
Abstract
Background
In today's age of genomic discovery, no attempt has been made to comprehensively sequence a gymnosperm genome. The largest genus in the coniferous family Pinaceae is Pinus, whose 110-120 species have extremely large genomes (c. 20-40 Gb, 2N = 24). The size and complexity of these genomes have prompted much speculation as to the feasibility of completing a conifer genome sequence. Conifer genomes are reputed to be highly repetitive, but there is little information available on the nature and identity of repetitive units in gymnosperms. The pines have extensive genetic resources, with approximately 329000 ESTs from eleven species and genetic maps in eight species, including a dense genetic map of the twelve linkage groups in Pinus taeda.
Results
We present here the Sanger sequence and annotation of ten P. taeda BAC clones and Genome Analyzer II whole genome shotgun (WGS) sequences representing 7.5% of the genome. Computational annotation of ten BACs predicts three putative protein-coding genes and at least fifteen likely pseudogenes in nearly one megabase of sequence. We found three conifer-specific LTR retroelements in the BACs, and tentatively identified at least 15 others based on evidence from the distantly related angiosperms. Alignment of WGS sequences to the BACs indicates that 80% of BAC sequences have similar copies (≥ 75% nucleotide identity) elsewhere in the genome, but only 23% have identical copies (99% identity). The three most common repetitive elements in the genome were identified and, when combined, represent less than 5% of the genome.
Conclusions
This study indicates that the majority of repeats in the P. taeda genome are 'novel' and will therefore require additional BAC or genomic sequencing for accurate characterization. The pine genome contains a very large number of diverged and probably defunct repetitive elements. This study also provides new evidence that sequencing a pine genome using a WGS approach is a feasible goal.
Similar content being viewed by others


Background
Ten years after the first plant genome sequence was completed [1], dozens more have been sequenced but to date no effort has been made to sequence the genome of a gymnosperm species. With 110-120 species spread throughout the Northern Hemisphere, the pines (Pinus) comprise the largest genus in the coniferous family Pinaceae of the gymnosperms. Known for their longevity and important ecological roles, pines have also undergone 200-300 million years of evolution separate from their distant angiosperm relatives [2]. A pine genome reference sequence would fill a great evolutionary gap, but it has long been questioned whether such an endeavor was even feasible.
Pine genomes are extremely large (c. 20-40 Gb) [3–6]. These genomes, however, show no evidence of recent polyploidy or chromosome duplication [7–9]. Pine chromosomes (2N = 24) are uniform in both number (2N = 24) and appearance, for they lack major distinguishing physical features [10]. They are also so large and dense that standard karyotyping probes such as rDNA, GC-rich and telomeric repeat sequences failed to produce differential banding patterns among the chromosomes and have complicated karyotyping efforts [11–18]. Pine chromosomes have Arabidopsis-type (A-type) telomere repeat sequences (TRS) at their terminal telomeres, as well as substantial centromeric and interstitial sites [19]. A reference karyotype and cytogenetic map was recently produced for Pinus taeda L. with improved chromosome spreading techniques and staining probes for two types of rDNA, the A-type TRS and centromeric AT-rich regions [20]. A leading commercial timber species native to the southeast United States, P. taeda is among the best-characterized pine genomes. There are currently a total of 328628 P. taeda expressed sequence tags (ESTs) in NCBI databases, the results of at least five major sequencing projects in root, needle, lignifying and embryonic tissues under varying conditions. These EST sequences were subsequently clustered into 18921 P. taeda Unigenes [21]. The current genetic map includes 373 markers across twelve linkage groups [22]. Thus, P. taeda is ideal for additional genomic exploration among pines, conifers and gymnosperms.
Several studies report on the complexity of the pine genome. A reassociation study estimated that the Pinus strobus L. genome contained 22-26% single-copy elements [23]. When the reassociation calculations were performed with a more accurate genome size estimate, single-copy sequences were estimated to occupy 14% of the genome, or 3100 Mb [3]. This very large single-copy fraction could be due to the presence of large complex gene families in pines, as was evidenced by southern hybridizations performed on P. taeda using single-to low-copy gene probes from angiosperms [24, 25]. Additionally, the single-copy fraction of the pine genome is enriched for repeats, as it was later shown that at least one fifth of low-copy sequences in P. taeda are retroelements and one third contain microsatellite repeats [26, 27].
Genomic exploration in conifers has not been limited to pines. Picea (spruces) is the most closely related genus to Pinus and contains 30-40 species with genome sizes similar to pines. Recent assembly and analysis of four bacterial artificial chromosome (BAC) sequences in Picea glauca (Moench) Voss revealed that only one targeted gene was present in each BAC assembly, despite averaging nearly 150 Kb in length [28, 29]. Assessment of the surrounding noncoding regions for similarity to repeat database elements revealed that high-complexity repeats comprise 22% and 18% of the two BAC assemblies, where authors noted a prevalence of retroelement-based elements in the results [28]. Repetitive content is thought to be similar in spruces and pines, but there are currently no comparable BAC resources for pine. While a BAC library has been reported for Pinus pinaster Ait., no BAC-length sequences have been published for that species [30].
Presented here is the first large-scale sequence survey of a pine genome. This study produced the annotated sequences of ten P. taeda BACs using standard Sanger sequencing and assembly methods, as well as 1.66 gigabases of the genome in whole genome shotgun (WGS) sequences from the Genome Analyzer II platform. The linear organization of coding and repetitive elements in ten contiguous genomic sequences is presented visually through computational annotation, similarity to repeat database elements, and several additional innovative repeat analyses. By aligning the WGS reads to the BAC sequences, variation in whole-genome coverage based on alignment stringency is shown. The WGS sequences produced evidence that the three most common repetitive elements in the pine genome together constitute less than 5% of the sequence, and that there appears to be a large number of previously unknown repeat families. While the pine genome is largely repetitive, most of the repeats are highly diverged from one another. Therefore, the main barrier to assembling the nuclear pine genome is not the content of the genome, but the cost associated with its large size.
Results
Sequencing and Assembly
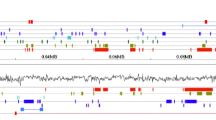
Ten P. taeda BAC clones were sequenced to an average depth of 10× coverage and assembled into contigs (Table 1). For ease of presentation, each BAC clone will be referenced by its lab designation, given in the leftmost column of Table 1. Among the sequenced clones, coverage ranged from 6× (for the longest clone, BAC3) to over 16× (BAC19). Nine assemblies were resolved into one scaffold that contains linkers and spacers as necessary, while BAC31 assembled into two unoriented contigs. The BAC sequences were deposited in Genbank (Accession nos. GU477256-GU477266). Three sets of whole genome shotgun sequencing were performed with read lengths of 40, 42, and 60 bp. These sets of WGS reads were deposited in the Short Read Archive and assigned accessions SRX017253, SRX017254 and SRX017255, respectively. The GC content of the BACs is similar to the shotgun reads, and the GC content of the P. taeda genome falls within typical ranges of angiosperm species (Table 1). The first BAC sequenced, BAC12, is visualized in Figure 1. All ten BACs, their predicted genes and all repeat analyses are shown in Additional file 1, Figure S1 or at http://dendrome.ucdavis.edu/treegenes/gbrowse.

Pinus taeda BAC12 (clone Pt314B2) illustrates several new trends found in the pine genome. (A) The length of BAC12 is shown along the horizontal axis. Shown above the axis are tracks of annotated genes (dicot and monocot parameters), similarity hits to Repbase [RM (blastx); DNA transposons; Non-LTR retroelements; ERV (endogenous retroviruses); LTR retroelements, copia; LTR retroelements, gypsy], and other elements identified in this study (simple repeats, tandem repeats, ORF elements, pairs of direct repeats, and regions of similarity among BACs). The bottom two tracks indicate WGS coverage at ≥ 75% identity and at ≥ 99% identity (B) Genes were annotated with both dicot and monocot parameters. The annotations generally differed in gene structure. (C) Coverage is similar between coverage tracks for active and relatively abundant retroelements in the pine genome such as this nested PtIFG7.
