
Abstract
Background
Pseudomonas fluorescens is a genetically and physiologically diverse species of bacteria present in many habitats and in association with plants. This species of bacteria produces a large array of secondary metabolites with potential as natural products. P. fluorescens isolate WH6 produces Germination-Arrest Factor (GAF), a predicted small peptide or amino acid analog with herbicidal activity that specifically inhibits germination of seeds of graminaceous species.
Results
We used a hybrid next-generation sequencing approach to develop a high-quality draft genome sequence for P. fluorescens WH6. We employed automated, manual, and experimental methods to further improve the draft genome sequence. From this assembly of 6.27 megabases, we predicted 5876 genes, of which 3115 were core to P. fluorescens and 1567 were unique to WH6. Comparative genomic studies of WH6 revealed high similarity in synteny and orthology of genes with P. fluorescens SBW25. A phylogenomic study also placed WH6 in the same lineage as SBW25. In a previous non-saturating mutagenesis screen we identified two genes necessary for GAF activity in WH6. Mapping of their flanking sequences revealed genes that encode a candidate anti-sigma factor and an aminotransferase. Finally, we discovered several candidate virulence and host-association mechanisms, one of which appears to be a complete type III secretion system.
Conclusions
The improved high-quality draft genome sequence of WH6 contributes towards resolving the P. fluorescens species, providing additional impetus for establishing two separate lineages in P. fluorescens. Despite the high levels of orthology and synteny to SBW25, WH6 still had a substantial number of unique genes and represents another source for the discovery of genes with implications in affecting plant growth and health. Two genes are demonstrably necessary for GAF and further characterization of their proteins is important for developing natural products as control measure against grassy weeds. Finally, WH6 is the first isolate of P. fluorescens reported to encode a complete T3SS. This gives us the opportunity to explore the role of what has traditionally been thought of as a virulence mechanism for non-pathogenic interactions with plants.
Similar content being viewed by others


Background
Pseudomonas fluorescens is a diverse species of bacteria that is found throughout natural habitats and associated with plants. Contributing to their diverse lifestyles is their ability to produce an equally diverse array of secondary metabolites that affect interactions with hosts and other inhabitants of their ecosystems. Some isolates benefit plants by producing growth promoting hormones or antimicrobial compounds to control against pathogens [1]. Others are deleterious and have the capacity to synthesize and secrete novel compounds that negatively affect growth of plants [2–4].
The physiological diversity of P. fluorescens is mirrored by its tremendous genetic diversity. However, the genetic diversity may reflect the possibility that P. fluorescens is not a single species, but rather a complex of at least two lineages. Molecular phylogenetic studies of 16 isolates suggested P. fluorescens should be represented by the P. chlororaphis and P. fluorescens lineages [5]. Alternatively or additionally, P. fluorescens may have an open pan genome [6, 7]. Finished genome sequences are available for the SBW25, Pf-5, and Pf0-1 isolates of P. fluorescens [8, 9]. Their genomes exceed 6.4 megabases and their relatively large sizes are not unexpected for free-living bacteria [10]. Comparative analyses of the three isolates of P. fluorescens revealed substantial variation in diversity of genome content and heterogeneity in genome organization [9]. Each genome has 1,000 to nearly 1,500 unique genes when compared to each other.
Plant-associated isolates of P. fluorescens potentially have mechanisms for interacting with plants. Many Gram-negative bacteria use a type III secretion system (T3SS) to interact with their hosts [11]. The T3SS is the most complex of the bacterial secretion systems and is typically encoded by a large cluster of genes arranged as a single superoperon. Its function is to inject type III effector proteins directly into host cells [11, 12]. These type III effectors are important host-range determinants of plant pathogenic bacteria because they perturb and potentially elicit plant defenses [13].
It is unclear as to how prevalent T3SS-encoding regions are in P. fluorescens. Nearly 60% of a surveyed collection of P. fluorescens strains had a homolog of rscN, which encodes the ATPase of the T3SS [14]. However, it is not known whether all genes necessary to complete the T3SS are present in these isolates. Of the three completed genomes, genes encoding the T3SS are present only in SBW25. Several important or typically conserved genes are missing or truncated in the T3SS-encoding locus of SBW25 [15]. Despite the cryptic appearance of the T3SS, when constitutively expressing the transcriptional regulator of the T3SS, SBW25 could deliver a heterologous type III effector into plant cells, suggesting the T3SS may still be functional [15].
The role of the T3SS for the lifestyle of P. fluorescens is still unclear. In SBW25, despite the cryptic T3SS, single mutants of some but not all the remaining T3SS-encoding genes were reduced in fitness in the rhizosphere of sugar beets [16]. This is not unheard of, as mutants of seemingly cryptic T3SS in pathogens are compromised in virulence [17]. However, in the case of SBW25, the T3SS mutants were also compromised in growth in vitro [16]. A T3SS mutant of the biocontrol isolate P. fluorescens KD was compromised in its ability to protect cucumbers against damping-off disease caused by Pythium ultimum [18]. This may be a result of KD requiring a functional T3SS to elicit host defenses, thereby indirectly protecting against P. ultimum or potentially as a direct mechanism against the pathogen.
We are interested in exploiting P. fluorescens for control of grassy weeds. We have previously reported the selection, isolation, and characterization of five strains of P. fluorescens that inhibit germination of seeds of grassy weeds [19]. Further characterizations led to the identification of Germination-Arrest Factor (GAF) produced by these isolates. GAF is a small, extremely hydrophilic secreted herbicide that reacts with ninhydrin and possesses an acid group, suggestive of a small peptide or amino acid analog [4, 20]. In particular, the high specificity of GAF towards grasses and inhibitory activity at only certain developmental stages during seed germination provides promise for its potential as a natural herbicide for the control of grassy weeds in grass seed production and turf management settings.
We selected P. fluorescens WH6 as the archetypal GAF-producing isolate. WH6 was extracted from the rhizosphere of Poa sp. and Triticum aestivum at the Hyslop Research Farm in Benton County, Oregon, USA [19]. We sequenced and developed an improved high-quality draft sequence for WH6 using a hybrid Illumina and 454-based sequencing approach. This standard is considered sufficient for our purposes of assessing gene inventory and comparing genome organization [21].
Comparative genomic analysis showed a high number of orthologous genes and strong similarity in genome organization between WH6 and SBW25. Phylogenomic analysis supported this observation and placed WH6 in the same lineage as SBW25, or the proposed P. fluorescens lineage. The high similarity in orthology and genome organization is in contrast to previous observations of P. fluorescens and in comparisons of WH6 to Pf-5 or Pf0-1 [9]. From a non-saturating Tn5-mutagenesis screen of WH6, we previously identified two mutants compromised in GAF activity (WH6-2::Tn5 and WH6-3::Tn5; [4]). Mapping of DNA sequences flanking the two mutants revealed genes encoding proteins with potential functions in regulation and biosynthesis of GAF. Finally, inspection of the WH6 genome revealed several candidate host-association mechanisms, including what appears to be a complete type III and two complete type VI secretion systems.
Results and Discussion
Sequencing and developing an improved, high-quality draft genome sequence
We used an Illumina and a 454 FLX GS LR70 to sequence the genome of WH6 (Table 1). The theoretical coverage using all filtered reads was estimated to be 316× assuming a genome size of approximately 6.5 megabases. We employed a number of steps to meet the standards of an improved, high-quality draft genome sequence of WH6 for comparative purposes. We used Velvet 0.7.55, combinations of short-reads, and a variety of parameter settings to de novo assemble the short reads to generate approximately 75 different assemblies [22]. We developed and used ad hoc Perl scripts with an associated visualization tool to compare each of the different assemblies to each other. This step enabled us to eliminate entire assemblies with large contigs not supported by any other assembly.
We identified a single high-quality de novo assembly based on nearly 24 million reads from all three sequencing methods (Table 1). The Velvet parameters were hash length of 31, expected coverage of 104, and a coverage cutoff of 20. Actual coverage of this assembly based on the total number of used reads was 65 ~ 120×, which was less than one-third the theoretical coverage. This assembly had 189 contigs greater than one kb and a total of 256 contigs greater than 100 bp. The largest contig was 264 kb and the N50 number and size were 26 contigs and 78 kb, respectively.
We used experimental and in silico approaches to improve the draft assembly by reducing the number of physical gaps. Of the 189 contigs greater than one kb, 139 contigs (74%) had significant homology to a reference sequence shared by the end of another contig. These 139 contigs potentially flanked 111 physical gaps (See Additional file 1: Table S1). We were able to amplify across 86 (77.5%) of the gaps using PCR. Physical gaps were subsequently resolved by reassembling the nearly 24 million short-reads with the 86 Sanger reads. Of the remaining scaffolds, we associated more based on in silico evidence. Some contigs shared long-range synteny to a reference genome (see below) and their ends had fifteen or more basepairs of sequence with 100% overlap to each other. This phenomenon is a result of Velvet failing to extend the contig because of low coverage. Secondly, some contigs could be paired together because their ends had partial coding regions with homology to a common reference gene. In total, nineteen more contigs were associated, resulting in a final assembly of 115 scaffolds greater than 100 bp. The largest scaffold was 814 kb and the N50 number and size were 8 and 203 kb, respectively.
