
Abstract
Background
Rainbow trout are important fish for aquaculture and recreational fisheries and serves as a model species for research investigations associated with carcinogenesis, comparative immunology, toxicology and evolutionary biology. However, to date there is no genome reference sequence to facilitate the development of molecular technologies that utilize high-throughput characterizations of gene expression and genetic variation. Alternatively, transcriptome sequencing is a rapid and efficient means for gene discovery and genetic marker development. Although a large number (258,973) of EST sequences are publicly available, the nature of rainbow trout duplicated genome hinders assembly and complicates annotation.
Results
High-throughput deep sequencing of the Swanson rainbow trout doubled-haploid transcriptome using 454-pyrosequencing technology yielded ~1.3 million reads with an average length of 344 bp, a total of 447 million bases. De novo assembly of the sequences yielded 151,847 Tentative Consensus (TC) sequences (average length of 662 bp) and 224,391 singletons. A combination assembly of both the 454-pyrosequencing ESTs and the pre-existing sequences resulted in 161,818 TCs (average length of 758 bp) and 261,071 singletons. Gene Ontology analysis of the combination assembly showed high similarities to transcriptomes of other fish species with known genome sequences.
Conclusion
The 454 library significantly increased the suite of ESTs available for rainbow trout, allowing improved assembly and annotation of the transcriptome. Furthermore, the 454 sequencing enables functional genome research in rainbow trout, providing a wealth of sequence data to serve as a reference transcriptome for future studies including identification of paralogous sequences and/or allelic variation, digital gene expression and proteomic research.
Similar content being viewed by others
Background
Rainbow trout (Oncorhynchus mykiss) are widely distributed fish species cultured in regions with cold and cool water temperatures around the world. They are important food and sport fish and the most cultivated cold freshwater fish in the US [1–3]. In addition, rainbow trout is one of the most rigorously studied fishes in many research areas including carcinogenesis, toxicology, comparative immunology, disease ecology, physiology and nutrition. For over a century, rainbow trout has been used as a model research species; more is known about the biology of rainbow trout than any other fish species. Rainbow trout is a member of the Salmonidae family. During the last decade more than 26,000 scientific papers have been published on salmonids. Rainbow trout can also serve as a surrogate for research needed on other economically important salmonid species such as Atlantic and Pacific salmon and char species [4]. This wealth of knowledge and interest in rainbow trout has guided increased attention to developing and using genomics tools, as evidenced by the considerable accumulation of genomic resources in recent years from several groups around the world [5, 6]. However, the rainbow trout genome sequence is not available yet, and this lack of information hinders the contemporary approaches to identification/characterization of important genes and genetic markers for aquaculture applications and biomedical research. Until the reference genome sequence becomes available, transcriptome sequencing is a fast and efficient means for gene discovery and genetic marker development. Transcriptome sequencing enables functional genomics studies including global gene expression, single nucleotide polymorphism (SNP) discovery, quantitative trait loci analysis and genome scans of diversity [7]. Rainbow trout is a significant fish model in terms of expressed sequence tags (ESTs) [8]. International large-scale EST sequencing projects for rainbow trout have generated large number of ESTs (258,973) that are publicly available through the Rainbow Trout Gene Index database [8–10].
As a member of the family Salmonidae, rainbow trout has descended from a tetraploid ancestor[11]. A whole genome duplication event occurred 25-100 mya and the genome is estimated to be 1/3 of the way toward re-diploidization [11, 12]. About 50% of protein-coding loci examined in salmonid species show duplicate gene expression [13]. The nature of duplicated rainbow trout genome hinders assembly and complicates annotation of the EST sequences as well as identification of SNPs [14]. Attempts to assemble and annotate the rainbow trout ESTs yielded 40,320 Tentative Consensus (TC) sequences, 49,408 singletons and 26,861 UniGenes [15, 16]. Previous efforts aimed at SNP discovery for rainbow trout using ESTs were unsuccessful, mainly, due to difficulties parsing allelic variation from the high frequency of duplicated genes [14]. To better characterize the rainbow trout transcriptome and improve discovery of allelic variations, a haploid transcriptome sequence is needed. Doubled haploid clonal lines, fish that have two identical copies of each chromosome, were established in salmonids, using androgenesis and gynogenesis; whole chromosome set manipulation techniques [17, 18]. In androgenesis, eggs are gamma-irradiated to destroy DNA and then fertilized with untreated sperm. Then, a temperature or pressure shock is used to interrupt mitosis, resulting in completely homozygous doubled haploid fish. Similarly, in gynogenesis, sperm are treated with ultraviolet radiation to induce development of untreated eggs before the temperature or pressure shock treatment. To create a clonal line, completely homozygous and identical doubled haploid, population, a single homozygous fish is reproduced through a second generation of chromosome set manipulation [17, 18].