Gene Content
The BAC library was probed for sequences similar to ten P. taeda genes known to be involved in the phenylpropanoid pathway and lignin biosynthesis (see Methods for probes). The probes were derived from EST contigs amplified from genomic DNA and either differ significantly from the published mRNA or lack corresponding mRNA sequence altogether. The BACs contained thirteen regions with similarity to six probes; cinnamyl-alcohol dehydrogenase (cad), caffeoyl-CoA O-methyltransferase (ccoaomt), caffeate O-methyltransferase (comt), LIM transcription factor 1(ptlim1), S-adenosyl methionine synthetase 1 (sam1) and S-adenosyl methionine synthetase 2 (sam2) (Additional file 2, Table S1). The regions of similarity with these genes averaged 211 bp with 87% nucleotide identity. Four probes did not significantly match any BAC sequence. Based on 100% nucleotide identity with its probe, it appears that a novel ccoaomt may have been captured in BAC19. The other nine BACs contained regions with partial similarity to the probes. In order to determine the coding potential of these regions and to discover untargeted genes in the BACs, the MAKER automated annotation software was run with both dicot and monocot parameters (Table 2) [31]. The runs indicate that approximately 1% of the sequence in these BAC clones may be protein-coding. Eight of the ten BACs contain predicted genes, while BAC3 and BAC37 contain no predicted genes. The two runs produced similar results, with dicot and monocot genes occupying the same or overlapping regions, but exon-intron structure generally differed between the two runs (Figure 1B; Additional file 3, Table S2). On average, the dicot-like genes occupy less genomic space but produce longer mRNAs and peptides than the monocot-derived genes. The dicot-like genes contained an average of 2.9 exons (average 219 bp) and the monocot-like genes showed an average of 2.8 exons (average 187 bp). Both runs predicted four small introns per gene, with average lengths of 350 and 842 bp, respectively. The dicot run predicted a total of 18 genes, two of which lack consensus start or stop codons (Additional file 2, Table S1). The monocot run also predicted 18 genes but seven genes lack consensus start or stop codons, making them gene fragments. All gene predictions employ consensus splice signals at exon-intron junctions. Fourteen of the 18 total predicted genes were annotated against protein or nucleotide genes sequences from organisms other than P. taeda. The results of the MAKER runs, including gene annotations and supporting evidence, can be viewed in Additional file1, Figure S1.
Of the 18 predicted genes, twelve have similarities to Interpro domains [32] with an E-value < 1e-05 (Additional file 4, Table S3). The putative ccoaomt gene on BAC19 is only one of several genes that, if they function as predicted, could be homologues to genes involved in lignin biosynthesis. Two additional ccoaomt genes (BAC20, BAC31), three comt genes (BAC12, BAC17, BAC40), a LIM transcription factor (BAC15), and one sam1 (BAC21) are predicted in regions of similarity to their probes (Additional file 1, Figure S1). The set of predicted genes also includes untargeted genes such as a member of the 4cl family (BAC12), a glycosyl transferase gene (BAC15), a SMARCA3 helicase (BAC21), three kinases (BAC40) and a reverse transcriptase that likely belongs to a transposon (BAC17). Three predicted genes do not show any similarity to Interpro (BAC40). Analysis of upstream promoter regions shows that while most of these twelve genes do contain promoter elements, many of the predicted genes lack appropriately located TATA or CAAT boxes that are generally required for transcription (Additional file 5, Table S4). Based on the presence of consensus start and stop codons, significant Interpro hits, and > 97% nucleotide identity with P. taeda ESTs, only three methyltransferases (comt in BAC12, ccoaomt in BAC19 and ccoaomt on BAC20) may be novel protein-coding genes. The other 15 predicted genes are most likely inactive pseudogenes.
Repeat Content
As can be seen in Additional Figure 1, the noncoding component of the ten pine BACs is composed of highly diverse repetitive elements. The MAKER output reports that BACs showed similarity to nearly 600 complex Repbase elements in less than one megabase of pine genomic sequence [33] (Table 2; Additional file 1, Figure S1). These include simple sequence repeats such as GC-and AT-rich areas and complex repeats such as LTR and non-LTR retrotransposons, DNA transposons (including hAT, MuDR and Helitrons), endogenous retroviruses (ERV), and other repetitive elements as defined by Repbase. The sources of the accessions with similarity to the pine BACs vary widely, including five gymnosperm species, 15 dicot and eight monocot species, 11 animals, three fungal species, a moss and one multicellular alga. Only 4% of all hits to complex Repbase repeats are to gymnosperm-derived repetitive elements, reflecting the relative lack of genomic resources for this clade.
Conifer-specific LTR retroelements
Similarity to gymnosperm accessions was used to identify conifer-specific LTR retroelements in the BACs. The IFG7 gypsy-like retroelement in Pinus radiata D. Don [Genbank: AJ004945] and PpRT1 in P. pinaster Aiton [Genbank: DQ394069] are known to be pervasive in the pine family [34, 35], and four P. taeda BACs show strong similarity (80-93% identity over an average 3900 bp) to IFG7 (Figure 1C). These four IFG7-like sequences are over 90% similar to each other, so they represent four partial copies of a novel P. taeda equivalent to IFG7 that is designated PtIFG7 (Table 2; Additional file 1, Figure S1). The high level of sequence conservation suggests that the retroelement may be relatively young and still active. The fact that PtIFG7 is actively transcribed is further supported by the fact that WGS coverage of the PtIFG7 elements is similar at 75% and 99% identity (Figure 1B) and, ultimately, that the sequence shows 100% nucleotide identity with at least seven P. taeda ESTs. Note that while PtIFG7 is present in four BACs, they are each interrupted by other elements or truncated by the end of the BAC assembly (Table 2; Additional file 1, Figure S1).
In BAC21, a nearly full-length match to PtIFG7 shows only 66-73% nucleotide identity. The region is flanked by 94% similar direct repeats and also contains 89 bp similar to the Gypsy8 element in Vitis vinifera L. Neither the LTRs nor VvGypsy8 are similar to PtIFG7. This novel relative of PtIFG7 is tentatively described as PtIFG7-2. As can be seen in Additional file 1, Figure S1, the LTRs of the putative PtIFG7-2 in BAC21 are tightly flanked by large (1100-1750 bp) similarity hits to copia-like elements from Medicago truncatula Gaertn., Populus trichocarpa Torr. & Gray and Oryza sativa L., so the PtIFG7-2 appears to be nested inside a larger uncharacterized copia-like element. The PtIFG7-2 element shows 100% nucleotide identity to a single EST, so it is possible that PtIFG7-2 is also actively transcribed in the genome. A second relative of the PtIFG7 element can be found in BAC37. Instead of one long similarity hit, however, this new PtIFG7-3 element is fragmented into five pieces and contains a number of unrelated repetitive elements and similarity hits. The pieces of the PtIFG7-3 element average 66% nucleotide similarity to PtIFG7 and 75% similarity to PtIFG7-2. Since the similarity seen here is below 80%, we consider PtIFG7-3 a distinct but related element to PtIFG7 and PtIFG7-2, although we note that classification of these elements into classes and families is confused by the level of divergence among them. One similarity hit each to LTR retroelements in Pinus elliottii Engelm. and P. thunbergii Parl. average only 230 bp in length and were uninformative.
