done for all genes.
References
Mooi FR: Bordetella pertussis and vaccination: the persistence of a genetically monomorphic pathogen. Infect Genet Evol. 2010, 10 (1): 36-49. 10.1016/j.meegid.2009.10.007.
Diavatopoulos DA, Cummings CA, van der Heide HG, van Gent M, Liew S, Relman DA, Mooi FR: Characterization of a highly conserved island in the otherwise divergent Bordetella holmesii and Bordetella pertussis genomes. J Bacteriol. 2006, 188 (24): 8385-8394. 10.1128/JB.01081-06.
van der Zee A, Groenendijk H, Peeters M, Mooi FR: The differentiation of Bordetella parapertussis and Bordetella bronchiseptica from humans and animals as determined by DNA polymorphism mediated by two different insertion sequence elements suggests their phylogenetic relationship. Int J Syst Bacteriol. 1996, 46 (3): 640-647. 10.1099/00207713-46-3-640.
Diavatopoulos DA, Cummings CA, Schouls LM, Brinig MM, Relman DA, Mooi FR: Bordetella pertussis, the Causative Agent of Whooping Cough, Evolved from a Distinct, Human-Associated Lineage of B. bronchiseptica. PLoSPathog. 2005, 1 (4): 373-383.
van der Zee A, Mooi F, Van Embden J, Musser J: Molecular evolution and host adaptation of Bordetella spp.: phylogenetic analysis using multilocus enzyme electrophoresis and typing with three insertion sequences. J Bacteriol. 1997, 179 (21): 6609-6617.
Parkhill J, Sebaihia M, Preston A, Murphy LD, Thomson N, Harris DE, Holden MT, Churcher CM, Bentley SD, Mungall KL, et al: Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. NatGenet. 2003, 35 (1): 32-40.
Crowcroft NS, Stein C, Duclos P, Birmingham M: How best to estimate the global burden of pertussis?. Lancet InfectDis. 2003, 3 (7): 413-418. 10.1016/S1473-3099(03)00669-8.
de Melker HE, Schellekens JF, Neppelenbroek SE, Mooi FR, Rumke HC, Conyn-van Spaendonck MA: Reemergence of pertussis in the highly vaccinated population of the Netherlands: observations on surveillance data. Emerg Infect Dis. 2000, 6 (4): 348-357. 10.3201/eid0604.000404.
He Q, Mertsola J: Factors contributing to pertussis resurgence. FutureMicrobiol. 2008, 3: 329-339.
Berbers GA, de Greeff SC, Mooi FR: Improving pertussis vaccination. HumVaccin. 2009, 5 (7): 497-503.
de Melker HE, Versteegh FG, Schellekens JF, Teunis PF, Kretzschmar M: The incidence of Bordetella pertussis infections estimated in the population from a combination of serological surveys. J Infect. 2006, 53 (2): 106-113. 10.1016/j.jinf.2005.10.020.
Cromer BA, Goydos J, Hackell J, Mezzatesta J, Dekker C, Mortimer EA: Unrecognized pertussis infection in adolescents. Am J Dis Child. 1993, 147 (5): 575-577.
Hodder SL, Cherry JD, Mortimer EA, Ford AB, Gornbein J, Papp K: Antibody responses to Bordetella pertussis antigens and clinical correlations in elderly community residents. ClinInfectDis. 2000, 31 (1): 7-14.
Ward JI, Cherry JD, Chang SJ, Partridge S, Keitel W, Edwards K, Lee M, Treanor J, Greenberg DP, Barenkamp S, et al: Bordetella Pertussis infections in vaccinated and unvaccinated adolescents and adults, as assessed in a national prospective randomized Acellular Pertussis Vaccine Trial (APERT). ClinInfectDis. 2006, 43 (2): 151-157.
Halperin SA: The control of pertussis - 2007 and beyond. NEnglJ Med. 2007, 356 (2): 110-113. 10.1056/NEJMp068288.
Mooi FR, van Oirschot H, Heuvelman K, van der Heide HG, Gaastra W, Willems RJ: Polymorphism in the Bordetella pertussis virulence factors P.69/pertactin and pertussis toxin in The Netherlands: temporal trends and evidence for vaccine-driven evolution. Infect Immun. 1998, 66 (2): 670-675.
Kallonen T, He Q: Bordetella pertussis strain variation and evolution postvaccination. Expert Rev Vaccines. 2009, 8 (7): 863-875. 10.1586/erv.09.46.