Recently, 454-pyrosequencing has become relatively rapid and cost-effective method for sequencing with progressively increasing depth and coverage. Therefore, it is a very suitable technique for de novo transcriptome sequencing for non-model species such as rainbow trout [7, 19–21]. The objectives of this study were to: 1) sequence and characterize the transcriptome of an individual from the Swanson doubled haploid rainbow trout clonal line using 454-pyrosequencing to produce a transcriptome reference sequence for identification of gene duplications, distinguishing true/false SNPs and future genomic/transcriptomic sequencing; and 2) use the 454-pyrosequencing and the Sanger-based EST data to characterize the rainbow trout transcriptome with improved assembly and annotation for digital gene expression and proteomic research in the future.
Results and Discussion
Sanger-based sequencing of the rainbow trout transcriptome
In the last few years, large-scale EST sequencing projects for rainbow trout have been undertaken in several laboratories worldwide. Rise and coworkers reported 14,544 ESTs (mostly 3'-sequences) from 6 rainbow trout cDNA libraries [10]. Govoroun and coworkers reported a total of 96,472 ESTs sequenced from three normalized and subtracted cDNA libraries including one highly complex pooled-tissue library from 14 different tissues [8]. A substantial number of ESTs were generated in our labs, currently representing 47% of the total number of available rainbow trout sequences (124,138 out of 258,973) (Rainbow Trout Gene Index database release 7.0) [9, 15]. Over 87,000 of these ESTs were sequenced from a normalized library constructed from mRNA pooled from several tissues including; muscle, brain, gill, spleen, liver, kidney and juvenile tissues [9, 10]. The normalization and pooling strategies were used to increase probability of identifying unique and diverse set transcripts. Clones were randomly picked and sequenced until library sequencing is saturated; low probability of discovering a new mRNA reached due to the redundancy effect. Collectively, the international efforts of sequencing the rainbow trout transcriptome have thus far generated 290,735 ESTs that are publicly available through the GenBank [22]. Table 1 shows summary statistics of the major rainbow trout EST projects listed in the Rainbow Trout Gene Index database [9, 15]. In this database, all EST sequence data were integrated and assembled producing 90,019 total unique sequences [8–10]. A total of 209,565 ESTs (81%) were clustered in 40,320 TCs and 49,408 ESTs (19%) were singletons. There is an average of 5.2 EST per TC (Rainbow Trout Gene Index database release 7.0) [23]. Most of the sequences were derived from 5'- or 3'-sequencing and thus are less likely to cover coding regions, which hinders the EST annotations. Table 2 shows summary statistics of the Sanger-based ESTs assembly compared to the below-mentioned de novo assembly of the 454-pyrosequencing and a combination assembly of both datasets.
454-Pyrosequencing of the rainbow trout transcriptome
A 454-pyrosequencing library was constructed from 14 tissues collected from a single immature male homozygous doubled-haploid fish from the Swanson clonal line, the same line used for BAC library construction (12). The 454 library was titrated in three one-sixteenth-plate regions to determine the optimal bead library concentration. After titration, the library was sequenced in two half-plate regions of a PicoTiterPlate. Great wealth of sequence data were obtained from the pyrosequencing runs, yielding six times more sequence data than all the currently available Sanger-based data. A total of 1,416,404 ESTs (average length 344 bp) and 447 million bases were obtained from 454-pyrosequencing. The primary sequence data were deposited in GenBank Short Read Archive (SRA) under accession number SRA009276.8. Of these, 1,290,292 (91%) passed quality standards (primer filtering; sequences that contain homopolymers and shorter than 100 bp were filtered out).