The improved, high-quality draft genome sequence had 67 sequence gaps totaling 258,650 Ns. There were 45 large sequence gaps with more than 300 Ns of which eight had more than 10,000 Ns each. We presumed these were artifacts of the Velvet assembly because the fragment size of our paired-end library was no larger than 300 bp. We corrected the sizes for 31 gaps to their corresponding length found in homologous reference sequences. In the other 14 cases, we simply reduced the number of Ns in the region to 300 bp, to reflect the maximum size of our paired-end library. Both approaches to correct the size of sequence gaps were validated using PCR of randomly selected regions (data not shown). In total, we reduced the number of Ns to 6,049 or ~2% of the original number of Ns.
The release of the finished genome sequence of SBW25 fortuitously coincided with our efforts of improving the draft genome sequence of WH6 [9]. We noted that nearly 90% of the homologous sequences we found in the NCBI nt dataset using our BLASTN-based approaches were to P. fluorescens SBW25. We therefore surmised that the genome of WH6 would be similar to the finished genome of P. fluorescens SBW25 and used it as a reference for Mauve Aligner to reorder the 115 WH6 scaffolds [9, 23].
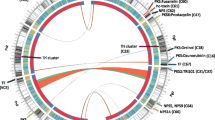
The genome of WH6 is presumed to be a single circular chromosome (Figure 1). A total of 53 scaffolds greater than one kb could be ordered using Mauve Aligner. The remaining 62 contigs could not be reordered and were excluded from our circular representation of the genome. These 62 contigs were all smaller than one kb and their sum total was only 13 kb. Attempts to use Pf0-1 or Pf-5 as a reference for Mauve Aligner were largely unsuccessful, supporting our observation that WH6 and SBW25 had higher synteny than previously detected in P. fluorescens and suggesting our WH6 de novo assembly was of high quality. We found no evidence of plasmids in the genome of WH6.

Circular representation of the improved, high-quality draft genome sequence of WH6. The outer scale designates the coordinates in half million base pair increments. The red ticks indicate physical gaps. Circles 2 and 3 show the predicted coding regions of WH6 on the positive and negative strands, respectively. Coding regions are colored to highlight orthologous (blue) and 1567 unique (red) coding regions of WH6 relative to the other sequenced P. fluorescens. Circles 4, 5, and 6 show orthologs (BLASTP e-value ≤ 1 × 10-7) of SBW25, Pf-5, and Pf0-1, respectively. The extent of homology relative to WH6 is depicted using a heat map of arbitrarily chosen bins; dark blue: orthologs with greater than 80% homology over the length of the gene; green: orthologs with between 60-80% homology over the length of the gene; pink: orthologs with between 20-60% homology over the length of the gene; white: no homology (less than 20% homology over the length of the gene). The positions of loci of interest are also denoted (see corresponding text for more details). Circles 7 and 8 show GC% (gold >60.6% average; gray < 60.6% average) and GC-skew.
This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession AEAZ00000000. The version described in this paper is the first version, AEAZ01000000. The WH6 genome sequence and its associated tools can also be accessed from our website at: http://changbugs.cgrb.oregonstate.edu/microbes/org_detail.html?org=WH6-G3.
One challenge with de novo assembly is dealing with repeated sequences [24]. Small repeated sequences are present in genomes of P. fluorescens but were not expected to have a large effect on our ability to assemble the WH6 genome because of the size of our paired-end fragments [9]. Larger repeats, however, could not be resolved. We only observed one rRNA operon in the genome of WH6. We suspect that WH6 has five rRNA operons similar to SBW25 and Pf-5, but they collapsed into one contig. There was approximately 5× more coverage for the contig containing the one rRNA operon of WH6 compared to the other contigs. Similarly, nonribosomal peptide synthases (NRPSs) are encoded by large genes with repeated modules [25, 26]. The modular domains either collapsed on one another in the assembly, or were assembled into short contigs that we could not extend. A large fraction of these partial NRPS-encoding genes were found in the small contigs that we could not reorder using Mauve Aligner. Here too, we noticed higher coverage than the other scaffolds.
Comparative and phylogenomic analyses of P. fluorescens
At a large scale, the genome of WH6 was similar to the genomes of the other P. fluorescens isolates (Table 2). The size of the genome is slightly smaller, which may be a consequence of the draft nature of our genome assembly. Nonetheless, the 5876 predicted coding sequences (CDSs) and 89.2% coding capacity were very similar.
Previous analyses of P. fluorescens found SBW25, Pf-5, and Pf0-1 to be divergent, with only ~61% of the genes shared and little long-range synteny [9]. We used HAL to carry out similar analyses to determine the effect of the WH6 genome on the phylogenetic relationship of the P. fluorescens species and potential changes to the size of its pan genome [27]. HAL uses a Markov Clustering algorithm based on e-values from reciprocal all-by-all BLASTP analysis to create clusters of orthologs. Core sequences from each species are concatenated and the super alignment is used in phylogenomic analysis. Using a core of 1966 translated sequences common to P. fluorescens, representative strains of P. syringae, and P. aeruginosa PAO1, HAL clustered the different species of Pseudomonas as expected [8, 9, 28–30]. Further, HAL clearly defined two separate lineages for P. fluorescens, placing WH6 with SBW25 (Figure 2).

Phylogenomic tree of eight Pseudomonas isolates based on a super alignment of 1966 translated sequences. P. fluorescens isolates: WH6, SBW25, Pf-5, and Pf0-1; P. syringae pathovars: tomato DC3000, phaseolicola 1448A, and syringae B728a; P. aeruginosa PAO1. Bootstrap support for nodes (r = 1000) were all 100. The scale bar indicates the number of amino acid substitutions per site.
Within the P. fluorescens species as presently defined, 3115 genes formed the core and represented 53%, 52.6%, 50.7%, and 54.3% of the genomes of WH6, SBW25, Pf-5, and Pf0-1, respectively (Figure 3). This was nearly a 10% reduction relative to previous analysis of three genomes [9]. A large fraction of the core genes was assigned to categories with general cellular processes such as energy production and conversion, amino acid transport and metabolism, translation, and transcription (Figure 4). Approximately 90% of the 3115 core genes clustered with orthologs sharing identical COG designations suggesting our automated annotation pipeline was accurate. There were some exceptions but their rarity and subtle differences did not warrant manual curation. For example, one cluster of orthologs had genes annotated as "arabinose efflux permeases" (COG2814) for genes from the published isolates of P. fluorescens but "permease of the major facilitator superfamily" (COG0477) for the ortholog of WH6.

Venn diagram comparing the gene inventories of four isolates of P. fluorescens. The numbers of shared and unique genes are shown. Comparisons were made using HAL.

Functional categories of the 3115 core genes of P. fluorescens and 1567 unique genes of WH6. The number of genes in each category are presented above each bar; black = core; orange = WH6. Categories: C) energy production and conversion; D) cell cycle control, cell division, chromosome partitioning; E) amino acid transport and metabolism; F) nucleotide transport and metabolism; G) carbohydrate transport and metabolism; H) coenzyme transport and metabolism; I) lipid transport and metabolism; J) translation, ribosomal structure and biogenesis; K) transcription; L) replication, recombination and repair; M) cell wall/membrane/envelope biogenesis; N) cell motility; O) posttranslational modification, protein turnover, chaperones; P) inorganic ion transport and metabolism; Q) secondary metabolites biosynthesis, transport and catabolism; R) general function; S) unknown function; T) signal transduction mechanisms; U) intracellular trafficking, secretion and vesicular transport; V) defense mechanisms; -) no COG designation.
A total of 4309 of the translated products of WH6 had an orthologous sequence in another isolate of P. fluorescens. Almost 69% of the WH6 genes had an orthologous sequence in SBW25, as compared to Pf-5 and Pf0-1 with 62% and 59%, respectively (Figures 1 and 3). We found similar levels of overlap using reciprocal BLASTP (data not shown). The 69% orthology between WH6 and SBW25 is much higher than previously observed between isolates of P. fluorescens [9]. These levels were still lower than those between different pathovars of P. syringae, which had greater than 80% orthology [29, 31, 32]. Therefore, the generalization that P. fluorescens have highly variable genomes still holds true.
The genomes of WH6 and SBW25 also showed extensive long-range synteny (Figure 5). This amount of synteny was unexpected given previous comparisons [9]. When compared to Pf-5 or Pf0-1, we found little long-range synteny, which tended to be near the origin of replication. Synteny rapidly degraded away from the origin with an increase in inversions between the genomes [9]. Taken together these lines of evidence all suggest WH6 and SBW25 to be similar and support, though perhaps prematurely, a redefinition of the P. fluorescens species [5, 9].