Two additional novel LTR retroelements were identified based on similarity to Picea elements (Additional file 1, Figure S1). BAC37 contains a 2300-bp region showing 84-87% identity with the PGGYPSYX1 (Spcl) retroelement in Picea glauca [Genbank: AF229252] [36]. The region is flanked by 90% identical LTRs, so this new gypsy-like element is tentatively described as PtGypsyX1. The PtGypsyX1 retroelement is also present twice in BAC3, and the three copies described here are 87-89% similar at the nucleotide level. A strong hit in BAC3 to the PGCOPIAX1 (Spdl) element from Picea glauca [Genbank: AF229251] is situated between direct repeats located about 10 Kb apart, a space shared with hits to copia-like elements in M. truncatula and V. vinifera. It remains unclear whether this LTR-flanked portion of BAC3 represents a single intact element, but the portion showing 79% nucleotide identity with the spruce element is tentatively identified as a fragment of the newly described PtCopiaX1 element. The PtGypsyX1 and PtCopiaX1 have different WGS coverage profiles between the thresholds (deep coverage at 75% identity, low coverage at 99%), suggesting that they are older than the PtIFG7 family. However, the PtGypsyX1 near position 60 Kb in BAC3 shows 100% nucleotide identity with six full-length P. taeda ESTs, so this element appears to be actively transcribing and proliferating in the genome. The PtCopiaX1 element in BAC3 has a relatively high WGS coverage in the LTRs and the portion corresponding to the assembled TPE1-containing element, but an internal portion (128-134 Kb) has a distinctly lower coverage than the PtCopiaX1 in which it is nested. This copy of the PtCopiaX1 element does not show similarity to any P. taeda ESTs, but the unrelated internal region (which happens to correspond with a putative ORF element) shows 100% nucleotide identity with two P. taeda ESTs. This particular copy of PtCopiaX1 is dead, and in this case appears to contain an active transcribed repetitive element of unknown origin.
Angiosperm-derived LTR retroelements
Sequences similar to Repbase or RepeatMasker database repeats contribute 23% to the total BAC assemblies, and contributions among BACs range from 19% in BAC40 to 33% in BAC31 (Table 2). If 1% of the BACs are considered coding (Table 2; Additional file 3, Table S2), this leaves approximately 75% of the BAC sequence uncategorized. To determine if the uncategorized sequence was single-copy or repetitive, WGS reads were aligned to the BACs and the alignment depth at each position was observed. The total coverage represented by the WGS was 0.036x of the genome, so most single-copy regions should appear unaligned. Repetitive regions, however, are expected to have multiple aligned reads. Alignments were categorized as either ≥ 99% identical or ≥ 75% identical. At the 99% threshold, most (77%) of the BAC sequence can be considered single-copy. There are a few regions, such as the newly characterized PtIFG7, which are repetitive at this stringent threshold (Figure 1C). At the 75% threshold, most (80%) of the BAC sequence is repetitive. Based on WGS coverage of the PtGypsyX1 elements in BAC3 and BAC37, it is estimated that the genome contains 65000 to 72000 copies with ≥ 75% similarity and 600 to 2000 copies with ≥ 99% similarity to the copies in the BACs. Using the PtCopiaX1 in BAC37, the copy number of this element is estimated to be around 84000 at the 75% threshold and just over 1000 at the 99% threshold. There also appear to be several new families of repeats as indicated by similarity to non-gymnosperms, with coverage profiles that appear to confirm and delineate repetitive units (indicated by red or green boxes in Additional file 1, Figure S1).
The average length of similarity between pine BAC sequence and non-gymnosperm Repbase accessions is 155 bp, far shorter than full-length transposons, and most hits were not suggestive of full-length repetitive elements in pine. The MAKER output also included 62 significant blastx hits to RepeatMasker coding sequences, averaging 753 bp of coding similarity to reverse transcriptases or polyproteins in angiosperm retroelements. Complicating novel retroelement identification is the fact that a single gymnosperm element often shows similarity to numerous angiosperm repeat accessions. Using the Repbase and RepeatMasker similarity hits coupled with the WGS coverage profiles, 15 novel partial or full-length pine LTR retroelement sequences were tentatively identified based on similarity to non-gymnosperm accessions (Additional file 1, Figure S1). Seven of these are gypsy-like elements and eight appear to be copia-like LTR retrotransposons. Informative Repbase accessions originate from Glycine max (L.) Merr., Populus trichocarpa, Oryza sativa, Vitis vinifera, Zea mays L., Cicer arietinum L. Lotus corniculatus L., and the novel pine retroelement sequences are indicated in Additional file 1, Figure S1. The 15 novel angiosperm-derived LTR retroelements tentatively identified in this study are also older and less frequent in the genome than PtIFG7, as evidenced by lower WGS coverage profiles and the presence of unrelated repetitive elements between the LTRs.
Direct, tandem and simple repeats
At least 161 direct repeats were found among the BACs, with individual BACs ranging from four pairs (BAC19) to 40 pairs (BAC3). As discussed above and illustrated in Additional file 1, Figure S1, about 30 pairs of direct repeats belong to identifiable LTR retroelements, and this is almost certainly an underestimate. Many of the direct repeats appear to simply be repeated sequences. As a case in point, the PtIFG7 element in BAC12 contains a 142-bp sequence that is also found about 16 Kb upstream of the PtIFG7 element (Figure 1). This small repeated sequence does not show similarity to any known sequence, and it is unclear how one copy was inserted into the PtIFG7 retroelement. The average nucleotide identity between direct repeats was 86% and ranged from 53% to 99%.
Tandem repeats, or minisatellites, have not to date been investigated in a gymnosperm genome. Tandem Repeat Finder identified 214 repeats of units 5-200 bp in length in the ten P. taeda BACs (Figure 1; Additional file 1, Figure S1) [37]. BAC12 shows a large defined peak in WGS coverage at the 50-bp tandem near the middle of the BAC (Figure 1). The common occurrence of tandem repeats in the BACs and the peak in WGS coverage profiles of the BAC12 tandem together offer evidence that tandem repeats may be dispersed throughout the genome and contribute to genome complexity. Simple repeats are difficult to quantify based on WGS coverage, but over 150 were identified among the ten P. taeda BACs (Table 2). Previous studies have shown that simple repeats and microsatellites are found throughout the pine genome [27, 38, 39]. Using the unmasked MAKER runs, there were also 70 putative ORF elements identified (Table 2). These are consistently predicted reading frames that are not near any putative protein-coding genes or pseudogenes in the BACs. As can be seen in Figure 1 and Additional file 1, Figure S1, the putative ORF elements typically fall within repetitive regions of the BACs as indicated by WGS coverage. They are not confirmed to belong to, or consist of, novel repetitive elements, but the suggestions are offered here as researchers begin exploring the evolutionary history of the conifer genome and identify more novel gymnosperm-specific families of repeats.
Genome-wide consensus repeats. To derive the full-length sequence of the most common repetitive elements in the genome, a highly permissive assembler was used to build the repeats from WGS reads (Table 3). Since no two reads are expected to truly overlap, the assembled elements are consensus sequences containing the most common base at each position. The most abundant element is 3896 bp long (with one LTR) and is 90% identical to the 1663-bp TPE1 [Genbank: Z50750], an internal copia-like sequence used to assay the occurrence of such retroelements in a variety of large gymnosperm genomes [40]. Alignments to WGS reads show that this element comprises approximately 1.6% of the P. taeda genome. The assembled TPE1-containing element is present in BAC3 in four fragments (> 90% identity to consensus) within the PtCopiaX1 element located there (Additional file 1, Figure S1). Although the PtCopiaX1 BAC3 was determined to be interrupted and inactive, the TPE1-containing consensus shows > 97% full-length nucleotide identity with at least five P. taeda ESTs. The second most common element in the genome is PtIFG7, of a similar size and abundance (3686 bp, 1.3% of the genome) to the TPE1-containing element. The third repeat assembled was a 50-nucleotide tandem repeat (cen-rpt), comprising 0.27% of the genome. This may correspond to the centromere repeat, and if so, represents the first example from a gymnosperm. The sequences of the three assembled elements are presented in Additional file 6, Table S5. A separate assay of the WGS reads found that 0.24% contains the A-type telomeric repeat (tel-rpt: TTTAGGG). This suggests that this telomeric repeat accounts for roughly 50-55 megabases of the genome.