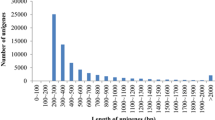
Sequences that passed quality standards were clustered and de novo assembled. A total of 1,065,901 reads were integrated into 151,847 TCs leaving 224,391 singletons; yielding a total of 376,238 unique sequences (Table 2). The de novo assembly sequence data were deposited in the Transcriptome Shotgun Assembly (TSA) database with the accession numbers EZ763335-EZ915181. The percentage of the assembled 454-pyrosequencing sequence reads was 83% compared to 81% of the Sanger-based EST data. The average length of the 454-pyrosequencing TCs was 662 bp, compared to 999 bp of the Sanger-based data. Distribution of 454-assembly showed a wide range of contig lengths with most contigs ranging between 300 and 900 bp (Figure 1). The number of contigs larger than 500 bp was 84,810 (56%). De novo assembly of pyrosequencing transcriptomic data from other non-model species yielded 25-88% ESTs assembled into contigs of average length 197-233 bp [7, 24, 25]. Successful assembly of rainbow trout pyrosequencing data into relatively long contigs is apparently due to the length of the individual reads and/or thorough coverage of the transcriptome.

Average length distribution of assemblies and pyrosequencing reads of the rainbow trout ESTs. Average lengths of the combination assembly contigs are more than those of the 454-pyrosequencing assembly over the 100-700 bp length range, indicating that addition of the Sanger-based data improved assembly of the short 454-pyrosequenceing reads up to 700 bp. However, over the 700-2000 bp length range, average contig lengths of the combination assembly are less than those of the 454-pyrosequencing assembly indicating that contigs longer than 700 bp of the combination assembly were mainly derived from the 454-pyroseqeuncing data, possibly because most of the 454-pyrosequencing reads were 400-600 bp. Number of the 454-pyrosequencing reads is divided by 10 for scaling.
The 454-pyrosequencing data were generated and assembled from a doubled-haploid rainbow trout clonal line to address problems associated with the nature of the rainbow trout duplicated genome that hinders assembly and complicates annotation [11, 14]. The 454-pyrosequencing data were assembled to serve as a reference sequence for future studies to identify gene duplications from allelic variations and to distinguish true/false SNPs. Sequence data were de novo assembled using the default parameters of the SeqMan NGen software (see Materials and Methods for details).
The 454-pyrosequencing library identified large number of new genes. A total of 282,357 gene transcripts were identified by subtracting the Sanger-based sequences from the 454-pyrosequencing sequence data. The subtraction was done by using BLASTn with an e-value below 1e-10, an alignment longer than 99 bp and 95% sequence identity within the aligned region (data not shown).
A combination of both the 454-pyrosequencing ESTs and the Sanger-based sequences offered the maximum amount of genetic information available for rainbow trout so far; 521 M bp, 1,380,311 ESTs and 422,889 unique sequences (Table 2). The combination assembly sequence data are available at the NAGRP Aquaculture Genome Projects [26]. With estimates of 2.4 to 3.0 × 109 bp genome size of the rainbow trout and 1% of the genome being mRNAs, the combined data provide an estimated 19× depth of coverage of the rainbow trout transcriptome [27, 28]. The combination assembly increased the number of TCs to 161,818 (average contig length of 758 bp) leaving 261,071 singletons. A comparison of the percentage of the assembled reads in the combination assembly (81%) to the 454-pyrosequencing and the Sanger-based assemblies (83% and 81%, respectively) indicates that combining the two sequence data did not improve the assembly percentage of each individual sequence dataset (Table 2).
Figure 1 shows contig length distribution of the two assemblies compared to 454-pyrosequence average read length distribution. The figure illustrates that over 100-700 bp length range, average contig lengths of the combination assembly are more than that of the 454-pyrosequencing assembly, indicating that adding the Sanger-based data improved assembly of the short 454-pyrosequences reads up to 700 bp. However, over 700-2000 bp length range, average contig lengths of the combination assembly are less than that of the 454-pyrosequencing assembly indicating that contigs longer than 700 bp of the combination assembly were mainly derived from the 454-pyroseqeuncing data, possibly because most of the 454-pyrosequencing reads were 400-600 bp. The relatively short read lengths of the 454 pyrosequencing compared to Sanger sequencing is a major concern of the 454-pyrosequencing [25, 29]. The 454/GS-Titanium sequencing platform, used in this study, has been used to address this issue and lead to successful assembly of the 454 pyrosequencing data, which make this technology suitable for de novo transcriptome sequencing of non-model species such as aquaculture species [7, 30].