Synteny plots comparing the organization of the WH6 genome to that of the three other isolates of P. fluorescens. Unique 25 mers from P. fluorescens isolates SBW25 (A), Pf-5 (B), and Pf0-1 (C) were compared to the improved, high quality draft genome sequence of WH6. The start positions of all matching pairs were plotted in an XY graph with the coordinates of the genomes of SBW25, Pf-5, and Pf0-1 along the x-axis and coordinates of the genome of WH6 along the y-axis. The termini are located at the approximate mid-way point for each comparison (red +). Genome scales are shown in one Mb increments.
It could be argued that the high level of synteny we found with SBW25 was an artifact of using SBW25 to reorder the WH6 scaffolds. Though we cannot exclude this possibility, we highlight several points that suggest otherwise. We used a de novo approach to assemble the genome of WH6. The long-range synteny to the SBW25 genome was observed within each and across the de novo assembled scaffolds of WH6 (Figure 5). Furthermore, synteny with SBW25 was also supported by our ability to use SBW25 to successfully and substantially reduce the number of WH6 scaffolds and improve the WH6 genome sequence (Figure 1). Finally, analysis of GC skew gave higher confidence in the reordering of WH6 scaffolds (Figure 1, track 8). Genomes often have a bias of guanine in the leading strand [33, 34]. Inversions of GC skew in regions distant from the replication origins and termini are indicative of a recent recombination event [35]. Barring these events, inversions of GC skew could also potentially indicate large-scale misassemblies or incorrect reordering of contigs. For the most part, the genome of WH6 showed the expected bias of guanine in the leading strand; there are perhaps two small inversions in GC skew flanked by physical gaps between scaffolds near the terminator. Our use of SBW25 as a reference for reordering scaffolds is therefore acceptable and the observed synteny between WH6 and SBW25 appeared to reflect true similarities in genome organization.
More than 30% of the WH6 coding regions were unique (Figures 1 and 3). Examinations of their annotated functions suggested greater diversity in metabolic and host-association functions such as carbohydrate transport and metabolism, inorganic ion transport and metabolism, secondary metabolite biosynthesis, transport and catabolism, intracellular trafficking, secretion and vesicular transport, as well as defense mechanisms (Figure 4).
Examples of CDSs unique to WH6 and enriched in these functional categories include 35 candidate permeases of the major facilitator superfamily, a large and diverse superfamily of secondary active transporters that control movement of substrates across membranes (COG0477; [36]). WH6 also had 12 unique CDSs that encode for putative TonB-dependent receptors, involved in uptake of iron and potentially other substrates (COG1629; [37]; see also section entitled, "Regulators of gene expression"). Restriction modification (RM) systems are widespread defense mechanisms that protect prokaryotes from attack by foreign DNA [38]. RM systems are diverse and can vary dramatically in numbers. WH6 has at least 30 CDSs with annotated functions or domains common to proteins of RM systems. PFWH6_5037-5039, for example, encode for a type I RM system that appears to be unique to WH6. Finally, other CDSs unique to WH6 and of direct interest to us are described in the following sections. The greater than 1500 genes unique to WH6 were dispersed throughout its genome with only a slight bias in location closer to the terminators (Figure 1). This bias was previously generalized for P. fluorescens [9].
Mapping GAF mutants
We previously identified two WH6 mutants from a non-saturating Tn5-mutagenesis screen for those affected in arresting the germination of Poa seeds [4]. We cloned, sequenced and mapped their flanking sequences to identify the disrupted genes. Mutant WH6-3::Tn5 had an insertion in PFWH6_3687. This CDS is annotated as a "predicted transmembrane transcriptional regulator (anti-σ factor)". Its closest homolog, with 94% similarity is PrtR encoded by P. fluorescens LS107d2 [39]. The Tn5 element had inserted at nucleotide position 417 within codon Asp139. Because loss of prtR led to a loss of GAF activity, PrtR is likely an activator rather than a repressor, as was the case in P. fluorescens LS107d2 [39].
Just upstream of prtR in WH6 is prtI, which encodes a candidate ECF σ70 factor. This arrangement is reminiscent of many sigma-anti-sigma factor pairs and suggests that the genes are potentially co-regulated and both may have roles in regulating GAF gene expression [40]. It is peculiar that we failed to identify an insertion in prtI but one obvious explanation is that our screen was not saturating. Regardless, it will be important to examine the necessity of PrtI for GAF activity to resolve its role.
Mutant WH6-2::Tn5 had an insertion in PFWH6_5256, a gene encoding a candidate aminotransferase class III. The identification of an aminotransferase as necessary for GAF supports our previous findings suggesting that GAF contains an amino group and may be a small peptide or amino acid analog [4]. Aminotransferases are pyridoxal phosphate (PLP)-dependent enzymes that catalyze the transfer of an amino group from a donor group (commonly an amino acid) to an acceptor molecule [41]. The Tn5 element had inserted at nucleotide position 1124 within codon Lys375. Based on comparisons to the acetyl ornithine aminotransferase family the insertion is distal to the conserved residues that compose pyridoxal 5'-phosphate binding sites, the conserved residues that compose inhibitor-cofactor binding pockets, and the catalytic residue [42]. Further characterization of WH6-2::Tn5 is necessary to examine its enzymatic properties and role in biosynthesis of GAF.
Regulators of gene expression
Bacteria with large genomes tend to have complex regulatory networks to integrate and respond to a multitude of environmental signals. The extracytoplasmic function (ECF) σ70 factors are a class of important transcriptional regulators of cell-surface signaling systems. Using a Hidden Markov Model (HMM) for ECF-encoding genes, we found 19, 26, 28, and 22 ECFs in WH6, SBW25, Pf-5 and Pf0-1, respectively [43]. Of the 19 identified in WH6, ten are part of the core set common to all four sequenced P. fluorescens isolates and included prtI and prtR, which we identified as necessary for GAF activity. Because we had previously shown that Pf-5 and Pf0-1 do not have GAF activity, these results suggest that the putative PrtI/R-regulon may be different between the different isolates of P. fluorescens [4]. Four of the 19 ECF-encoding genes were exclusive to the plant-associated strains WH6, SBW25, or Pf-5. Two of these were only shared with SBW25, of which one was rspL (see below). The other two lacked sufficient annotations to speculate on their functions. The remaining five ECFs were unique to WH6 and all are potentially co-expressed with genes encoding outer membrane receptors involved in iron perception or uptake (chuA, fhuA, and fhuE).
Virulence factors
Pseudomonads produce a wide-range of secondary metabolites with potential benefit or detriment to plants and microbes [25, 44]. Many are synthesized by non-ribosomal peptide synthases (NRPS) or polyketide synthases [25, 26, 44]. We found evidence for several NRPS-encoding genes. Because of their modular architecture, most NRPS-encoding genes of WH6 were fragmented and found on small contigs that failed to assemble or reorder. Therefore, it was not possible to determine the structure of the repeats or infer functions based on homology. We were, however, able to identify several other candidate toxins and virulence factors (Table 3).
We identified several secretion systems in WH6 unique to host-associated bacteria and/or necessary for full virulence of pathogenic bacteria. WH6 appears to encode a complete and functional type III secretion system (PFWH6_0718-0737; Figure 6a). We named its genes according to the nomenclature first proposed for SBW25 [15, 45]. There is strong homology and synteny between the T3SS-encoding regions of WH6 and P. syringae, raising the possibility of a recent acquisition of the T3SS-encoding locus by WH6, similar to KD [14]. Phylogenetic analyses of rscN, however, placed WH6 with the group 8 of biocontrol isolates of P. fluorescens (data not shown; [14]). Additionally, 15 kb of sequences on either side of the T3SS-encoding region of WH6 were highly syntenic to regions flanking the T3SS-encoding region of SBW25 with the exception of the type III effector gene, ropE. Together, these data argue against a recent acquisition of the T3SS-encoding region by WH6.

Schematic Representations and Comparisons of Type III and Type VI Secretion Systems. A) The type III secretion system of WH6 (top) compared to that of P. syringae (middle), and SBW25 (bottom). Co-transcribed genes and orthologous transcriptional units are colored similarly. *No detectable homology but inferred based on location in the T3SS-encoding locus. **Truncated coding regions. B) The type VI secretion system-1 of WH6 (top) compared to HSI-I of P. aeruginosa PAO1. C) The type VI secretion system-2 of WH6 (top) compared to a candidate type VI secretion system of P. syringae pv tomato DC3000 (bottom). Orthologous transcriptional units are colored similarly. For A-C, the directions of transcription are represented. Gray boxes highlight homologous regions.