Discussion
The purpose of this study was to explore the content and organization of a conifer genome and assess the feasibility of sequencing and assembling a reference pine genome. In the course of the study, several interesting aspects of the pine genome have emerged. Analyses of the structure and content of ten P. taeda BACs suggest that pseudogenes may be common in the pine genome and that isolated repetitive elements such as LTR retrotransposons can be discerned from a background of fragmented fossil repeats of unknown origin. The frequency of partially conserved coding regions in the genome is consistent with the numerous hybridizations observed to probes for single-copy genes [25]. In these ten BACs, apparent pseudogenes appear to occur five times more frequently than true potentially functional protein-coding genes, but the BACs were enriched for coding sequences and represent only 0.0042% of the genome. Whether this ratio extends to the entire genome remains to be seen. The common occurrence of pseudogenes in these BACs is consistent with the two conclusions from other studies: (1) most pine genes have many paralogues or pseudogenes and (2) pine genes are relatively compact. The accuracy of these and any other computational gene predictions in pine are limited by our incomplete knowledge of codon or promoter usage in pines. Gene identification in this complex genome must be achieved through deep transcriptome or genome sequencing, as well as experimental validation of expression. It is, however, critical to note the pine genes do appear to be quite compact relative to the dauntingly vast genome size (Additional file 3, Table S2). Thus, intermediate sequencing strategies that can leverage a deep transcriptome may efficiently assemble the primary functional genomic domains. Genetic localization of such gene islands can serve both as an experimentally valuable (if sparse) scaffold and a solid foundation for completion of a reference genome sequence.
Previous studies showed that the pine genome is highly repetitive, but not predominantly high C0T [41]. The current study indicates that the reason for this is that there are many diverged repeats, and no single conserved repetitive element constitutes more than 2% of the genome. Depending on how one conducts a hybridization experiment, one could see extremely different results. Under stringent conditions (99% identical), only 24% of the genome is repetitive. Under more permissive conditions (75% identical), 80% of the genome is repetitive. The present sequence survey of the pine genome supports the findings of hybridization studies by suggesting a massive 'low-copy' fraction and a very small 'high-copy' fraction containing a few repeat families that occur fewer than 100000 times in the 22-Gb genome. At this point, little else is known about LTR retroelement families and other types of repeats in pine except that they appear to be numerous and highly diverged. The IFG7 family of elements is currently thought to exist only in Pinus, while PtCopiaX1 looks to be shared with Picea. While it is clear that a significant amount of work remains in order to truly determine the age of any LTR retroelements in pine, the fact that PtCopiaX1 is about 85% identical with PGCOPIAX1 suggests that this element may have been present in the common ancestor of the two genera. Considering that Pinus and Picea diverged approximately 140 million years ago [42], this copia-like element could be ancient. Alternatively, the retroviral progenitor of PtGypsyX1 and PGGYPSYX1 may have inserted multiple times during the evolution of conifers. In either case, two aspects of the LTR retroelements described in the BACs are fundamentally different from those found in other plant genomes: age and degree of divergence.
Analyses of LTR retrotransposons have been performed in a wide variety of plant genomes including eudicot, monocot and gymnosperm species. The abundance and organization of these repetitive elements are loosely correlated with clade: eudicots generally have fewer and smaller repetitive elements than other clades, while the monocots are known for their high LTR retroelement activity and rapidly changing genomes. Now, conifer genomes can be distinguished from angiosperm genomes by the old age and high degree of divergence in both their intact and fragmented LTR retrotransposons compared to the younger LTR retroelements in angiosperm genomes. A rapid increase of retroelement density in Oryza sativa occurred around eight million years ago, but unequal homologous recombination subsequently removed two-thirds of that LTR retroelement sequence and left a genome consisting of only 26% retrotransposons [43]. The Sorghum bicolor L. (Moench) genome, twice the size of Oryza sativa, is composed of 55% retrotransposons that have mostly inserted in the last two million years [44]. The Zea mays genome, three times as large as Sorghum bicolor, contains roughly 80% retroelements, most of which are not present in the orthologous Sorghum bicolor [45]. This implies that most of the LTR retroelements in the Zea mays genome inserted since its divergence from the Sorghum genome about 16 million years ago [46]. A survey of two linked Triticum aestivum L. BACs identified eleven LTR retroelements, all determined to have inserted less than 10-14 million years ago [47]. The majority of LTR retroelements observed in these studies of grass genomes are relatively young and clearly distinguishable. In contrast, only two conifer-specific LTR retroelements in the BACs (PtGypsyX1 and PtCopiaX1) may be still active after 140 million years, and the IFG7 family of gypsy-like elements appears to be active in both subgenera of Pinus [34], which diverged approximately 110 million years ago [2].
The bulk of the pine genome remains an enigma. It appears to contain diverged fragments of an extremely diverse set of repetitive elements. The analyses performed here virtually exhaust similarity-based identification so future repeat discovery will require additional genomic sequence. In any case the implications of the ancient and diverged nature of the repeats that comprise a majority of the pine genome are both theoretical and practical. The conservative karyotype and the lack of rapid turnover of the vast and repetitive portion of the pine genome raises questions about the possible functional roles leading to evolutionary constraint, and about potentially unique mechanisms for the maintenance of genomic integrity. In terms of the challenging goal of a reference pine genome sequence, this predominance of a highly diverged repetitive component is critical since large amounts of identical dispersed repeats are inevitable sources of gaps in an assembly.
Conclusions
In this detailed analysis of ten Pinus taeda BACs, we identified three putative protein-coding genes and at least fifteen pseudogenes or gene fragments. Examining the BACs in the context of 34.3 million WGS reads and 600 similarity hits to repeat databases, we found that 9.1% of the BACs had high WGS coverage or significant similarity to one of three positively identifiable conifer-specific LTR retroelements (PtIFG7, PtGypsyX1 and PtCopiaX1). An additional 12% of the BACs contain relicts of LTR retrotransposons that were tentatively identified as copia-like or gypsy-like based on similarity to angiosperm repeats. Simple repeats and imperfect tandem repeats together represent less than 4% of the total BAC sequence. Direct repeats larger than 100 bp occupy nearly 30% of the BAC sequence, but two thirds of pairs are less than 90% similar (none are identical) and only a minority are clearly associated with a retroelement. The majority of the BAC assemblies were comprised of ancient repetitive sequences. This is in sharp contrast to the Oryza sativa, Sorghum bicolor and Zea mays genomes, where recently amplified and minimally diverged LTR retrotransposons occupy much of the intergenic space. There is no evidence of recent segmental or block duplications within the BACs.
These preliminary insights into the nature of repeats in the pine genome provide compelling evidence that sequencing a large pine genome such as P. taeda is certainly within reach. Sequencing a genome this large using a rigorous BAC-by-BAC approach, however, would entail an exorbitant time and monetary cost. Can P. taeda be sequenced and assembled from WGS reads? It is difficult to answer this definitively because no one has attempted to assemble a 22-Gb genome to date, but one can gain insight by comparing the repeat content to sequenced genomes. The P. taeda genome contains fewer repeats that are nearly identical (98-100%) than either Sorghum bicolor or Zea mays (Figure 2, see Methods for description of this computational comparison). The Sorghum bicolor genome was successfully assembled from WGS Sanger reads, and the Pinus genome contains fewer recently amplified, and highly similar, repeats than Sorghum [44]. The degree of divergence within the genome should facilitate assembly of a draft sequence for P. taeda. Based on the age and diversity of pine repeats, it may even be possible to assemble the genome using a whole genome shotgun strategy based on several platforms and a range of insert sizes. Regardless of strategy, the massive P. taeda genome will surely challenge the limits of contemporary sequencing technology.

Comparison of repeat content among twelve sequenced genomes and Pinus taeda.
Methods
BAC Clone Sequencing and Assembly
Loblolly pine BAC library Pt_7Ba (Clemson University Genomics Institute [CUGI], Clemson, SC) was screened with multiplexed 32P-labeled PCR amplicons from ten genes mapped to quantitative trait loci (QTL) associated with wood chemical traits in loblolly pine [48–50] (Additional file 6, Table S5). Hybridizations identified 256 positive clones, and a random 48-clone subset of the positives was obtained from CUGI. The BAC DNA was isolated with the Colony Fast-Screen| Kit (Epicentre Biotechnologies, Madison, WI) and sized relative to BAC-Tracker| Supercoiled DNA Ladder (Epicentre) using SYBR, Gold (Molecular Probes, Eugene, OR) and agarose gel electrophoresis. Subsequently, ten BACs were selected that showed single BAC insert bands and were different from each other. Glycerol stocks were sent to Beckman Coulter Genomics (Danvers, MA) for subclone library construction and sequencing.