Functional annotation and Gene Ontology analyses
Functional annotation of transcripts of the combination assembly was carried out by BLASTx search against the NCBI non-redundant protein database using the Blast2GO suite [31]. BLAST result accessions were used to retrieve associated gene names and Gene ontology (GO) terms. Gene ontology is widely used to standardize representation of genes across species and provides a controlled vocabulary of terms for describing gene product [32]. Biological processes constituted the largest class of GO assignment of the transcripts (17,694 counts, 39%), followed by cellular components (16,452, 36%) and molecular function (11,160, 25%). Level 2 GO assignments within the three categories are summarized in Figure 2. Of the biological process categories, 25% of the genes are associated with cellular processes and 25% are related to regulation of biological process. Within the molecular functional category, 46% are related to protein binding followed by 32% catalytic activity. Of the cellular components, 59% of the genes are related to the cell and 24% were related to organelles.

Gene Ontology (GO) assignment (2nd level GO terms) of the rainbow trout Sanger-based and 454-pyrosequncing combination assembly. Biological processes constitute majority of GO assignment of the transcripts (17,694 counts, 39%), followed by cellular components (16,452 counts, 36%) and molecular function (11,160 counts, 25%).
A BLASTx top-hit species distribution of gene annotations showed highest homology to Zebrafish (Danio rerio), followed by Atlantic salmon (Salmo salar) and Puffer fish (Tetraodon nigrovirdis) (Figure 3). In addition, the trout sequences had significant homologies to four more fish species including Japanese pufferfish (Takifugu rubripes), rainbow smelt (Osmerus mordax), Northern pike (Esox lucius) and European plaice (Pleuronectes platessa). These results indicate a high level of phylogenetic conservation of the rainbow trout gene content compared to these fish species. On the other hand, the model fish species Medaka fish (Oryzias latipes) and Stickleback fish (Gasterostreus aculeatus) showed very low homology to the trout sequences. It is worth mentioning that only 5.7% of the BLASTx top-hits matched rainbow trout protein sequences. This may be explained on the basis of the limited number of the rainbow trout proteins (6,915) that are currently available in the NCBI database compared to other fish species with known genome sequences (141,396 proteins in the case of Zebrafish) and suggests identification of large number of new genes by the 454-pyrosequencing library [22]. The results of BLASTx top-hit of fish species are provided in Additional file 1, Table S1.

BLASTx top-hit species distribution of gene annotations showing high homology to fish species with known genome sequences. Only 5.7% of the BLAST hits matched rainbow trout protein sequences due to the limited number of the rainbow trout proteins (6,915) that are currently available in the NCBI database (compared to 141,396 proteins in Zebrafish).
Based on the BLASTx top-hit species distribution of gene annotations, Zebrafish was chosen for a detailed Gene Ontology comparison (Figure 4). Distribution profile and percentage of clustered 2nd level Gene Ontology terms of the combination assembly showed high similarity to that of the Zebrafish suggesting an unbiased representation of the combination assembly to the rainbow trout transcriptome. Similarities of the rainbow trout and Zebrafish transcripts as indicated by distributions of clusters of different categories of the biological processes, cellular components and molecular functional implies that the combination assembly represents the rainbow trout tissue complexity and that normalization of the 454-pyrosequencing pooled-tissue library and the Sanger-based sequences were effective in reducing redundancy (Figure 4).

Gene Ontology (2nd level GO terms) comparison of the rainbow trout combination assembly showing high similarity to the transcriptome of Zebrafish (with known genome sequence).
Conclusion
A hybrid approach of the high throughput 454-pyrosequencing together with the traditional Sanger-based sequences was used to generate high-quality draft assemblies of the rainbow trout transcriptome. The 454-pyrosequencing data were generated and assembled from a Swanson doubled-haploid rainbow trout individual to address problems associated with the nature of the rainbow trout duplicated genome; to serves as a reference sequence for future studies to identify gene duplications from allelic variations and to distinguish true/false SNPs.