There were some differences between T3SS-encoding regions of WH6 and P. syringae. The rspR, rspZ, and rspV genes of WH6 were not present and we failed to detect any homology between the rspF/hrpF, rspA/hrpA, and rspG/hrpG genes. Data, however suggest these differences likely have little to no effect on T3SS function. HrpR and HrpS are highly similar and are functionally redundant. In some Erwinia strains, HrpS by itself is demonstrably sufficient for T3SS function [46, 47]. Deletion mutants of hrpZ are still functional and HrpV functions as a negative regulator of the T3SS [48–50]. HrpF and HrpA are homologous to each other and are structural components of the T3SS. They are the most polymorphic proteins encoded within the T3SS-cluster and the absence of significant homology between rspF and rspA to their counterparts of P. syringae was therefore not surprising [51, 52]. Our automated annotation approach failed to identify rspG but upon visual inspection, we noted a small CDS that encodes a potential product of 63 amino acids. BLAST searches failed to detect homology to hrpG, but given its position in the T3SS-encoding region and similarity in size to the translated product of hrpG, we have annotated it as rspG. In total, these data support the notion that WH6 encodes a complete and functional T3SS, although, its role in the lifestyle of WH6 remains unknown.
Candidate type III effectors of WH6
We used a homology-based approach to search for type III effector genes in the genome of WH6. Our database of type III effectors included those from T3SS-using phytopathogens and some mammal pathogens. We only identified one translated sequence with homology to PipB from Salmonella, and another with homology to HopI1 from P. syringae (e-value < 1 × 10-7, > 33% identity; [53]). However, neither appeared to be strong candidates for a type III effector. We identified a homolog of pipB in the genome of Pf-5, which does not encode the T3SS. HopI1 encodes a J domain and its homolog in WH6 was annotated as the molecular chaperone, dnaJ [54]. These results suggest that if WH6 does encode type III effectors, they are very divergent in sequence. SBW25, in contrast, had at least five genes with homology to known type III effectors, of which two were expressed to sufficiently high levels and delivered by a heterologous T3SS-encoding bacterium [55].
Computational approaches have been successfully used to identify candidate type III effectors from P. syringae, based in part on identifying a cis-regulatory element upstream of their genes and also some genes of the T3SS [56–58]. This so-called hrp-box is recognized by HrpL, an extracytoplasmic function (ECF) σ70 factor encoded within the T3SS-encoding region of P. syringae [59]. We therefore used a Hidden Markov Model (HMM) trained using 38 known HrpL-regulated genes of P. syringae pv tomato DC3000 to mine the genome of WH6 for hrp-boxes [56, 60].
We found 115 hrp-boxes in the genome of WH6 (bit score ≥ 3.0) but only 24 were within 500 bp of a CDS. Two were located upstream of rspF and rscR in the T3SS-encoding region, with bit scores of 7.9 and 3.2, respectively. We also identified a hrp-box upstream of rspJ but it had a lower bit score of 1.2. Fifteen of the CDSs downstream of candidate hrp-boxes had annotated functions not typically associated with type III effectors and we did not list them as possible candidates (data not shown). The remaining eight CDSs downstream of hrp-boxes were annotated as hypothetical proteins and the five with the highest bit scores for their corresponding hrp-boxes were not present in the genomes of Pf-5 and Pf0-1; all but PFWH6_1942 were unique to WH6 (Table 4). Further investigation of their first amino-terminal residues indicated that three have characteristics suggestive of T3SS-dependent secretion [56, 61, 62].
Our two computational approaches yielded very few candidate type III effectors. One possible explanation is that because RspL and HrpL have only 50% identity (70% similarity), they recognize slightly different cis-regulatory sequences and our HMM was not adequately trained for the cis-regulatory sequence recognized by RspL. This is an unlikely scenario. Three sequences with strong similarity to the hrp-box of P. syringae were found in the T3SS-encoding regions for WH6 and SBW25 [15]. Additionally, it has been observed that all HrpL-dependent phytopathogenic bacteria share an identical motif in the hrp-box despite having as little as 52% similarity [63]. Furthermore, in σ70 factors, DNA binding specificity is conferred by the helix-turn-helix domain 4.2 [64, 65]. Domain 4.2 of the WH6 RspL is highly similar (82.5%) to the corresponding domain of HrpL. An alternative explanation is that WH6 encodes very few type III effectors with little homology to those that have been identified. This is not unheard of. P. aeruginosa for example, has only three type III effectors [66, 67].
Type VI secretion systems
The type VI secretion system (T6SS) is another secretion apparatus that is common to host-associated bacteria. Computational approaches suggest the T6SS may also be in P. fluorescens [68, 69]. We found evidence for two complete and functional T6SSs in WH6. We have named these two systems T6SS-1 (Figure 6b; PFWH6_5796-5812) and T6SS-2 (Figure 6c; PFWH6_3251-3270). It is not uncommon for organisms to possess multiple T6SSs that are of different lineages and acquired independently [68]. Additionally, in other strains that have been characterized, different T6SSs appear to be independently regulated, suggesting each T6SS may have functions specific to different aspects of the lifestyle of the bacterium [70]. Whether this is also the case with WH6 awaits further characterization.
T6SS-1 belongs to the group A lineage and shares homology and synteny to HSI-I of P. aeruginosa PAO1 [68]. We therefore named the corresponding genes in WH6 according to the nomenclature established for HSI-I (Figure 6b). Synteny extended beyond the T6SS-encoding region and included the tagQRST genes bordering ppkA. We did not, however, find evidence for tagJ1 in WH6 [68, 71]. T6SS-2 is a group B secretion system [68]. Less is understood about the group B secretion systems but T6SS-2 showed strong homology and synteny to a corresponding T6SS encoded in the genome of the phytopathogen P. syringae pv tomato DC3000 (Figure 6c; [28]).
There are few proteins that are demonstrable type VI effectors. Three homologs of VgrG and Hcp have been shown to be secreted by the T6SS but both likely have functions for the T6SS itself [72–74]. We found four vgrG genes, of which only one was associated with T6SS-1. The other three genes were found elsewhere in the genome. Whether products from these latter three are secreted proteins of the T6SS or merely homologous in sequence is unknown. Both T6SSs of WH6 had a homolog of hcp. Recently, three additional proteins from P. aeruginosa PAO1 were shown to be secreted by the T6SS, but their orthologs were not found in WH6 [75].
Conclusions
P. fluorescens is a genetically and physiologically diverse species found in many habitats. We sequenced the genome of the isolate WH6 because it produces Germination-Arrest Factor (GAF), an herbicide that specifically arrests seed germination of graminaceous species. Comparisons of the WH6 genome to genomes of SBW25, Pf-5, and Pf0-1 helped better define this species, with WH6 and SBW25 forming one lineage. Comparative studies revealing substantial similarity in gene inventory and synteny supported its placement and the argument of at least two major lineages of P. fluorescens [5].
With the genome sequence, we were able to deduce potential functions for two genes necessary for GAF activity. One encoded a candidate anti-sigma factor. Our previous results suggest that PrtR is an activator and suggests it has a role in regulating expression of genes necessary for GAF. The second gene encoded a candidate aminotransferase, which tentatively supports our previous speculation that GAF is a small peptide or amino acid analog. Further studies are required to confirm their functions. A less labor-intensive and saturating screen will also be necessary for a fuller understanding of the pathway controlling GAF expression and biosynthesis. The genome sequence will certainly facilitate such future endeavors.
We also identified a number of mechanisms that potentially affect plant health and some typically associated with host-associated bacteria. One of the more extensively characterized mechanisms is the type III secretion system. WH6 appears to encode the necessary repertoire of genes for a complete and functional T3SS. We also identified two T6SSs in WH6. Further studies are necessary to identify the role these secretion systems and their effectors play in the lifestyle of WH6.
Methods
Sequencing DNA flanking Tn5-insertions
To determine the sites of Tn5 insertion, genomic DNA from the two GAF mutants, WH6-2::Tn5 and WH6-3::Tn5 was digested with Bam HI or Pst I, respectively. We used Southern blotting with a biotinylated probe of the TetR gene from pUTmini-Tn5 gfp to identify the fusion fragments between the TetR gene and flanking WH6 DNA [76]. DNA fragments of corresponding size were cloned into pBluescript SK+ (Stratagene, La Jolla, CA), transformed into E. coli DH5∝, selected based on tetracycline resistance, isolated, and sequenced outwards using primers to the TetR gene.
P. fluorescens WH6 Genome Sequencing
We used the ZR Fungal/Bacterial DNA Kit to isolate genomic DNA from P. fluorescens WH6 grown overnight in LB at 28°C (Zymo Research, Orange, CA). Purity and concentration were determined using a Nanodrop ND1000 (Thermo Scientific, Waltham, MA). For Illumina-based sequencing, we prepared the DNA according to the instructions of the manufacturer and sequenced the DNA fragments on the Illumina GA I and II using 36-cycle (4 channels) and paired-end 76-cycle (1 channel) sequencing, respectively (Illumina, San Diego, CA). The Sanger and Illumina sequencing was done at the Center for Genome Research and Biocomputing Core Labs (CGRB; Oregon State University, Corvallis, OR). We also sequenced genomic DNA using the 454 FLX GS LR70 (454, Branford, CT). Preparation and sequencing by 454 was done at the Consortium for Comparative Genomics (University of Colorado Health Sciences Center, Denver, CO).