For each BAC, a shotgun library was prepared from a single clone inoculated to 500 mL of LB with 12.5 μg/mL chloramphenicol. High molecular weight DNA was produced using the Qiagen (Valencia, CA) Large-Construct Kit. The DNA was randomly sheared using a Genemachines Hydroshear (Genomic Solutions, Ann Arbor, MI). The sheared DNA was end-repaired with Epicentre End-It| End-Repair Kit and size selected for inserts from 2 to 4 kilobases to produce libraries with average insert sizes of 2 Kb, 3 Kb and 3.5 Kb. The insert DNA was ligated to pUC19 high copy plasmid vector (Fermentas, Glen Burie, MD). The ligations were transformed into DH10B T1r E.coli cells (Invitrogen, Carlsbad, CA) and plated on LB agar with appropriate carbenicillin, X-gal and IPTG concentrations. Transformation mixes were quality controlled via enzyme digest and arrayed into 384-well plates containing LB freezing medium. Subclone DNA templates were sequenced in 384-well format, using BigDye® Version 3.1 reactions on ABI3730xl instruments (Applied Biosystems, Foster City, CA) with the forward and reverse reactions (paired ends) being done in the same plate to maximize the paired end rate. Thermal cycling was performed using 384-well Thermocyclers (Applied Biosystems). Sequencing reactions were purified using Agencourt's CleanSeq® dye-terminator removal kit.
All reads were processed using PHRED base calling software and constantly monitored against quality metrics using the PHRED Q20 [51, 52]. The quality scores for each run were monitored through Agencourt's Galaxy LIMS system. A passing read was defined as an average high quality PHRED score of 20 or higher for at least 100 bases. Typical average read-lengths extended 500-600 bp. The Arachne Whole Genome Assembler [53], coupled with Agencourt's LIMS system, was used to assemble the BAC sequences. Assemblies were viewed in CONSED [54, 55].
Computational Annotation of BAC Assemblies
Annotations for the P. taeda contigs were prepared using the program MAKER, a genome annotation pipeline that identifies repetitive elements, aligns EST and protein homology evidence, prepares ab initio gene predictions, calculates quality control metrics, and synthesizes these data into final genome annotations.
The EST/cDNA sequences used by MAKER were derived from P. taeda and were combined with EST/cDNA sequences from all other Pinaceae species found in dbEST [56]. The UniProt/Swiss-Prot [57, 58] protein database was used as the protein homology database for the MAKER run. Repeat elements were identified using a MAKER internal transposable element database, the RepBase repeat library in conjunction with RepeatMasker, and pre-computed repeats from the program CENSOR [33] passed to MAKER via the algorithm's GFF3-passthrough option.
The total length of the preliminary contig set (923817 bp) was too short to accurately train the ab inito gene predictors specifically for the P. taeda genome. Instead a hybrid approach was taken by using existing training parameters from both monocot and dicot plant species to produce gene predictions in separate MAKER runs. Because MAKER uses evidence alignments to produce "hints" which are then sent to the ab initio gene prediction algorithms that can accept them, prediction algorithms that run inside the MAKER pipeline are capable of producing improved gene models even when the training parameters are imperfect. After producing a pool of possible ab initio and "hint-based" gene predictions, MAKER chooses those that are best supported by EST and protein homology evidence alignments using internal quality control metrics [31, 59] and promotes them to the status of genome annotations.
MAKER was first run using the ab initio gene prediction algorithms SNAP [60], Augustus [61, 62], and GeneMark [63] trained for Arabidopsis thaliana and FGENESH [64] trained for a generic dicot species (the exact species was not specified in the FGENESH documentation). The second run of MAKER was performed using SNAP and GeneMark trained for Oryza sativa in conjunction with Augustus trained for Zea mays. Both sets of MAKER-produced gene models were saved in GFF3 [65] format and simple intron/exon structure statistics were calculated against them using the program Eval [61, 66] The MAKER runs were viewed and evaluated using the Apollo Genome Annotation Curation Tool [67]. The peptide sequences corresponding to both sets of MAKER gene predictions were searched for conserved protein domains using Interproscan with default parameters [68] against the Interpro protein signature database.
High-throughput Whole Genome Shotgun Sequencing
Whole genome shotgun sequencing was performed on diploid DNA from the same individual used to construct the BAC library, using the high-throughput Illumina Genome Analyzer II sequencing platform. Genomic DNA library construction was carried out using the Illumina genomic DNA sample preparation kit according to manufacturer's instructions, except that paired end specific oligonucleotides were used instead of the single read oligonucleotides. Starting material was 80 ul of pine genomic DNA at a concentration of 62.5 ng/ul sonicated in a Diagenode Bioruptor for 15 cycles of 30" on maximum power then 30" rest. Following paired end adapter ligation, fragments of approximate size 400-425 bp were gel purified and PCR amplified using the paired end Illumina library PCR primers (primers 1.0 and 2.0). After AMPure purification (Beckman Coulter Genomics), the sample was applied to an Agilent Bioanalyzer for quantitation. Based on the bioanalyzer-reported sample concentration, the library was applied to a flow cell at 5 pM using v1 cluster reagents. Sequencing was performed on an Illumina Genome Analyzer II using version 2 sequencing reagents for 40, 42 and 60 cycles. Basecalling was carried out using the Illumina GA Pipeline v1.3. The WGS sequencing was carried out at the University of California, Davis, Genome Center.
Additional Element Characterization in BAC Assemblies
As previously described, the MAKER automated annotation pipeline was customized for both gene prediction and repeat identification in the ten P. taeda BAC assemblies. MAKER reported simple sequence repeats, as well as similarity to Repbase accessions and the MAKER internal transposable element database. Since only a handful of complex repetitive elements have been characterized in conifers, it is expected that this similarity-based repeat landscape described by MAKER is incomplete.
Several additional methods were included to complete the identification of putative repetitive elements in the BAC assemblies. Tandem Repeats Finder was used to locate tandemly duplicated units of 5-200 bp, Gepard [69] was used to produce dotplots in order to visualize longer direct and inverse repeats within each BAC, and discontiguous megablast (word size 11, match/mismatch = +1/-1, gap open/extension cost= 2/2) was used within each BAC to delineate direct repeats of minimum length 100 bp that span at least 500 bp of putatively noncoding sequence. The resulting pairs of direct repeats are presented in this paper as potential long terminal repeats of uncharacterized LTR retrotransposons. The results of MAKER run with dicot parameters on unmasked pine BACs were also examined for evidence of nongenic open reading frames (ORFs) that may correspond to 'novel' complex repetitive elements such as DNA transposons or LTR retrotransposons.
Regions were identified where at least two MAKER gene-finding tools predicted ORFs, but the sequence failed to show enough similarity to EST and protein databases to be annotated as protein-coding genes. Each putative nongenic ORF element shows significant similarity to at least one known repetitive element and is described using the longest ORF (minimum length 240 bp) among similar predictions.
Whole Genome Shotgun Sequence Analysis
Two consensus transposons and a putative centromeric tandem repeat were assembled from a pool of 40 and 42-bp WGS reads using nugtohs.pl (unpublished). In order to assess genome-wide occurrence of putative genic and repetitive elements in the BAC assemblies, 60-bp WGS reads were aligned to each BAC sequence with BLASTN and post-processed with a Perl script. This produced two WGS-coverage maps of each BAC; one coverage map optimized for WGS-to-BAC alignments showing 99% nucleotide identity (score threshold 55) and one map optimized to count alignments at or above 75% nucleotide identity (score threshold 24). The coverage maps are reported in hits per base pair in .sgr formats that were initially analyzed using the Integrated Genome Browser [70]. Genome-wide copy number of BAC elements were computed by averaging hits per base pair along the length of each element and calculating the ratio of this value to the estimated genome coverage (0.036×) provided by the 60-bp reads.