The 454-pyrosequencing yielded a great wealth of sequence data. Assembly of the 454-pyrosequencing data together with the Sanger-based EST sequencing data provides 19× depth of coverage of the rainbow trout transcriptome, allowing robust functional annotation and gene ontology designation of the rainbow trout transcripts, and providing a reference sequence for digital gene expression, proteomic studies and QTL mapping.
While the released assembly sequences significantly increased the suite of transcriptome sequences available for rainbow trout that enables functional genome research, next-generation sequence assemblies may contain incorrectly assembled contigs, special attention should be given to this fact.
Methods
Production of doubled haploid rainbow trout
The doubled haploid trout from the Swanson clonal line were produced at Washington State University (WSU) as described previously[17, 18] Homozygous rainbow trout were produced by androgenesis and by gynogenesis by blockage of the first cleavage [33]. Fish were raised to sexual maturity in recirculating systems at WSU. Clones of homozygous individuals were produced by collecting sperm from homozygous males and doing another cycle of androgenesis as described above, or by stripping the eggs from homozygous androgenetically or gynogenetically produced females and performing gynogenesis by retention of the second polar body [34]. The eggs were incubated in Heath trays at 12°C until hatching, then transferred to re-circulating systems for grow out.
Tissue collection and RNA isolation
Tissues were collected from a single immature (~2-year-old, ~250 g) male homozygous doubled-haploid fish from the Swanson River clonal line[18]. Tissues were flash frozen in liquid nitrogen and shipped on dry ice from WSU to WVU then stored at -80°C until total RNA isolation. Total RNA was isolated using TRIzol™ (Invitrogen, Carlsbad, CA) from 14 tissues including white muscle, red muscle, testis, spleen, kidney, head kidney, pituitary, stomach, brain, heart, intestine, gill, skin and liver. Equal masses of total RNAs from various tissues were pooled and used to construct the 454-pyrosequencing library.
454-cDNA Library construction
Library preparation and 454-pyrosequencing and sequence assemblies were done at Roy J. Carver Biotechnology Center (University of Illinois at Urbana-Champaign) as described before [35]. Briefly, messenger RNA was isolated from total RNA using the Oligotex mRNA Mini kit (Qiagen, CA). First and second strand cDNA were synthesized from 200 ng of mRNA using the SuperScript® Double-Stranded cDNA Synthesis Kit (Invitrogen, CA) with 100 μM random hexamer primers (Fermentas, USA). Double-stranded cDNA was cleaned up with a QIAquick Minelute PCR purification column (Qiagen, CA). Double-stranded DNA was nebulized with the nebulization kit supplied with the GS Titanium Library Preparation kit (Roche/454 Life Sciences, CT) following their recommendations (30 psi for 1 minute) and cleaned up with a QIAquick PCR minelute column and eluted in 50 ul EB. Nebulized cDNA was blunt-ended (25 μl water, 10 μl 10× T4 DNA Ligase buffer (NEB), 4 μl 10 mM dNTP mix, 5 μl T4 DNA polymerase (3 U/μl) (NEB), 1 μl Klenow polymerase (5 U/μl) (NEB), and 5 μl Polynucleotide kinase (10 U/μl) (NEB) and cleaned up with a Qiaquick PCR minelute column and eluted in 32 ul EB. A dA-overhang was added at 3' end of cDNA by adding the following to the blunt-ended cDNA: 5 μl 10× buffer 2 (NEB), 10 μl 1 mM dATP and 3 μl Klenow exo-minus polymerase (5 U/μl) (NEB). The reaction was incubated at 37°C for 30 minutes and then cleaned up with a QIAquick MiniElute column and eluted in 10 μl EB. The cDNA was adaptored with Titanium adaptors (Roche/454 Life Sciences, CT) by adding 9 μl water, 25 μl 2× Rapid Ligase buffer (Enzymatics, MA) 5 μl (50 μM) Titanium adapter A/B mix and 1 μl T4 DNA Ligase (600 U/μl (Enzymatics, MA) and incubated the ligation reaction at room temperature for 15 minutes. The reaction was cleaned up using a Qiaquick MiniElute column (Qiagen), eluting the cDNA in 20 μl EB. Adaptored cDNA was run on a E-GEL EX 2% agarose (Invitrogen, CA) following the manufacturer instructions and cDNAs in the size range of 400-800 bp were excised from the gel and purified with a Qiagen's Gel Extraction kit and the cDNA was eluted in 30 μl EB. One μl of the gel-purified cDNA was used as template for amplification in 50 μl PCR reactions containing 10 μl 5× Phusion Buffer HF (NEB), 25 μM Adapter A_For primer (5'CCATCTCATCCCTGCGTGTCTCCGACTCAGACGAGTGCGT3'), 25 μM Adapter B_For primer (5'CCTATCCCCTGTGTGCCTTGGCAGTCTCAGT3'), 3% DMSO, 10 mM dNTPs and 1 U Phusion polymerase (Finnzymes/NEB, USA). The PCR conditions were as follows: 98°C for 30 seconds, followed by 15 cycles with 98°C for 10 seconds, 68°C for 30 seconds and 72°C for 30 seconds, with a final extension of 72°C for 5 minute and cleaned up with a Qiaquick minelute PCR column.