Short-read assembly
For Illumina-derived reads, the last four and six bases were trimmed from the 36 mer and 76 mer reads, respectively. We filtered out all Illumina-derived short reads that had ambiguous bases. For the paired-end reads, both reads were filtered out if one read of a pair had ambiguous bases. We used Velvet 0.7.55 to de novo assemble the reads [22]. We assembled short reads from the different sequencing platforms independently, as well as in combination. We wrote ad hoc shell scripts to test different Velvet parameters of hash length, coverage cutoff, and expected coverage. In total, we generated approximately 75 different genome assemblies of WH6. Shell scripts are available for download (http://changlab.cgrb.oregonstate.edu).
Improvements to the high-quality draft assembly
We developed ad hoc Perl scripts to use BLASTN to compare between each of the WH6 assemblies and used congruency in contigs from the various assemblies to cull those with potential misassemblies (see next section for description of scripts; data were visualized using blast_draw.pl). We used Tablet 1.10.01.28 to inspect the remaining genome assemblies for depth of coverage and potential misassemblies [77]. Finally, we used Mauve Aligner 2.3 and the genome sequence of P. fluorescens SBW25 as a reference to reorder WH6 contigs greater than 100 bp from our assembly with highest confidence [9, 23]. Default settings were used for Mauve Aligner 2.3.
Physical and sequence gap closure
To identify contigs that potentially flanked a physical gap, we wrote and used Contig_end_blast_A.pl, to extract 300 bp of sequence from the ends of each contig greater than one kb in size and use the contig ends as queries in a BLASTN search against the NCBI nt database. We also wrote and used Contig_end_blast_B.pl to find contig ends that shared significant homology (e-value ≤ 0.02) to the same reference sequence but aligned to different regions no more than one kb apart. The contigs corresponding to these ends were thus predicted to be physically linked in the genome of WH6. PCR using contig-specific primers and subsequent Sanger sequencing were used to close the physical gaps (See Additional file 2: Table S2). To correct the sizes for sequence gaps larger than 300 bp, we used a similar approach. PCR was used to validate our corrections for sequence gaps.
Contig_end_blast_A.pl, Contig_end_blast_B.pl, and blast_draw.pl are available for download from our website at: http://changlab.cgrb.oregonstate.edu.
Genome Annotation
We used a custom pipeline to annotate the improved high-quality draft assembly of WH6 as previously described [78]. The only exceptions were that we used Glimmer 3.02 rather than Glimmer 2 to predict coding regions and gene models were trained using the "long-orfs" option ([79]; http://www.cbcb.umd.edu/software/glimmer/).
Bioinformatic analyses
For analysis of synteny, we first parsed the genomes of SBW25, Pf-5 and Pf0-1 into all possible 25 mers and identified their unique 25 mer sequences. Next, we used CASHX to align all unique 25 mers from each of three genomes to both strands of a formatted database from the WH6 genome sequence [80]. Only perfect matches were allowed. We identified the corresponding genome coordinates for each 25 mer and the matching 25 mer in the WH6 genome and used R to plot the start coordinates of each matching pair in an XY graph [81].
Phylogenomic relationships were determined using HAL ([27]; http://aftol.org/pages/Halweb3.htm). HAL uses an all-by-all reciprocal BLASTP to create a similarity matrix from e-values. These are then used to group proteins into related clusters using a Markov Clustering algorithm. Clusters containing one protein sequence from each genome that identified each other as best hits were extracted, concatenated within each proteome, and used to infer phylogenetic relationships. Phylogenetic trees were visualized using the Archaeopteryx & Forester Java application ([82]; http://www.phylosoft.org/archaeopteryx/).
Hidden Markov Models (HMMs) for hrp-boxes were trained from a set of 38 confirmed hrp-boxes in the P. syringae pv tomato DC3000 genome [28, 56, 57, 60]. The HMM for the extracytoplasmic function σ70 factors was downloaded from http://www.g2l.bio.uni-goettingen.de/software/f_software.html. Searches were done using HMMER 2.3.2 (http://hmmer.janelia.org/).
Circular diagrams were plotted using DNAplotter ([83]; http://www.sanger.ac.uk/Software/ Artemis/circular/).
References
Haas D, Defago G: Biological control of soil-borne pathogens by fluorescent pseudomonads. Nat Rev Microbiol. 2005, 3 (4): 307-319. 10.1038/nrmicro1129.
Flores-Vargas RD, O'Hara GW: Isolation and characterization of rhizosphere bacteria with potential for biological control of weeds in vineyards. J Appl Microbiol. 2006, 100 (5): 946-954. 10.1111/j.1365-2672.2006.02851.x.
Li Y, Sun A, Zhuang X, Xu L, Chen S, Li M: Research progress on microbial herbicides. Crop Protection. 2003, 22: 247-252. 10.1016/S0261-2194(02)00189-8.
Armstrong D, Azevedo M, Mills D, Bailey B, Russell B, Groenig A, Halgren A, Banowetz G, McPhail K: Germination-Arrest Factor (GAF): 3. Determination that the herbicidal activity of GAF is associated with a ninhydrin-reactive compound and counteracted by selected amino acids. Biological Control. 2009, 51 (1): 181-190. 10.1016/j.biocontrol.2009.06.004.
Yamamoto S, Kasai H, Arnold DL, Jackson RW, Vivian A, Harayama S: Phylogeny of the genus Pseudomonas: intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes. Microbiology. 2000, 146 (Pt 10): 2385-2394.
Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R: The microbial pan-genome. Curr Opin Genet Dev. 2005, 15 (6): 589-594. 10.1016/j.gde.2005.09.006.
Tettelin H, Riley D, Cattuto C, Medini D: Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol. 2008, 11 (5): 472-477. 10.1016/j.mib.2008.09.006.
Paulsen IT, Press CM, Ravel J, Kobayashi DY, Myers GS, Mavrodi DV, DeBoy RT, Seshadri R, Ren Q, Madupu R, Dodson RJ, Durkin AS, Brinkac LM, Daugherty SC, Sullivan SA, Rosovitz MJ, Gwinn ML, Zhou L, Schneider DJ, Cartinhour SW, Nelson WC, Weidman J, Watkins K, Tran K, Khouri H, Pierson EA, Pierson LS, Thomashow LS, Loper JE: Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nat Biotechnol. 2005, 23 (7): 873-878. 10.1038/nbt1110.
Silby MW, Cerdeno-Tarraga AM, Vernikos GS, Giddens SR, Jackson RW, Preston GM, Zhang XX, Moon CD, Gehrig SM, Godfrey SA, Knight CG, Malone JG, Robinson Z, Spiers AJ, Harris S, Challis GL, Yaxley AM, Harris D, Seeger K, Murphy L, Rutter S, Squares R, Quail MA, Saunders E, Mavromatis K, Brettin TS, Bentley SD, Hothersall J, Stephens E, Thomas CM, Parkhill J, Levy SB, Rainey PB, Thomson NR: Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens. Genome Biol. 2009, 10 (5): R51-10.1186/gb-2009-10-5-r51.
Merhej V, Royer-Carenzi M, Pontarotti P, Raoult D: Massive comparative genomic analysis reveals convergent evolution of specialized bacteria. Biol Direct. 2009, 4: 13-10.1186/1745-6150-4-13.
Galan JE, Wolf-Watz H: Protein delivery into eukaryotic cells by type III secretion machines. Nature. 2006, 444 (7119): 567-573. 10.1038/nature05272.
Grant SR, Fisher EJ, Chang JH, Mole BM, Dangl JL: Subterfuge and manipulation: type III effector proteins of phytopathogenic bacteria. Annu Rev Microbiol. 2006, 60: 425-449. 10.1146/annurev.micro.60.080805.142251.
Jones JD, Dangl JL: The plant immune system. Nature. 2006, 444 (7117): 323-329. 10.1038/nature05286.
Rezzonico F, Defago G, Moenne-Loccoz Y: Comparison of ATPase-encoding type III secretion system hrcN genes in biocontrol fluorescent Pseudomonads and in phytopathogenic proteobacteria. Appl Environ Microbiol. 2004, 70 (9): 5119-5131. 10.1128/AEM.70.9.5119-5131.2004.
Preston GM, Bertrand N, Rainey PB: Type III secretion in plant growth-promoting Pseudomonas fluorescens SBW25. Mol Microbiol. 2001, 41 (5): 999-1014. 10.1046/j.1365-2958.2001.02560.x.
Jackson RW, Preston GM, Rainey PB: Genetic characterization of Pseudomonas fluorescens SBW25 rsp gene expression in the phytosphere and in vitro. J Bacteriol. 2005, 187 (24): 8477-8488. 10.1128/JB.187.24.8477-8488.2005.