Hozbor D, Mooi F, Flores D, Weltman G, Bottero D, Fossati S, Lara C, Gaillard ME, Pianciola L, Zurita E, et al: Pertussis epidemiology in Argentina: trends over 2004-2007. J Infect. 2009, 59 (4): 225-231. 10.1016/j.jinf.2009.07.014.
King AJ, Berbers G, van Oirschot HF, Hoogerhout P, Knipping K, Mooi FR: Role of the polymorphic region 1 of the Bordetella pertussis protein pertactin in immunity. Microbiology. 2001, 147 (Pt 11): 2885-2895.
Watanabe M, Nagai M: Effect of acellular pertussis vaccine against various strains of Bordetella pertussis in a murine model of respiratory infection. J Health Science. 2002, 48 (6): 560-564. 10.1248/jhs.48.560.
Gzyl A, Augustynowicz E, Gniadek G, Rabczenko D, Dulny G, Slusarczyk J: Sequence variation in pertussis S1 subunit toxin and pertussis genes in Bordetella pertussis strains used for the whole-cell pertussis vaccine produced in Poland since 1960: efficiency of the DTwP vaccine-induced immunity against currently circulating B. pertussis isolates. Vaccine. 2004, 22 (17-18): 2122-2128. 10.1016/j.vaccine.2003.12.006.
Fingermann M, Fernandez J, Sisti F, Rodriguez ME, Gatti B, Bottero D, Graieb A, Gaillard ME, Ayala SG, Mooi FR, et al: Differences of circulating Bordetella pertussis population in Argentina from the strain used in vaccine production. Vaccine. 2006, 24 (17): 3513-3521. 10.1016/j.vaccine.2006.02.026.
Mooi FR, van Loo IHM, van Gent M, He Q, Heuvelman CJ, Bart MJ, de Greeff SC, Diavatopoulos DA, Teunis PF, Nagelkerke NJ, et al: Bordetella pertussis Strains with Increased Toxin Production Associated with Pertussis Resurgence. Emerg Infect Dis. 2009, 15 (8): 1206-1213. 10.3201/eid1508.081511.
Carbonetti NH, Artamonova GV, Van Rooijen N, Ayala VI: Pertussis Toxin Targets Airway Macrophages to Promote Bordetella pertussis Infection of the Respiratory Tract. Infect Immun. 2007, 75 (4): 1713-1720. 10.1128/IAI.01578-06.
Kirimanjeswara GS, Agosto LM, Kennett MJ, Bjornstad ON, Harvill ET: Pertussis toxin inhibits neutrophil recruitment to delay antibody-mediated clearance of Bordetella pertussis. JClinInvest. 2005, 115 (12): 3594-3601.
van Loo IH, Mooi FR: Changes in the Dutch Bordetella pertussis population in the first 20 years after the introduction of whole-cell vaccines. Microbiology. 2002, 148 (Pt 7): 2011-2018.
Gogol EB, Cummings CA, Burns RC, Relman DA: Phase variation and microevolution at homopolymeric tracts in Bordetella pertussis. BMCGenomics. 2007, 8: 122-
Maharjan RP, Gu C, Reeves PR, Sintchenko V, Gilbert GL, Lan R: Genome-wide analysis of single nucleotide polymorphisms in Bordetella pertussis using comparative genomic sequencing. Res Microbiol. 2008, 159 (9-10): 602-608. 10.1016/j.resmic.2008.08.004.
Holt KE, Parkhill J, Mazzoni CJ, Roumagnac P, Weill FX, Goodhead I, Rance R, Baker S, Maskell DJ, Wain J, et al: High-throughput sequencing provides insights into genome variation and evolution in Salmonella Typhi. Nat Genet. 2008, 40 (8): 987-993. 10.1038/ng.195.
Monot M, Honore N, Garnier T, Araoz R, Coppee JY, Lacroix C, Sow S, Spencer JS, Truman RW, Williams DL, et al: On the origin of leprosy. Science. 2005, 308 (5724): 1040-1042. 10.1126/science/1109759.
Cummings CA, Brinig MM, Lepp PW, van de Pas S, Relman DA: Bordetella species are distinguished by patterns of substantial gene loss and host adaptation. J Bacteriol. 2004, 186 (5): 1484-1492. 10.1128/JB.186.5.1484-1492.2004.