Normalization of cDNA library
The cDNA library was normalized according to the protocol described in the Trimmer Direct Kit (Evrogen, Russia). In brief, 300 ng of cDNA were incubated at 95°C for 5 minutes followed by incubation at 68°C for 4 hours in the hybridization buffer included in the kit (50 mM Hepes, pH7.5 and 0.5 M NaCl). After the incubation, the reaction was treated with ¼ units of duplex specific nuclease (DSN). The normalized cDNA was then amplified from 1 μl of DSN-treated cDNA by PCR reactions (10 cycles) described above and gel purified for the fragment size of 400-800 bp as described above.
454/GS-Titanium sequencing
After library construction, the samples were quantified using a Qubit fluorometer (Invitrogen, CA) and average fragment sizes were determined by analyzing 1 μl of the samples on the Bioanalyzer (Agilent, CA) using a DNA 7500 chip. The library was diluted to 1 × 106molecules/μl. emPCR was set up using Titanium emPCR kits (Roche) at 0.25 copies of DNA molecules per bead (8.75 μl of 1 × 106 molecules/μl into 35 million beads). Processing was done per Roche protocols for the remaining emPCR breaking, enrichment, and PTP loading steps.
Bioinformatics analysis
The sff sequence files were processed by using tools from Roche [36] and in-house Java scripts. Sequences that contain homopolymers (in which 60% over the entire length of the read is represented by one nucleotide) and that are shorter than 100 bp were filtered out. Modified sequence data were De novo assembled using the software SeqMan NGen version 2.0 (trial version) from DNAStar [37] at default parameters (Match size, 40 bp; minimum match percentage, 90%; mismatch penalty, 25; gap penalty, 25). Similarly, a combination assembly was done using both the 454-pyrosequencing ESTs and the pre-existing Sanger sequences downloaded from the rainbow trout gene index [15].
References
Harvey DJ: Aquaculture Outlook. Electronic Outlook Report from the Economic Research Service. 2006, [http://www.ers.usda.gov]
Wolf K, Rumsey G: The representative research animal: why rainbow trout? Salmo gairdneri?. Z Angew Ichthyol. 1985, 3: 131-138. 10.1111/j.1439-0426.1985.tb00422.x.
Behnke RJ: Native Trout of Western North America. American Fisheries Society Monograph 6, American Fisheries Society, Bethesda, MD. 1992
Thorgaard GH, Bailey GS, Williams D, Buhler DR, Kaattari SL, Ristow SS, Hansen JD, Winton JR, Bartholomew JL, Nagler JJ: Status and opportunities for genomics research with rainbow trout. Comp Biochem Physiol B Biochem Mol Biol. 2002, 133 (4): 609-646. 10.1016/S1096-4959(02)00167-7.
Davidson W, Chevalet C, Rexroad Iii CE, Omholt S: Salmonid Genomic Sequencing Initiative: the Case for Sequencing the Genomics of Atlantic Salmon (Salmo Salar) and Rainbow Trout (Oncorynchus Mykiss). Government Publication/report. 2006
Koop BF, von Schalburg KR, Leong J, Walker N, Lieph R, Cooper GA, Robb A, Beetz-Sargent M, Holt RA, Moore R: A salmonid EST genomic study: genes, duplications, phylogeny and microarrays. BMC Genomics. 2008, 9: 545-545. 10.1186/1471-2164-9-545.