Assessment of Pine Genome for Sequencing and Assembly
To assess the P. taeda genome for sequencing and assembly, the repeat content of the genome was compared to twelve previously sequenced genomes: Caenorhabditis briggsae [71], Drosophila melanogaster [72], Chlamydomonas reinhardtii [73], Arabidopsis thaliana [1], Oryza sativa [74], Vitis vinifera [75], Physcomitrella patens [76], Populus trichocarpa [77], Sorghum bicolor [44], Malus x domestica (Troggio, unpublished), Zea mays [78], and Homo sapiens [79, 80]. Whole genome shotgun reads of each species were retrieved from the NCBI Trace Archive and converted to 60-bp lengths. 0.036× genome equivalents of these "reads" were then aligned to 920000 bp (similar to the total P. taeda BAC sequence) of randomly-selected regions of each genome using BLAST. Alignments were categorized into three nucleotide identity groups: 70-84%, 85-97% and 98-100%. The genomic sampling was conducted 10 times and averaged.
To simultaneously visualize all elements that were identified in the BAC assemblies, the program gff2ps was used [81]. The following data were formatted into GFF files and used to create Figure 1 and Additional file 1, Figure S1: MAKER dicot and monocot runs on masked and unmasked sequence; simple repeats; tandem repeats; direct repeats (potential LTRs); nongenic ORF elements; and coverage maps of each BAC at 75% identity and 99% identity. The coverage maps are shown in these figures as histograms of average hits per base pair in 50-bp windows. The GFF files are available for interactive browsing or download at http://dendrome.ucdavis.edu/treegenes/gbrowse, where a modified version of the GMOD project GBrowse was implemented in the TreeGenes database to display the annotations [82, 83].
References
Arabidopsis Genome Initiative: Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000, 408 (6814): 796-815. 10.1038/35048692.
Eckert AJ, Hall BD: Phylogeny, historical biogeography, and patterns of diversification for Pinus (Pinaceae): Phylogenetic tests of fossil-based hypotheses. Mol Phylogenet Evol. 2006, 40 (1): 166-182. 10.1016/j.ympev.2006.03.009.
Wakamiya I, Newton RJ, Johnston JS, Price HJ: Genome Size and Environmental Factors in the Genus Pinus. American Journal of Botany. 1993, 80 (11): 1235-1241. 10.2307/2445706.
Rabinowicz PD: Constructing gene-enriched plant genomic libraries using methylation filtration technology. Methods Mol Biol. 2003, 236: 21-36.
Rabinowicz PD, Citek R, Budiman MA, Nunberg A, Bedell JA, Lakey N, O'Shaughnessy AL, Nascimento LU, McCombie WR, Martienssen RA: Differential methylation of genes and repeats in land plants. Genome Res. 2005, 15 (10): 1431-1440. 10.1101/gr.4100405.
Bogunic F, Muratovic E, Brown SC, Siljak-Yakovlev S: Genome size and base composition of five Pinus species from the Balkan region. Plant Cell Reports. 2003, 22 (1): 59-63. 10.1007/s00299-003-0653-2.
Khoshoo TN: Polyploidy in Gymnosperms. Evolution. 1959, 13 (1): 24-39. 10.2307/2405943.
Khoshoo TN: Chromosome Numbers in Gymnosperms. Silvae Genetica. 1961, 10: 1-7. full_text.
Ohri D, Khoshoo TN: Genome Size in Gymnosperms. Plant Systematics and Evolution. 1986, 153 (1-2): 119-132. 10.1007/BF00989421.
Sax K, Sax HJ: Chromosome number and morphology in the conifers. J Arnold Arboretum. 1933, 14: 356-375.
MacPherson P, Filion WG: Karyotype analysis and the distribution of constitutive heterochromatin in five species of Pinus. J Hered. 1981, 72 (3): 193-198.
Jacobs MD, Gardner RC, Murray BG: Cytological characterization of heterochromatin and rDNA in Pinus radiata and P. taeda. Plant Systematics and Evolution. 2000, 223 (1): 71-79. 10.1007/BF00985327.
Hizume M, Shibata F, Matsusaki Y, Garajova Z: Chromosome identification and comparative karyotypic analyses of four Pinus species. Theor Appl Genet. 2002, 105 (4): 491-497. 10.1007/s00122-002-0975-4.
Doudrick RL, Heslop-Harrison JS, Nelson CD, Schmidt T, Nance WL, Schwarzacher T: Karyotype of Slash Pine (Pinus elliottii var. elliottii) using patterns of fluorescence in situ hybridization and fluorochrome banding. J Hered. 1995, 86 (4): 289-296.
Borzan Z: Karyotype analysis in Pinus: A contribution to the standardization of the karyotype analysis and review of some applied techniques. Silvae genetica. 1978, 27: 3-4-144.
Drewry A: G-banded chromosomes in Pinus resinosa. J Hered. 1982, 73 (4): 305-306.
Saylor LC: Karyotype analysis of the genus Pinus, subgenus Strobus. Silvae genetica. 1983, 32 (3-4): 119-124.
Hizume M, Ohgiku A, Tanaka A: Chromosome banding in the genus Pinus. Journal of Plant Research. 1989, 102 (1): 25-36.
Fuchs J, Brandes A, Schubert I: Telomere Sequence Localization and Karyotype Evolution in Higher-Plants. Plant Systematics and Evolution. 1995, 196 (3-4): 227-241. 10.1007/BF00982962.
Islam-Faridi MN, Nelson CD, Kubisiak TL: Reference karyotype and cytomolecular map for loblolly pine (Pinus taeda L.). Genome. 2007, 50 (2): 241-251. 10.1139/G06-153.
Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S, Geer LY, Helmberg W, Kapustin Y, Kenton DL, Khovayko O, Lipman DJ, Madden TL, Maglott DR, Ostell J, Pruitt KD, Schuler GD, Schriml LM, Sequeira E, Sherry ST, Sirotkin K, Souvorov A, Starchenko G, Suzek TO, Tatusov R, Tatusova TA: Database resources of the National Center for Biotechnology. Nucleic Acids Res. 2006, D173-D180. 10.1093/nar/gkj158. 34 Database
Eckert AJ, Pande B, Ersoz ES, Wright MH, Rashbrook VK, Nicolet CM, Neale DB: High-throughput genotyping and mapping of single nucleotide polymorphisms in loblolly pine (Pinus taeda L.). Tree Genetics & Genomes. 2009, 5 (1): 225-234.
Kriebel HB: DNA-Sequence Components of the Pinus-Strobus Nuclear Genome. Canadian Journal of Forest Research-Revue Canadienne De Recherche Forestiere. 1985, 15 (1): 1-4. 10.1139/x85-001.
Kinlaw CS, Gerttula SM, Carter MC: Lipid transfer protein genes of loblolly pine are members of a complex gene family. Plant Mol Biol. 1994, 26 (4): 1213-1216. 10.1007/BF00040702.
Kinlaw CS, Neale DB: Complex gene families in pine genomes. Trends in Plant Science. 1998, 2 (9): 356-359. 10.1016/S1360-1385(97)84624-9.
Elsik CG, Williams CG: Retroelements contribute to the excess low-copy-number DNA in pine. Molecular and General Genetics. 2000, 264 (1-2): 47-55. 10.1007/s004380000279.
Elsik CG, Williams CG: Families of clustered microsatellites in a conifer genome. Mol Genet Genomics. 2001, 265 (3): 535-542. 10.1007/s004380100443.
Hamberger B, Hall D, Yuen M, Oddy C, Hamberger B, Keeling CI, Ritland C, Ritland K, Bohlmann J: Targeted isolation, sequence assembly and characterization of two white spruce (Picea glauca) BAC clones for terpenoid synthase and cytochrome P450 genes involved in conifer defence reveal insights into a conifer genome. BMC Plant Biol. 2009, 9: 106-10.1186/1471-2229-9-106.
Keeling CI, Dullat HK, Yuen M, Ralph SG, Jancsik S, Bohlmann J: Identification and Functional Characterization of Monofunctional ent-Copalyl Diphosphate and ent-Kaurene Synthases in White Spruce Reveal Different Patterns for Diterpene Synthase Evolution for Primary and Secondary Metabolism in Gymnosperms. Plant Physiology. 2010, 152 (3): 1197-1208. 10.1104/pp.109.151456.
Bautista R, Villalobos DP, Diaz-Moreno S, Canton FR, Canovas FM, Claros MG: Toward a Pinus pinaster bacterial artificial chromosome library. Annals of Forest Science. 2007, 64 (8): 855-864. 10.1051/forest:2007060.
Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, Moore B, Holt C, Alvarado AS, Yandell M: MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008, 18 (1): 188-196. 10.1101/gr.6743907.
Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L, Finn RD, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Laugraud A, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowell J, Mistry J, Mitchell A, Mulder N, Natale D, Orengo C, Quinn AF: InterPro: the integrative protein signature database. Nucleic Acids Res. 2009, D211-215. 10.1093/nar/gkn785. 37 Database
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J: Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005, 110 (1-4): 462-467. 10.1159/000084979.
Kossack DS, Kinlaw CS: IFG, a gypsy-like retrotransposon in Pinus (Pinaceae), has an extensive history in pines. Plant Mol Biol. 1999, 39 (3): 417-426. 10.1023/A:1006115732620.
Rocheta M, Cordeiro J, Oliveira M, Miguel C: PpRT1: the first complete gypsy-like retrotransposon isolated in Pinus pinaster. Planta. 2007, 225 (3): 551-562. 10.1007/s00425-006-0370-5.
L'homme Y, Seguin A, Tremblay FM: Different classes of retrotransposons in coniferous spruce species. Genome. 2000, 43 (6): 1084-1089. 10.1139/gen-43-6-1084.
Benson G: Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999, 27 (2): 573-580. 10.1093/nar/27.2.573.
Elsik CG, Minihan VT, Hall SE, Scarpa AM, Williams CG: Low-copy microsatellite markers for Pinus taeda L. Genome. 2000, 43 (3): 550-555. 10.1139/gen-43-3-550.
Schmidt A, Doudrick RL, Heslop-Harrison JS, Schmidt T: The contribution of short repeats of low sequence complexity to large conifer genomes. Theoretical and Applied Genetics. 2000, 101 (1-2): 7-14. 10.1007/s001220051442.
Kamm A, Doudrick RL, Heslop-Harrison JS, Schmidt T: The genomic and physical organization of Ty1-copia-like sequences as a component of large genomes in Pinus elliottii var. elliottii and other gymnosperms. Proc Natl Acad Sci USA. 1996, 93 (7): 2708-2713. 10.1073/pnas.93.7.2708.
Plomion C, Chagne D, Pot D, Kumar S, Wilcox PL, Burdon RD, Prat D, Peterson DG, Paiva J, Chaumeil P, Vendramin GG, Sebastiani F, Nelson CD, Echt CS, Savolainen O, Kubisiak TL, Cervera MT, de Maria N, Islam-Faridi MN: Pines. Genome Mapping and Molecular Breeding in Plants. Edited by: Kole C. 2007, Berlin: Springer-Verlag, 7: 29-92. full_text.
Millar CI: Early evolution of pines. Ecology and Biogeography of Pinus. Edited by: Richardson DM. 1998, Cambridge, MA: Cambridge University Press, 69-91.
Ma J, Devos KM, Bennetzen JL: Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 2004, 14 (5): 860-869. 10.1101/gr.1466204.
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, Schmutz J, Spannagl M, Tang HB, Wang XY, Wicker T, Bharti AK, Chapman J, Feltus FA, Gowik U, Grigoriev IV, Lyons E, Maher CA, Martis M, Narechania A, Otillar RP, Penning BW, Salamov AA, Wang Y, Zhang LF: The Sorghum bicolor genome and the diversification of grasses. Nature. 2009, 457 (7229): 551-556. 10.1038/nature07723.
SanMiguel P, Gaut BS, Tikhonov A, Nakajima Y, Bennetzen JL: The paleontology of intergene retrotransposons of maize. Nature Genetics. 1998, 20 (1): 43-45. 10.1038/1695.
SanMiguel P, Bennetzen JL: Evidence that a recent increase in maize genome size was caused by the massive amplification of intergene retrotransposons. Annals of Botany. 1998, 82: 37-44. 10.1006/anbo.1998.0746.
SanMiguel PJ, Ramakrishna W, Bennetzen JL, Busso CS, Dubcovsky J: Transposable elements, genes and recombination in a 215-kb contig from wheat chromosome 5A(m). Funct Integr Genomics. 2002, 2 (1-2): 70-80. 10.1007/s10142-002-0056-4.
Brown GR, Bassoni DL, Gill GP, Fontana JR, Wheeler NC, Megraw RA, Davis MF, Sewell MM, Tuskan GA, Neale DB: Identification of quantitative trait loci influencing wood property traits in loblolly pine (Pinus taeda L.). III. QTL Verification and candidate gene mapping. Genetics. 2003, 164 (4): 1537-1546.
Brown GR, Gill GP, Kuntz RJ, Langley CH, Neale DB: Nucleotide diversity and linkage disequilibrium in loblolly pine. Proc Natl Acad Sci USA. 2004, 101 (42): 15255-15260. 10.1073/pnas.0404231101.
Sewell MM, Davis MF, Tuskan GA, Wheeler NC, Elam CC, Bassoni DL, Neale DB: Identification of QTLs influencing wood property traits in loblolly pine (Pinus taeda L.). II. Chemical wood properties. Theor Appl Genet. 2002, 104 (2-3): 214-222. 10.1007/s001220100697.
Ewing B, Green P: Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8 (3): 186-194.
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8 (3): 175-185.
Batzoglou S, Jaffe DB, Stanley K, Butler J, Gnerre S, Mauceli E, Berger B, Mesirov JP, Lander ES: ARACHNE: a whole-genome shotgun assembler. Genome Res. 2002, 12 (1): 177-189. 10.1101/gr.208902.
Gordon D: Viewing and editing assembled sequences using Consed. Curr Protoc Bioinformatics. 2003, Chapter 11 (Unit 11.2):
Gordon D, Abajian C, Green P: Consed: a graphical tool for sequence finishing. Genome Res. 1998, 8 (3): 195-202.
Boguski MS, Lowe TMJ, Tolstoshev CM: Dbest - Database for Expressed Sequence Tags. Nature Genetics. 1993, 4 (4): 332-333. 10.1038/ng0893-332.
Bairoch A, Apweiler R: The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28 (1): 45-48. 10.1093/nar/28.1.45.
Bairoch A, Bougueleret L, Altairac S, Amendolia V, Auchincloss A, Puy GA, Axelsen K, Baratin D, Blatter MC, Boeckmann B, Bollondi L, Boutet E, Quintaje SB, Breuza L, Bridge A, deCastro E, Coral D, Coudert E, Cusin I, Dobrokhotov P, Dornevil D, Duvaud S, Estreicher A, Famiglietti L, Feuermann M, Gehant S, Farriol-Mathis N, Ferro S, Gasteiger E, Gateau A: The universal protein resource (UniProt). Nucleic Acids Res. 2007, 35: D193-D197. 10.1093/nar/gkl929.
Eilbeck K, Moore B, Holt C, Yandell M: Quantitative measures for the management and comparison of annotated genomes. BMC Bioinformatics. 2009, 10: 67-10.1186/1471-2105-10-67.
Korf I: Gene finding in novel genomes. BMC Bioinformatics. 2004, 5: 59-10.1186/1471-2105-5-59.
Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B: AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34: W435-W439. 10.1093/nar/gkl200.
Stanke M, Diekhans M, Baertsch R, Haussler D: Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008, 24 (5): 637-644. 10.1093/bioinformatics/btn013.
Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M: Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005, 33 (20): 6494-6506. 10.1093/nar/gki937.
Salamov AA, Solovyev VV: Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000, 10 (4): 516-522. 10.1101/gr.10.4.516.
Bao W, O'Malley DM, Whetten R, Sederoff RR: A Laccase Associated with Lignification in Loblolly Pine Xylem. Science. 1993, 260 (5108): 672-674. 10.1126/science.260.5108.672.
Keibler E, Brent MR: Eval: A software package for analysis of genome annotations. BMC Bioinformatics. 2003, 4: 50-10.1186/1471-2105-4-50.