Ideses D, Gophna U, Paitan Y, Chaudhuri RR, Pallen MJ, Ron EZ: A degenerate type III secretion system from septicemic Escherichia coli contributes to pathogenesis. J Bacteriol. 2005, 187 (23): 8164-8171. 10.1128/JB.187.23.8164-8171.2005.
Rezzonico F, Binder C, Defago G, Moenne-Loccoz Y: The type III secretion system of biocontrol Pseudomonas fluorescens KD targets the phytopathogenic Chromista Pythium ultimum and promotes cucumber protection. Mol Plant Microbe Interact. 2005, 18 (9): 991-1001. 10.1094/MPMI-18-0991.
Banowetz GM, Azevedo MD, Armstrong DJ, Halgren AB, Mills DI: Germination-Arrest Factor (GAF): Biological properties of a novel, naturally-occurring herbicide produced by selected isolates of rhizosphere bacteria. Biological Control. 2008, 46 (3): 380-390. 10.1016/j.biocontrol.2008.04.016.
Banowetz GM, Azevedo MD, Armstrong DJ, Mills DI: Germination arrest factor (GAF): Part 2. Physical and chemical properties of a novel, naturally occurring herbicide produced by Pseudomonas fluorescens WH6. Biological Control. 2009, 50: 103-110. 10.1016/j.biocontrol.2009.03.011.
Chain PS, Grafham DV, Fulton RS, Fitzgerald MG, Hostetler J, Muzny D, Ali J, Birren B, Bruce DC, Buhay C, Cole JR, Ding Y, Dugan S, Field D, Garrity GM, Gibbs R, Graves T, Han CS, Harrison SH, Highlander S, Hugenholtz P, Khouri HM, Kodira CD, Kolker E, Kyrpides NC, Lang D, Lapidus A, Malfatti SA, Markowitz V, Metha T, Nelson KE, Parkhill J, Pitluck S, Qin X, Read TD, Schmutz J, Sozhamannan S, Sterk P, Strausberg RL, Sutton G, Thomson NR, Tiedje JM, Weinstock G, Wollam A, Detter JC: Genomics. Genome project standards in a new era of sequencing. Science. 2009, 326 (5950): 236-237. 10.1126/science.1180614.
Zerbino DR, Birney E: Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18 (5): 821-829. 10.1101/gr.074492.107.
Rissman AI, Mau B, Biehl BS, Darling AE, Glasner JD, Perna NT: Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics. 2009, 25 (16): 2071-2073. 10.1093/bioinformatics/btp356.
Pop M, Salzberg SL: Bioinformatics challenges of new sequencing technology. Trends Genet. 2008, 24 (3): 142-149.
Gross H, Loper JE: Genomics of secondary metabolite production by Pseudomonas spp. Nat Prod Rep. 2009, 26 (11): 1408-1446. 10.1039/b817075b.
Wilkinson B, Micklefield J: Chapter 14. Biosynthesis of nonribosomal peptide precursors. Methods Enzymol. 2009, 458: 353-378. full_text.
Robbertse B, Reeves JB, Schoch CL, Spatafora JW: A phylogenomic analysis of the Ascomycota. Fungal Genet Biol. 2006, 43 (10): 715-725. 10.1016/j.fgb.2006.05.001.
Buell CR, Joardar V, Lindeberg M, Selengut J, Paulsen IT, Gwinn ML, Dodson RJ, Deboy RT, Durkin AS, Kolonay JF, Madupu R, Daugherty S, Brinkac L, Beanan MJ, Haft DH, Nelson WC, Davidsen T, Zafar N, Zhou L, Liu J, Yuan Q, Khouri H, Fedorova N, Tran B, Russell D, Berry K, Utterback T, Van Aken SE, Feldblyum TV, D'Ascenzo M, Deng WL, Ramos AR, Alfano JR, Cartinhour S, Chatterjee AK, Delaney TP, Lazarowitz SG, Martin GB, Schneider DJ, Tang X, Bender CL, White O, Fraser CM, Collmer A: The complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. tomato DC3000. Proc Natl Acad Sci USA. 2003, 100 (18): 10181-10186. 10.1073/pnas.1731982100.
Feil H, Feil WS, Chain P, Larimer F, DiBartolo G, Copeland A, Lykidis A, Trong S, Nolan M, Goltsman E, Thiel J, Malfatti S, Loper JE, Lapidus A, Detter JC, Land M, Richardson PM, Kyrpides NC, Ivanova N, Lindow SE: Comparison of the complete genome sequences of Pseudomonas syringae pv. syringae B728a and pv. tomato DC3000. Proc Natl Acad Sci USA. 2005, 102 (31): 11064-11069. 10.1073/pnas.0504930102.
Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV: Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000, 406 (6799): 959-964. 10.1038/35023079.
Joardar V, Lindeberg M, Jackson RW, Selengut J, Dodson R, Brinkac LM, Daugherty SC, Deboy R, Durkin AS, Giglio MG, Madupu R, Nelson WC, Rosovitz MJ, Sullivan S, Crabtree J, Creasy T, Davidsen T, Haft DH, Zafar N, Zhou L, Halpin R, Holley T, Khouri H, Feldblyum T, White O, Fraser CM, Chatterjee AK, Cartinhour S, Schneider DJ, Mansfield J, Collmer A, Buell CR: Whole-genome sequence analysis of Pseudomonas syringae pv. phaseolicola 1448A reveals divergence among pathovars in genes involved in virulence and transposition. J Bacteriol. 2005, 187 (18): 6488-6498. 10.1128/JB.187.18.6488-6498.2005.
Studholme DJ, Ibanez SG, MacLean D, Dangl JL, Chang JH, Rathjen JP: A draft genome sequence and functional screen reveals the repertoire of type III secreted proteins of Pseudomonas syringae pathovar tabaci 11528. BMC Genomics. 2009, 10: 395-10.1186/1471-2164-10-395.
Arakawa K, Tomita M: The GC Skew Index: A Measure of Genomic Compositional Asymmetry and the Degree of Replicational Selection. Evol Bioinform Online. 2007, 3: 159-168.
Rocha EP: The replication-related organization of bacterial genomes. Microbiology. 2004, 150 (Pt 6): 1609-1627. 10.1099/mic.0.26974-0.
Parkhill J, Sebaihia M, Preston A, Murphy LD, Thomson N, Harris DE, Holden MT, Churcher CM, Bentley SD, Mungall KL, Cerdeno-Tarraga AM, Temple L, James K, Harris B, Quail MA, Achtman M, Atkin R, Baker S, Basham D, Bason N, Cherevach I, Chillingworth T, Collins M, Cronin A, Davis P, Doggett J, Feltwell T, Goble A, Hamlin N, Hauser H, Holroyd S, Jagels K, Leather S, Moule S, Norberczak H, O'Neil S, Ormond D, Price C, Rabbinowitsch E, Rutter S, Sanders M, Saunders D, Seeger K, Sharp S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Unwin L, Whitehead S, Barrell BG, Maskell DJ: Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet. 2003, 35 (1): 32-40. 10.1038/ng1227.
Law CJ, Maloney PC, Wang DN: Ins and outs of major facilitator superfamily antiporters. Annu Rev Microbiol. 2008, 62: 289-305. 10.1146/annurev.micro.61.080706.093329.
Blanvillain S, Meyer D, Boulanger A, Lautier M, Guynet C, Denance N, Vasse J, Lauber E, Arlat M: Plant carbohydrate scavenging through tonB-dependent receptors: a feature shared by phytopathogenic and aquatic bacteria. PLoS One. 2007, 2 (2): e224-10.1371/journal.pone.0000224.
Tock MR, Dryden DT: The biology of restriction and anti-restriction. Curr Opin Microbiol. 2005, 8 (4): 466-472. 10.1016/j.mib.2005.06.003.
Burger M, Woods RG, McCarthy C, Beacham IR: Temperature regulation of protease in Pseudomonas fluorescens LS107d2 by an ECF sigma factor and a transmembrane activator. Microbiology. 2000, 146 (Pt 12): 3149-3155.
Hughes KT, Mathee K: The anti-sigma factors. Annu Rev Microbiol. 1998, 52: 231-286. 10.1146/annurev.micro.52.1.231.
Yoshimura T, Jhee KH, Soda K: Stereospecificity for the hydrogen transfer and molecular evolution of pyridoxal enzymes. Biosci Biotechnol Biochem. 1996, 60 (2): 181-187. 10.1271/bbb.60.181.
Marchler-Bauer A, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Tasneem A, Thanki N, Yamashita RA, Zhang D, Zhang N, Bryant SH: CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009, D205-210. 10.1093/nar/gkn845. 37 Database
Staron A, Sofia HJ, Dietrich S, Ulrich LE, Liesegang H, Mascher T: The third pillar of bacterial signal transduction: classification of the extracytoplasmic function (ECF) sigma factor protein family. Mol Microbiol. 2009, 74 (3): 557-581. 10.1111/j.1365-2958.2009.06870.x.