Heikkinen E, Kallonen T, Saarinen L, Sara R, King AJ, Mooi FR, Soini JT, Mertsola J, He Q: Comparative Genomics of Bordetella pertussis Reveals Progressive Gene Loss in Finnish Strains. PLoS ONE. 2007, 2 (9): e904-10.1371/journal.pone.0000904.
Bouchez V, Caro V, Levillain E, Guigon G, Guiso N: Genomic content of Bordetella pertussis clinical isolates circulating in areas of intensive children vaccination. PLoSONE. 2008, 3 (6): e2437-
King AJ, van Gorkom T, Pennings JL, van der Heide HG, He Q, Diavatopoulos D, Heuvelman K, van Gent M, van Leeuwen K, Mooi FR: Comparative genomic profiling of Dutch clinical Bordetella pertussis isolates using DNA microarrays: identification of genes absent from epidemic strains. BMCGenomics. 2008, 9 (1): 311-
King AJ, van Gorkom T, van der Heide HG, Advani A, van der Lee S: Changes in the genomic content of circulating Bordetella pertussis strains isolated from the Netherlands, Sweden, Japan and Australia: adaptive evolution or drift?. BMC Genomics. 2010, 11 (1): 64-10.1186/1471-2164-11-64.
Caro V, Bouchez V, Guiso N: Is the Sequenced Bordetella pertussis strain Tohama I representative of the species?. J Clin Microbiol. 2008, 46 (6): 2125-2128. 10.1128/JCM.02484-07.
Stibitz S: The bvg regulon. Bordetella Molecular Microbiology. 2007, Horizon Bioscience, Norfolk, UK, 47-67.
Novichkov PS, Wolf YI, Dubchak I, Koonin EV: Trends in prokaryotic evolution revealed by comparison of closely related bacterial and archaeal genomes. J Bacteriol. 2009, 191 (1): 65-73. 10.1128/JB.01237-08.
Rocha EP, Smith JM, Hurst LD, Holden MT, Cooper JE, Smith NH, Feil EJ: Comparisons of dN/dS are time dependent for closely related bacterial genomes. J TheorBiol. 2006, 239 (2): 226-235. 10.1016/j.jtbi.2005.08.037.
Yang Z, Bielawski JP: Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 2000, 15 (12): 496-503. 10.1016/S0169-5347(00)01994-7.
van Loo IH, Heuvelman KJ, King AJ, Mooi FR: Multilocus sequence typing of Bordetella pertussis based on surface protein genes. JClinMicrobiol. 2002, 40 (6): 1994-2001.
Packard ER, Parton R, Coote JG, Fry NK: Sequence variation and conservation in virulence-related genes of Bordetella pertussis isolates from the UK. J MedMicrobiol. 2004, 53 (Pt 5): 355-365.
Panina EM, Mattoo S, Griffith N, Kozak NA, Yuk MH, Miller JF: A genome-wide screen identifies a Bordetella type III secretion effector and candidate effectors in other species. Mol Microbiol. 2005, 58 (1): 267-279. 10.1111/j.1365-2958.2005.04823.x.
French CT, Panina EM, Yeh SH, Griffith N, Arambula DG, Miller JF: The Bordetella T3SS effector BteA contains a conserved N-terminal motif that guides bacterial virulence factors to lipid rafts. Cell Microbiol. 2009
Willems R, Paul A, van der Heide HG, ter Avest AR, Mooi FR: Fimbrial phase variation in Bordetella pertussis: a novel mechanism for transcriptional regulation. EMBO J. 1990, 9 (9): 2803-2809.
van der Woude MW, Baumler AJ: Phase and antigenic variation in bacteria. ClinMicrobiolRev. 2004, 17 (3): 581-611. table
van Gent M, de Greeff SC, van der Heide HG, Mooi FR: An investigation into the cause of the 1983 whooping cough epidemic in the Netherlands. Vaccine. 2009, 27 (13): 1898-1903. 10.1016/j.vaccine.2009.01.111.
Winfield MD, Groisman EA: Phenotypic differences between Salmonella and Escherichia coli resulting from the disparate regulation of homologous genes. Proc Natl Acad Sci USA. 2004, 101 (49): 17162-17167. 10.1073/pnas.0406038101.
Li M, Diep BA, Villaruz AE, Braughton KR, Jiang X, DeLeo FR, Chambers HF, Lu Y, Otto M: Evolution of virulence in epidemic community-associated methicillin-resistant Staphylococcus aureus. Proc Natl Acad Sci USA. 2009, 106 (14): 5883-5888. 10.1073/pnas.0900743106.