Vera JC, Wheat CW, Fescemyer HW, Frilander MJ, Crawford DL, Hanski I, Marden JH: Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Mol Ecol. 2008, 17 (7): 1636-1647. 10.1111/j.1365-294X.2008.03666.x.
Govoroun M, Le Gac F, Guiguen Y: Generation of a large scale repertoire of Expressed Sequence Tags (ESTs) from normalised rainbow trout cDNA libraries. BMC Genomics. 2006, 7: 196-196. 10.1186/1471-2164-7-196.
Rexroad CE, Lee Y, Keele JW, Karamycheva S, Brown G, Koop B, Gahr SA, Palti Y, Quackenbush J: Sequence analysis of a rainbow trout cDNA library and creation of a gene index. Cytogenet Genome Res. 2003, 102 (1-4): 347-354. 10.1159/000075773.
Rise ML, von Schalburg KR, Brown GD, Mawer MA, Devlin RH, Kuipers N, Busby M, Beetz-Sargent M, Alberto R, Gibbs AR: Development and application of a salmonid EST database and cDNA microarray: data mining and interspecific hybridization characteristics. 2004, 14 (3): 478-490.
Allendorf FW, Thorgaard GH: Tetraploidy and the evolution of salmonid fishes. Evolutionary Genetics of Fishes. 1984, BJ T: Plenum Press, New York, 1-53.
Palti Y, Gahr SA, Hansen JD, Rexroad CE: Characterization of a new BAC library for rainbow trout: evidence for multi-locus duplication. Anim Genet. 2004, 35 (2): 130-133. 10.1111/j.1365-2052.2004.01112.x.
Bailey GS, Poulter RT, Stockwell PA: Gene duplication in tetraploid fish: model for gene silencing at unlinked duplicated loci. Proc Natl Acad Sci USA. 1978, 75 (11): 5575-5579. 10.1073/pnas.75.11.5575.
Castano C, Smith TP, Wiedmann RT, Vallejo RL, Salem M, Yao J, Rexroad CE: Single nucleotide polymorphism discovery in rainbow trout by deep sequencing of a reduced representation library. BMC Genomics. 2009, 10:559:
The DFCI R.trout Gene Index. [http://compbio.dfci.harvard.edu/tgi/cgi-bin/tgi/gimain.pl?gudb=r_trout]
The NCBI UniGene database, Oncorhynchus mykiss (UniGene Build #31). [http://www.ncbi.nlm.nih.gov/UniGene/UGOrg.cgi?TAXID=8022]
Robison BD, Wheeler PA, Thorgaard GH: Variation in development rate among clonal lines of rainbow trout (Oncorhynchus mykiss). Aquaculture. 1999, 173: 131-141. 10.1016/S0044-8486(98)00481-5.
Young WP, Wheeler PA, Fields RD, Thorgaard GH: DNA fingerprinting confirms isogenicity of androgenetically-derived rainbow trout lines. J Hered. 1996, 87: 77-81.
Weber AP, Weber KL, Carr K, Wilkerson C, Ohlrogge JB: Sampling the Arabidopsis transcriptome with massively parallel pyrosequencing. Plant Physiol. 2007, 144 (1): 32-42. 10.1104/pp.107.096677.
BrÃutigam A, Shrestha RP, Whitten D, Wilkerson CG, Carr KM, Froehlich JE, Weber AP: Low-coverage massively parallel pyrosequencing of cDNAs enables proteomics in non-model species: comparison of a species-specific database generated by pyrosequencing with databases from related species for proteome analysis of pea chloroplast envelopes. J Biotechnol. 2008, 136 (1-2): 44-53. 10.1016/j.jbiotec.2008.02.007.
Garcia-Reyero N, Griffitt RJ, Liu L, Kroll KJ, Farmerie WG, Barber DS, Denslow ND: Construction of a robust microarray from a non-model species largemouth bass, Micropterus salmoides (Lacèpede), using pyrosequencing technology. Journal of Fish Biology. 2008, 72: 2354-2376. 10.1111/j.1095-8649.2008.01904.x.