Lindeberg M, Myers CR, Collmer A, Schneider DJ: Roadmap to new virulence determinants in Pseudomonas syringae: insights from comparative genomics and genome organization. Mol Plant Microbe Interact. 2008, 21 (6): 685-700. 10.1094/MPMI-21-6-0685.
Bogdanove AJ, Beer SV, Bonas U, Boucher CA, Collmer A, Coplin DL, Cornelis GR, Huang HC, Hutcheson SW, Panopoulos NJ, Van Gijsegem F: Unified nomenclature for broadly conserved hrp genes of phytopathogenic bacteria. Mol Microbiol. 1996, 20 (3): 681-683. 10.1046/j.1365-2958.1996.5731077.x.
Hutcheson SW, Bretz J, Sussan T, Jin S, Pak K: Enhancer-binding proteins HrpR and HrpS interact to regulate hrp-encoded type III protein secretion in Pseudomonas syringae strains. J Bacteriol. 2001, 183 (19): 5589-5598. 10.1128/JB.183.19.5589-5598.2001.
Wei Z, Kim JF, Beer SV: Regulation of hrp genes and type III protein secretion in Erwinia amylovora by HrpX/HrpY, a novel two-component system, and HrpS. Mol Plant Microbe Interact. 2000, 13 (11): 1251-1262. 10.1094/MPMI.2000.13.11.1251.
Alfano JR, Bauer DW, Milos TM, Collmer A: Analysis of the role of the Pseudomonas syringae pv. syringae HrpZ harpin in elicitation of the hypersensitive response in tobacco using functionally non-polar hrpZ deletion mutations, truncated HrpZ fragments, and hrmA mutations. Mol Microbiol. 1996, 19 (4): 715-728. 10.1046/j.1365-2958.1996.415946.x.
Ortiz-Martin I, Thwaites R, Mansfield JW, Beuzon CR: Negative regulation of the Hrp type III secretion system in Pseudomonas syringae pv. phaseolicola. Mol Plant Microbe Interact. 2010, 23 (5): 682-701. 10.1094/MPMI-23-5-0682.
Preston G, Deng WL, Huang HC, Collmer A: Negative regulation of hrp genes in Pseudomonas syringae by HrpV. J Bacteriol. 1998, 180 (17): 4532-4537.
Deng WL, Preston G, Collmer A, Chang CJ, Huang HC: Characterization of the hrpC and hrpRS operons of Pseudomonas syringae pathovars syringae, tomato, and glycinea and analysis of the ability of hrpF, hrpG, hrcC, hrpT, and hrpV mutants to elicit the hypersensitive response and disease in plants. J Bacteriol. 1998, 180 (17): 4523-4531.
Ramos AR, Morello JE, Ravindran S, Deng WL, Huang HC, Collmer A: Identification of Pseudomonas syringae pv. syringae 61 type III secretion system Hrp proteins that can travel the type III pathway and contribute to the translocation of effector proteins into plant cells. J Bacteriol. 2007, 189 (15): 5773-5778. 10.1128/JB.00435-07.
Knodler LA, Vallance BA, Hensel M, Jackel D, Finlay BB, Steele-Mortimer O: Salmonella type III effectors PipB and PipB2 are targeted to detergent-resistant microdomains on internal host cell membranes. Mol Microbiol. 2003, 49 (3): 685-704. 10.1046/j.1365-2958.2003.03598.x.
Jelenska J, Yao N, Vinatzer BA, Wright CM, Brodsky JL, Greenberg JT: A J domain virulence effector of Pseudomonas syringae remodels host chloroplasts and suppresses defenses. Curr Biol. 2007, 17 (6): 499-508. 10.1016/j.cub.2007.02.028.
Vinatzer BA, Jelenska J, Greenberg JT: Bioinformatics correctly identifies many type III secretion substrates in the plant pathogen Pseudomonas syringae and the biocontrol isolate P. fluorescens SBW25. Mol Plant Microbe Interact. 2005, 18 (8): 877-888. 10.1094/MPMI-18-0877.
Chang JH, Urbach JM, Law TF, Arnold LW, Hu A, Gombar S, Grant SR, Ausubel FM, Dangl JL: A high-throughput, near-saturating screen for type III effector genes from Pseudomonas syringae. Proc Natl Acad Sci USA. 2005, 102 (7): 2549-2554. 10.1073/pnas.0409660102.
Ferreira AO, Myers CR, Gordon JS, Martin GB, Vencato M, Collmer A, Wehling MD, Alfano JR, Moreno-Hagelsieb G, Lamboy WF, DeClerck G, Schneider DJ, Cartinhour SW: Whole-genome expression profiling defines the HrpL regulon of Pseudomonas syringae pv. tomato DC3000, allows de novo reconstruction of the Hrp cis clement, and identifies novel coregulated genes. Mol Plant Microbe Interact. 2006, 19 (11): 1167-1179. 10.1094/MPMI-19-1167.
Fouts DE, Abramovitch RB, Alfano JR, Baldo AM, Buell CR, Cartinhour S, Chatterjee AK, D'Ascenzo M, Gwinn ML, Lazarowitz SG, Lin NC, Martin GB, Rehm AH, Schneider DJ, van Dijk K, Tang X, Collmer A: Genomewide identification of Pseudomonas syringae pv. tomato DC3000 promoters controlled by the HrpL alternative sigma factor. Proc Natl Acad Sci USA. 2002, 99 (4): 2275-2280. 10.1073/pnas.032514099.
Innes RW, Bent AF, Kunkel BN, Bisgrove SR, Staskawicz BJ: Molecular analysis of avirulence gene avrRpt2 and identification of a putative regulatory sequence common to all known Pseudomonas syringae avirulence genes. J Bacteriol. 1993, 175 (15): 4859-4869.
Schechter LM, Vencato M, Jordan KL, Schneider SE, Schneider DJ, Collmer A: Multiple approaches to a complete inventory of Pseudomonas syringae pv. tomato DC3000 type III secretion system effector proteins. Mol Plant Microbe Interact. 2006, 19 (11): 1180-1192. 10.1094/MPMI-19-1180.
Guttman DS, Vinatzer BA, Sarkar SF, Ranall MV, Kettler G, Greenberg JT: A functional screen for the type III (Hrp) secretome of the plant pathogen Pseudomonas syringae. Science. 2002, 295 (5560): 1722-1726. 10.1126/science.295.5560.1722.
Petnicki-Ocwieja T, Schneider DJ, Tam VC, Chancey ST, Shan L, Jamir Y, Schechter LM, Janes MD, Buell CR, Tang X, Collmer A, Alfano JR: Genomewide identification of proteins secreted by the Hrp type III protein secretion system of Pseudomonas syringae pv. tomato DC3000. Proc Natl Acad Sci USA. 2002, 99 (11): 7652-7657. 10.1073/pnas.112183899.
Nissan G, Manulis S, Weinthal DM, Sessa G, Barash I: Analysis of promoters recognized by HrpL, an alternative sigma-factor protein from Pantoea agglomerans pv. gypsophilae. Mol Plant Microbe Interact. 2005, 18 (7): 634-643. 10.1094/MPMI-18-0634.
Harley CB, Reynolds RP: Analysis of E. coli promoter sequences. Nucleic Acids Res. 1987, 15 (5): 2343-2361. 10.1093/nar/15.5.2343.
Potvin E, Sanschagrin F, Levesque RC: Sigma factors in Pseudomonas aeruginosa. FEMS Microbiol Rev. 2008, 32 (1): 38-55. 10.1111/j.1574-6976.2007.00092.x.
Feltman H, Schulert G, Khan S, Jain M, Peterson L, Hauser AR: Prevalence of type III secretion genes in clinical and environmental isolates of Pseudomonas aeruginosa. Microbiology. 2001, 147 (Pt 10): 2659-2669.
Wolfgang MC, Kulasekara BR, Liang X, Boyd D, Wu K, Yang Q, Miyada CG, Lory S: Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 2003, 100 (14): 8484-8489. 10.1073/pnas.0832438100.
Bingle LE, Bailey CM, Pallen MJ: Type VI secretion: a beginner's guide. Curr Opin Microbiol. 2008, 11 (1): 3-8. 10.1016/j.mib.2008.01.006.
Shrivastava S, Mande SS: Identification and functional characterization of gene components of Type VI Secretion system in bacterial genomes. PLoS One. 2008, 3 (8): e2955-10.1371/journal.pone.0002955.
Mougous JD, Cuff ME, Raunser S, Shen A, Zhou M, Gifford CA, Goodman AL, Joachimiak G, Ordonez CL, Lory S, Walz T, Joachimiak A, Mekalanos JJ: A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science. 2006, 312 (5779): 1526-1530. 10.1126/science.1128393.
Hsu F, Schwarz S, Mougous JD: TagR promotes PpkA-catalysed type VI secretion activation in Pseudomonas aeruginosa. Mol Microbiol. 2009, 72 (5): 1111-1125. 10.1111/j.1365-2958.2009.06701.x.