Osborne SE, Walthers D, Tomljenovic AM, Mulder DT, Silphaduang U, Duong N, Lowden MJ, Wickham ME, Waller RF, Kenney LJ, et al: Pathogenic adaptation of intracellular bacteria by rewiring a cis-regulatory input function. Proc Natl Acad Sci USA. 2009, 106 (10): 3982-3987. 10.1073/pnas.0811669106.
Perez JC, Groisman EA: Transcription factor function and promoter architecture govern the evolution of bacterial regulons. Proc Natl Acad Sci USA. 2009, 106 (11): 4319-4324. 10.1073/pnas.0810343106.
Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM: Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007, 8 (7): R143-10.1186/gb-2007-8-7-r143.
Guindon S, Delsuc F, Dufayard JF, Gascuel O: Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol. 2009, 537: 113-137. full_text.
Mooi FR, He Q, van Oirschot H, Mertsola J: Variation in the Bordetella pertussis virulence factors pertussis toxin and pertactin in vaccine strains and clinical isolates in Finland. Infect Immun. 1999, 67 (6): 3133-3134.
Mooi FR, Hallander H, Wirsing von Konig CH, Hoet B, Guiso N: Epidemiological typing of Bordetella pertussis isolates: recommendations for a standard methodology. EurJ ClinMicrobiolInfectDis. 2000, 19 (3): 174-181.
Mastrantonio P, Spigaglia P, van Oirschot H, van der Heide HG, Heuvelman K, Stefanelli P, Mooi FR: Antigenic variants in Bordetella pertussis strains isolated from vaccinated and unvaccinated children. Microbiology. 1999, 145 (Pt 8): 2069-2075. 10.1099/13500872-145-8-2069.
Tsang RS, Lau AK, Sill ML, Halperin SA, Van Caeseele P, Jamieson F, Martin IE: Polymorphisms of the fimbria fim3 gene of Bordetella pertussis strains isolated in Canada. JClinMicrobiol. 2004, 42 (11): 5364-5367.
Herrou J, Debrie AS, Willery E, Renaud-Mongenie G, Locht C, Mooi F, Jacob-Dubuisson F, Antoine R: Molecular evolution of the two-component system BvgAS involved in virulence regulation in Bordetella. PLoS One. 2009, 4 (9): e6996-10.1371/journal.pone.0006996.
Brinig MM, Cummings CA, Sanden GN, Stefanelli P, Lawrence A, Relman DA: Significant gene order and expression differences in Bordetella pertussis despite limited gene content variation. J Bacteriol. 2006, 188 (7): 2375-2382. 10.1128/JB.188.7.2375-2382.2006.
Acknowledgements
This project was supported by the RIVM (SOR project S/230446).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
MB carried out the molecular genetic studies, analyzed the data and contributed to discussions and writing of the manuscript. MvG carried out the molecular genetic studies, analyzed the data and contributed to discussions and writing of the manuscript. HvdH analyzed the data. JB analyzed the data and contributed to discussions and writing of the manuscript. PH contributed to discussions and writing of the manuscript. JP contributed to discussions and writing of the manuscript. FM conceived the project and wrote the first draft of the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2010_10061_MOESM1_ESM.XLSX
Additional file 1: Supplementary table S1A & S1B & S1C.xlsx. Contains a list of SNPs, small insertions and deletions found in the analyzed genomes. (XLSX 127 KB)
12864_2010_10061_MOESM2_ESM.PPTX
Additional file 2: Supplementary figure S1.jpg. Shows large regions of differences (RDs) between six Dutch B. pertussis strains and Tohama I (PPTX 170 KB)
12864_2010_10061_MOESM3_ESM.XLSX
Additional file 3: Supplementary table S2.xlsx. Shows large Regions of Difference (RDs) between the analyzed genomes. (XLSX 15 KB)
12864_2010_10061_MOESM5_ESM.XLSX
Additional file 5: Supplementary table S4.xlsx. Shows allelic polymorphisms found in pathogenicity-associated genes (XLSX 22 KB)
12864_2010_10061_MOESM6_ESM.XLSX
Additional file 6: Supplementary table S5.xlsx. Shows alleles which distinguish strains isolated before and after the introduction of vaccination (XLSX 28 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article
Cite this article
Bart, M.J., van Gent, M., van der Heide, H.G. et al. Comparative genomics of prevaccination and modern Bordetella pertussis strains. BMC Genomics 11, 627 (2010). https://doi.org/10.1186/1471-2164-11-627
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-11-627