The NCBI database. [http://www.ncbi.nlm.nih.gov/]
The DFCI R.trout Gene Index release 7.0. [http://compbio.dfci.harvard.edu/tgi/cgi-bin/tgi/T_release.pl?gudb=r_trout]
Cheung F, Win J, Lang JM, Hamilton J, Vuong H, Leach JE, Kamoun S, Andre Levesque C, Tisserat N, Buell CR: Analysis of the Pythium ultimum transcriptome using Sanger and Pyrosequencing approaches. BMC Genomics. 2008, 9: 542-542. 10.1186/1471-2164-9-542.
Kristiansson E, Asker N, Forlin L, Larsson DG: Characterization of the Zoarces viviparus liver transcriptome using massively parallel pyrosequencing. BMC Genomics. 2009, 10: 345-10.1186/1471-2164-10-345.
Aquaculture Genome Projects. [http://www.animalgenome.org/aquaculture/salmonids/rainbowtrout/EST_WV.html]
Palti Y, Luo MC, Hu Y, Genet C, You FM, Vallejo RL, Thorgaard GH, Wheeler PA, Rexroad CE: A first generation BAC-based physical map of the rainbow trout genome. BMC Genomics. 2009, 10 (1): 462-462. 10.1186/1471-2164-10-462.
Dunwell JM, Moya-León MA, Herrera R: Transcriptome analysis and crop improvement (a review). Biol Res. 2001, 34 (3-4): 153-164. 10.4067/S0716-97602001000300003.
Ronaghi M, Uhlén M, Nyrén P: A Sequencing Method Based on Real-Time Pyrophosphate. Science. 1998, 281 (5): 363+365-10.1126/science.281.5375.363.
Quinn NL, Levenkova N, Chow W, Bouffard P, Boroevich KA, Knight JR, Jarvie TP, Lubieniecki KP, Desany BA, Koop BF: Assessing the feasibility of GS FLX Pyrosequencing for sequencing the Atlantic salmon genome. BMC Genomics. 2008, 9: 404-404. 10.1186/1471-2164-9-404.
Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A: High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36 (10): 3420-3435. 10.1093/nar/gkn176.
Gene Ontology C: The Gene Ontology project in 2008. Nucleic Acids Res. 2008, 36: D440-D444. 10.1093/nar/gkm883.
Scheerer PD, Thorgaard GH, Allendorf FW: Genetic analysis of androgenetic rainbow trout. J Exp Zool. 1991, 260: 382-390. 10.1002/jez.1402600312.
Thorgaard GH: Chromosome set manipulation and sex control in fish. Fish Physiology. Edited by: Hoar WS, Randall DJ, Donaldson EM. 1983, New York: Academic Press, IXB: 405-434. full_text.
Ali S, Wrigh CW, Lane A, Vlach E, Hetrick L, Bachman S, Vullaganti DR, Thimmapuram J, Drnevich J, Band M: Development of Adapter Free-Transcriptome Sequencing Procedures on the GS FLX/Titanium Next-Generation DNA Sequencing System. PlosONE. 2010,
Roche Applied Science. [https://www.roche-applied-science.com]
DNAStar. [http://www.dnastar.com]
Acknowledgements
This project was supported by National Research Initiative Competitive Grant No.2007-35205-17914 from the USDA Cooperative State Research, Education, and Extension Service; and USDA-ARS Cooperative Agreement No. 58-1930-5-537. It is published with the approval of the Director of the West Virginia Agriculture and Forestry Experiment Station as scientific paper No. 3080. We thank Paul Wheeler for providing tissues from the Swanson doubled haploid trout.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
MS designed the project, was responsible for generation of the pyrosequence data, data analysis, and drafted the manuscript; CR was responsible for the Sanger-based data, shared in conception of the project; JW assisted with pyrosequence library preparation; GT developed the Swanson doubled haploid line and provided tissues as a source of RNA for pyrosequence data; JY contributed to overall project design and is the corresponding author for manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2010_9955_MOESM1_ESM.XLS
Additional file 1: BLASTx top-hit fish species. Top-hit from BLASTx search matching sequences from fish species (XLS 12 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article
Cite this article
Salem, M., Rexroad, C.E., Wang, J. et al. Characterization of the rainbow trout transcriptome using Sanger and 454-pyrosequencing approaches. BMC Genomics 11, 564 (2010). https://doi.org/10.1186/1471-2164-11-564
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-11-564