Pukatzki S, Ma AT, Revel AT, Sturtevant D, Mekalanos JJ: Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc Natl Acad Sci USA. 2007, 104 (39): 15508-15513. 10.1073/pnas.0706532104.
Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ: Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci USA. 2006, 103 (5): 1528-1533. 10.1073/pnas.0510322103.
Wu HY, Chung PC, Shih HW, Wen SR, Lai EM: Secretome analysis uncovers an Hcp-family protein secreted via a type VI secretion system in Agrobacterium tumefaciens. J Bacteriol. 2008, 190 (8): 2841-2850. 10.1128/JB.01775-07.
Hood RD, Singh P, Hsu F, Guvener T, Carl MA, Trinidad RR, Silverman JM, Ohlson BB, Hicks KG, Plemel RL, Li M, Schwarz S, Wang WY, Merz AJ, Goodlett DR, Mougous JD: A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe. 2010, 7 (1): 25-37. 10.1016/j.chom.2009.12.007.
Matthysse AG, Stretton S, Dandie C, McClure NC, Goodman AE: Construction of GFP vectors for use in Gram-negative bacteria other than Escherichia coli. FEMS Microbiol Lett. 1996, 145 (1): 87-94. 10.1111/j.1574-6968.1996.tb08561.x.
Milne I, Bayer M, Cardle L, Shaw P, Stephen G, Wright F, Marshall D: Tablet--next generation sequence assembly visualization. Bioinformatics. 2010, 26 (3): 401-402. 10.1093/bioinformatics/btp666.
Giovannoni SJ, Hayakawa DH, Tripp HJ, Stingl U, Givan SA, Cho JC, Oh HM, Kitner JB, Vergin KL, Rappe MS: The small genome of an abundant coastal ocean methylotroph. Environ Microbiol. 2008, 10 (7): 1771-1782. 10.1111/j.1462-2920.2008.01598.x.
Delcher AL, Harmon D, Kasif S, White O, Salzberg SL: Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27 (23): 4636-4641. 10.1093/nar/27.23.4636.
Fahlgren N, Sullivan CM, Kasschau KD, Chapman EJ, Cumbie JS, Montgomery TA, Gilbert SD, Dasenko M, Backman TW, Givan SA, Carrington JC: Computational and analytical framework for small RNA profiling by high-throughput sequencing. Rna. 2009, 15 (5): 992-1002. 10.1261/rna.1473809.
R Development Core Team: R: A language and environment for statistical computing. 2007, R Foundation for Statistical Computing. Vienna, Austria, ISBN 3-900051-07-0, [http://www.R-project.org]
Zmasek CM, Eddy SR: ATV: display and manipulation of annotated phylogenetic trees. Bioinformatics. 2001, 17 (4): 383-384. 10.1093/bioinformatics/17.4.383.
Carver T, Thomson N, Bleasby A, Berriman M, Parkhill J: DNAPlotter: circular and linear interactive genome visualization. Bioinformatics. 2009, 25 (1): 119-120. 10.1093/bioinformatics/btn578.
Alfano JR, Charkowski AO, Deng WL, Badel JL, Petnicki-Ocwieja T, van Dijk K, Collmer A: The Pseudomonas syringae Hrp pathogenicity island has a tripartite mosaic structure composed of a cluster of type III secretion genes bounded by exchangeable effector and conserved effector loci that contribute to parasitic fitness and pathogenicity in plants. Proc Natl Acad Sci USA. 2000, 97 (9): 4856-4861. 10.1073/pnas.97.9.4856.
Chen C, Beattie GA: Characterization of the osmoprotectant transporter OpuC from Pseudomonas syringae and demonstration that cystathionine-beta-synthase domains are required for its osmoregulatory function. J Bacteriol. 2007, 189 (19): 6901-6912. 10.1128/JB.00763-07.
Chen C, Beattie GA: Pseudomonas syringae BetT is a low-affinity choline transporter that is responsible for superior osmoprotection by choline over glycine betaine. J Bacteriol. 2008, 190 (8): 2717-2725. 10.1128/JB.01585-07.
Tan Y, Donovan WP: Deletion of aprA and nprA genes for alkaline protease A and neutral protease A from Bacillus thuringiensis: effect on insecticidal crystal proteins. J Biotechnol. 2001, 84 (1): 67-72. 10.1016/S0168-1656(00)00328-X.
Bowen D, Rocheleau TA, Blackburn M, Andreev O, Golubeva E, Bhartia R, ffrench-Constant RH: Insecticidal toxins from the bacterium Photorhabdus luminescens. Science. 1998, 280 (5372): 2129-2132. 10.1126/science.280.5372.2129.
Lee JS, Heo YJ, Lee JK, Cho YH: KatA, the major catalase, is critical for osmoprotection and virulence in Pseudomonas aeruginosa PA14. Infect Immun. 2005, 73 (7): 4399-4403. 10.1128/IAI.73.7.4399-4403.2005.
Yu J, Penaloza-Vazquez A, Chakrabarty AM, Bender CL: Involvement of the exopolysaccharide alginate in the virulence and epiphytic fitness of Pseudomonas syringae pv. syringae. Mol Microbiol. 1999, 33 (4): 712-720. 10.1046/j.1365-2958.1999.01516.x.
Hettwer U, Jaeckel FR, Boch J, Meyer M, Rudolph K, Ullrich MS: Cloning, nucleotide sequence, and expression in Escherichia coli of levansucrase genes from the plant pathogens Pseudomonas syringae pv. glycinea and P. syringae pv. phaseolicola. Appl Environ Microbiol. 1998, 64 (9): 3180-3187.
Koczan JM, McGrath MJ, Zhao Y, Sundin GW: Contribution of Erwinia amylovora exopolysaccharides amylovoran and levan to biofilm formation: implications in pathogenicity. Phytopathology. 2009, 99 (11): 1237-1244. 10.1094/PHYTO-99-11-1237.
Ellison DW, Miller VL: Regulation of virulence by members of the MarR/SlyA family. Curr Opin Microbiol. 2006, 9 (2): 153-159. 10.1016/j.mib.2006.02.003.
Johnson TL, Abendroth J, Hol WG, Sandkvist M: Type II secretion: from structure to function. FEMS Microbiol Lett. 2006, 255 (2): 175-186. 10.1111/j.1574-6968.2006.00102.x.
Meyers DJ, Berk RS: Characterization of phospholipase C from Pseudomonas aeruginosa as a potent inflammatory agent. Infect Immun. 1990, 58 (3): 659-666.
Bronstein PA, Marrichi M, Cartinhour S, Schneider DJ, DeLisa MP: Identification of a twin-arginine translocation system in Pseudomonas syringae pv. tomato DC3000 and its contribution to pathogenicity and fitness. J Bacteriol. 2005, 187 (24): 8450-8461. 10.1128/JB.187.24.8450-8461.2005.
Caldelari I, Mann S, Crooks C, Palmer T: The Tat pathway of the plant pathogen Pseudomonas syringae is required for optimal virulence. Mol Plant Microbe Interact. 2006, 19 (2): 200-212. 10.1094/MPMI-19-0200.
Arrebola E, Cazorla FM, Codina JC, Gutierrez-Barranquero JA, Perez-Garcia A, de Vicente A: Contribution of mangotoxin to the virulence and epiphytic fitness of Pseudomonas syringae pv. syringae. Int Microbiol. 2009, 12 (2): 87-95.
Acknowledgements
We thank Mark Dasenko and Chris Sullivan of the Center for Genome Research and Biocomputing (CGRB) for Illumina sequencing and computational support, as well as Don Chen and Philip Hillebrand for their assistance. We thank Dr. Joey Spatafora for providing us HAL before publication, Dr. Joyce Loper and Jason Cumbie for their valuable advice. Finally, we thank two anonymous reviewers for their helpful comments in improving this manuscript. This research was supported in part by General Research Funds to JHC and the National Research Initiative Competitive Grant no. 2008-35600-18783 from the USDA's National Institute of Food and Agriculture, Microbial Functional Genomics Program to JHC, and by grants from the USDA CSREES Grass Seed Cropping Systems for Sustainable Agriculture Special Grant Program and from the OSU Agricultural Research Foundation to DJA and DIM.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
JAK prepared the DNA for sequencing, assembled and analyzed the genome sequence of WH6, as well as drafted the manuscript. SAG annotated the genome. ABH sequenced the DNA flanking the Tn5 insertions, did the preliminary analyses of gene functions, and helped draft the manuscript. ALC assisted with analyzing the different assemblies. DIM, GMB, DJA, and JHC conceived of the study and drafted the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kimbrel, J.A., Givan, S.A., Halgren, A.B. et al. An improved, high-quality draft genome sequence of the Germination-Arrest Factor-producing Pseudomonas fluorescens WH6. BMC Genomics 11, 522 (2010). https://doi.org/10.1186/1471-2164-11-522
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-11-522