Lewis SE, Searle SM, Harris N, Gibson M, Lyer V, Richter J, Wiel C, Bayraktaroglir L, Birney E, Crosby MA, Kaminker JS, Matthews BB, Prochnik SE, Smithy CD, Tupy JL, Rubin GM, Misra S, Mungall CJ, Clamp ME: Apollo: a sequence annotation editor. Genome Biol. 2002, 3 (12): RESEARCH0082-10.1186/gb-2002-3-12-research0082.
Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R: InterProScan: protein domains identifier. Nucleic Acids Res. 2005, 33: W116-W120. 10.1093/nar/gki442.
Krumsiek J, Arnold R, Rattei T: Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics. 2007, 23 (8): 1026-1028. 10.1093/bioinformatics/btm039.
Nicol JW, Helt GA, Blanchard SG, Raja A, Loraine AE: The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics. 2009, 25 (20): 2730-2731. 10.1093/bioinformatics/btp472.
Stein LD, Bao ZR, Blasiar D, Blumenthal T, Brent MR, Chen NS, Chinwalla A, Clarke L, Clee C, Coghlan A, Coulson A, D'Eustachio P, Fitch DHA, Fulton LA, Fulton RE, Griffith-Jones S, Harris TW, Hillier LW, Kamath R, Kuwabara PE, Mardis ER, Marra MA, Miner TL, Minx P, Mullikin JC, Plumb RW, Rogers J, Schein JE, Sohrmann M, Spieth J: The genome sequence of Caenorhabditis briggsae: A platform for comparative genomics. Plos Biol. 2003, 1 (2): 166-10.1371/journal.pbio.0000045.
Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, George RA, Lewis SE, Richards S, Ashburner M, Henderson SN, Sutton GG, Wortman JR, Yandell MD, Zhang Q, Chen LX, Brandon RC, Rogers YHC, Blazej RG, Champe M, Pfeiffer BD, Wan KH, Doyle C, Baxter EG, Helt G, Nelson CR: The genome sequence of Drosophila melanogaster. Science. 2000, 287 (5461): 2185-2195. 10.1126/science.287.5461.2185.
Merchant SS, Prochnik SE, Vallon O, Harris EH, Karpowicz SJ, Witman GB, Terry A, Salamov A, Fritz-Laylin LK, Marechal-Drouard L, Marshall WF, Qu LH, Nelson DR, Sanderfoot AA, Spalding MH, Kapitonov VV, Ren QH, Ferris P, Lindquist E, Shapiro H, Lucas SM, Grimwood J, Schmutz J, Cardol P, Cerutti H, Chanfreau G, Chen CL, Cognat V, Croft MT, Dent R: The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science. 2007, 318 (5848): 245-251. 10.1126/science.1143609.
Goff SA, Ricke D, Lan TH, Presting G, Wang RL, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, Hadley D, Hutchinson D, Martin C, Katagiri F, Lange BM, Moughamer T, Xia Y, Budworth P, Zhong JP, Miguel T, Paszkowski U, Zhang SP, Colbert M, Sun WL, Chen LL, Cooper B, Park S, Wood TC, Mao L, Quail P: A draft sequence of the rice genome (Oryza sativa L. ssp japonica). Science. 2002, 296 (5565): 92-100. 10.1126/science.1068275.
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeal F, Hugueney P, Dasilva C, Horner D, Mica E, Joblot D, Poulain J, Bruyere C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V: The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007, 449 (7161): 463-U465. 10.1038/nature06148.
Rensing SA, Lang D, Zimmer AD, Terry A, Salamov A, Shapiro H, Nishiyama T, Perroud PF, Lindquist EA, Kamisugi Y, Tanahashi T, Sakakibara K, Fujita T, Oishi K, Shin-I T, Kuroki Y, Toyoda A, Suzuki Y, Hashimoto S, Yamaguchi K, Sugano S, Kohara Y, Fujiyama A, Anterola A, Aoki S, Ashton N, Barbazuk WB, Barker E, Bennetzen JL, Blankenship R: The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science. 2008, 319 (5859): 64-69. 10.1126/science.1150646.
Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, Schein J, Sterck L, Aerts A, Bhalerao RR, Bhalerao RP, Blaudez D, Boerjan W, Brun A, Brunner A, Busov V, Campbell M, Carlson J, Chalot M, Chapman J, Chen GL, Cooper D, Coutinho PM, Couturier J, Covert S, Cronk Q: The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science. 2006, 313 (5793): 1596-1604. 10.1126/science.1128691.
Messing J, Bharti AK, Karlowski WM, Gundlach H, Kim HR, Yu Y, Wei FS, Fuks G, Soderlund CA, Mayer KFX, Wing RA: Sequence composition and genome organization of maize. Proc Natl Acad Sci USA. 2004, 101 (40): 14349-14354. 10.1073/pnas.0406163101.
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C: Initial sequencing and analysis of the human genome. Nature. 2001, 409 (6822): 860-921. 10.1038/35057062.
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amadatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XQH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang JH, Miklos GLG, Nelson C, Broder S, Clark AG, Nadeau C, McKusick VA, Zinder N: The sequence of the human genome. Science. 2001, 291 (5507): 1304-1351. 10.1126/science.1058040.
Abril JF, Guigo R: gff2ps: visualizing genomic annotations. Bioinformatics. 2000, 16 (8): 743-744. 10.1093/bioinformatics/16.8.743.
Donlin MJ: Using the Generic Genome Browser (GBrowse). Curr Protoc Bioinformatics. 2009, Chapter 9 (Unit 9.9):
Wegrzyn JL, Lee JM, Tearse BR, Neale DB: TreeGenes: A Forest Tree Genome Database. Int J Plant Genomics. 2008, 2008: 412875-
Acknowledgements
We acknowledge Douglas R. Cook and Jinliang Gao for assistance with BAC library screening, Charles Nicolet for technical support in WGS sequencing and Michela Troggio for sharing the unpublished apple genome. For scripting and programming support, we acknowledge Keith Bradnam, Minyoung Choi, Ben Figueroa, and John Liechty. Andrew J. Eckert, Jeffrey Ross-Ibarra and Monika J. Lipinski provided editorial comments. Funding for this study was provided by the National Science Foundation (grant number 0501763) to C.H. Langley and D.B. Neale.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
ASK conceived and designed analyses, performed analyses and drafted the manuscript. JLW assisted with bioinformatic analysis and implemented the web browser. GP made major contributions to the design of the study and to graphic representation of results. CH worked with MY to customize the MAKER annotation pipeline for this project. GEB performed the BAC library screening. CAL performed the promoter analysis on predicted genes. JH was responsible for the sequencing, assembly and finishing of BAC assemblies. CHL, along with DBN, conceived of the project and obtained funding to perform this study. IK performed all bioinformatic analyses of the whole genome shotgun sequences used in this study and assisted with drafting the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2010_3014_MOESM1_ESM.PDF
Additional file 1: Figure S1. Ten P. taeda BAC assemblies with gene predictions, repeat identification and WGS coverage profiles of the BACs, as described in the text. (PDF 1 MB)
12864_2010_3014_MOESM2_ESM.XLS
Additional file 2: Table S1. Ten genic amplicons used to probe the P. taeda BAC library for sequences similar to coding sequences. (XLS 34 KB)
12864_2010_3014_MOESM3_ESM.DOC
Additional file 3: Table S2. Summary of eighteen genes and gene fragments predicted by MAKER in ten P. taeda BAC sequences. (DOC 70 KB)
12864_2010_3014_MOESM4_ESM.DOC
Additional file 4: Table S3. Twelve of the peptides predicted by MAKER showed significant similarity to Interpro (E-value > 1e-05). (DOC 56 KB)
12864_2010_3014_MOESM5_ESM.DOC
Additional file 5: Table S4. Promoter analysis of twelve predicted genes that showed similarity to Interpro (E value > 1e-05). (DOC 46 KB)
12864_2010_3014_MOESM6_ESM.DOC
Additional file 6: Table S5. Consensus sequences of the three most common sequence elements in the P. taeda genome, assembled from WGS reads. (DOC 46 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kovach, A., Wegrzyn, J.L., Parra, G. et al. The Pinus taeda genome is characterized by diverse and highly diverged repetitive sequences. BMC Genomics 11, 420 (2010). https://doi.org/10.1186/1471-2164-11-420
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-11-420