
Abstract
Background
Plasmodium parasites are causative agents of malaria which affects >500 million people and claims ~2 million lives annually. The completion of Plasmodium genome sequencing and availability of PlasmoDB database has provided a platform for systematic study of parasite genome. Aminoacyl-tRNA synthetases (aaRS s) are pivotal enzymes for protein translation and other vital cellular processes. We report an extensive analysis of the Plasmodium falciparum genome to identify and classify aaRSs in this organism.
Results
Using various computational and bioinformatics tools, we have identified 37 aaRS s in P. falciparum. Our key observations are: (i) fraction of proteome dedicated to aaRS s in P. falciparum is very high compared to many other organisms; (ii) 23 out of 37 Pf-aaRS sequences contain signal peptides possibly directing them to different cellular organelles; (iii) expression profiles of Pf-aaRSs vary considerably at various life cycle stages of the parasite; (iv) several PfaaRSs posses very unusual domain architectures; (v) phylogenetic analyses reveal evolutionary relatedness of several parasite aaRS s to bacterial and plants aaRSs; (vi) three dimensional structural modelling has provided insights which could be exploited in inhibitor discovery against parasite aaRSs.
Conclusion
We have identified 37 Pf-aaRSs based on our bioinformatics analysis. Our data reveal several unique attributes in this protein family. We have annotated all 37 Pf-aaRSs based on predicted localization, phylogenetics, domain architectures and their overall protein expression profiles. The sets of distinct features elaborated in this work will provide a platform for experimental dissection of this family of enzymes, possibly for the discovery of novel drugs against malaria.
Similar content being viewed by others


Background
Aminoacylation is the process of adding an aminoacyl group to the 3' end (CCA) of the tRNA molecule. tRNA is aminoacylated with a specific amino acid by aminoacyl-tRNA synthetase (aaRS s). aaRS s are responsible for attaching correct amino acid onto the cognate tRNA molecule in a two-step reaction. The amino acid is first activated with ATP forming an aminoacyladenylate intermediate. Once activated, this amino acid is transferred to the 3' end of its corresponding tRNA molecule to be processed during protein synthesis. All aaRSs require divalent cation MgCl2 for their aminoacylation reaction [1, 2].
Reaction:
-
1.
amino acid + ATP → aminoacyl-AMP + PPi
-
2.
aminoacyl-AMP + tRNA → aminoacyl-tRNA + AMP
The aaRS s are divided into two major classes based on structural topology of their active sites. Class I aaRS s represent 11 amino acids, including Arg, Cys, Gln, Glu, Ile, Leu, Lys, Met, Val, Trp and Tyr. Class II aaRS s includes 10 amino acids - Ala, Asp, Asn, Gly, His, Lys, Phe, Pro, Ser and Thr. Core domains of class I enzymes are characterized by a Rossmann fold which consists of α-helices and β-pleated sheets. This domain contains two conserved motifs ('HIGH' and 'KMSKS') which are directly involved in ATP binding. Catalytic domain of class II enzymes has a unique fold with a central core of anti-parallel β strands flanked by α helices [3]. There are three weakly conserved motifs, two of them are involved in ATP binding while the third one plays a role in homo dimerization. Class I enzymes bind ATP in an extended conformation while class II do so in a bent conformation. The two aaRS classes have different modes of aminoacylation - class I enzymes aminoacylate the 2'OH of the cognate tRNA whereas class II enzymes aminoacylate 3'OH of the tRNA (with the exception of PheRS) [4]. All known aaRS s are multidomain proteins with complex modular architectures [5]. In addition, eukaryotic aaRSs are distinguished by the presence of appended domains at either the N- or C-terminus which are generally absent from their bacterial/archaeal counterparts [6]. These appendages to the catalytic cores of several aaRSs are non-catalytic and instead function to mediate protein- protein interactions or act as general RNA-binding domains [7–9].
In mammalian cells, some aaRS s are present as a larger multi- aaRS complex (MSC) composed of nine synthetases (arginyl-, aspartyl-, glutamyl-, glutaminyl-, leucyl-, lysyl-, isoleucyl-, methionyl- and prolyl-tRNA synthetases) [10–12]. The MSC is composed of a mixture of class I and class II aaRS s along with three non- aaRS proteins p38, p43 and p18. It is not clear why certain aaRS s exist as a complex while some are in free form. MSC might help in efficient protein synthesis by preventing mixing of charged tRNAs with cellular pool and by increasing local concentration of tRNA near the site of protein synthesis [13].
The accuracy of tRNA aminoacylation reaction is critical in ensuring fidelity in protein translation [14]. To achieve this accuracy, some aaRS enzymes possess a proofreading (editing) mechanism that hydrolyzes tRNAs aminoacylated with the non-cognate amino acid [15]. For example, editing domains may be found attached to alanyl-tRNA synthetase (AlaRS), leucyltRNA synthetase (LeuRS) and so on [16–21]. In other cases, the editing domain is not attached to aaRS but rather functions as an individual protein [22, 23]. For example, YbaK protein from Haemophilus influenza is capable of efficiently editing Cys-tRNAPro[24]. ThrRS has been shown to have another editing domain called NTD which can cleave the bond between D-amino acid and tRNA [25].
Recently it has been shown that aaRS s are not only involved in protein synthesis but also perform many non-catalytic and non-canonical roles in RNA processing/trafficking, apoptosis, rRNA synthesis, angiogenesis and inflammation [26–30]. These versatile properties of aaRS s are the outcome of their differential cellular localization, nucleic acid binding properties, protein-protein interactions and collaboration (fusion) with additional domains. In case of malaria parasite, apicoplast proteins and pathways have already received particular attention as drug targets [31]. In this work we present a study of aaRS s from P. falciparum - the most virulent agent of human malaria. Our aim for this study was to use bioinformatics tools to (a) discover special and unusual modules present in parasite aaRSs which are potentially absent from human homologues, and (b) to identify potential new drug targets based on this protein family.
Results and Discussion
Sequence extraction and analysis
We exploited current annotation available in PlasmoDB [32] to identify the repertoire of aaRS s in P. falciparum genome. According to Enzyme Commission (EC) 37 proteins in PlasmoDB (see additional file 1) are annotated as belonging to the EC group 6.1.1. (EC number provided for aaRS s). Although in many cases current annotations allow an assignment to Class I or II of aaRS s, for some annotations are still preliminary. Due to this, we used Hidden Markov Models (HMMs) for identifying aaRSs in P. falciparum. For each aaRS a set of known sequences was utilized to construct 20 HMMs (see methods for details). For each database search a score distribution was obtained and 4 cutoffs were considered to identify aaRS. Results are reported in Table 1. We observed that 2 proteins annotated as belonging to EC group 6.1.1.- in PlasmoDB are not found by HMMs - PF14_0401 annotated as MetRS is instead a generic tRNA binding protein as elucidated in the genome re-annotation process, while the second one (PFC0470w) is still mis-annotated as ValRS. A total of 18 Pf-aaRS s can be classified within the 10 aaRS s that define class I. All members of this class are represented in the P. falciparum proteome. The annotations of these sequences are summarized in additional file 1. Similar to class I Pf-aaRSs, the class II Pf-aaRS s have a total of 18 sequences for 10 different amino acid synthetases. Four genes are present in P. falciparum for PheRS but these likely encode for 1 heterodimeric and 2 monomeric versions of PheRS.
In order to carry out comparative analyses of aaRS s of P. falciparum with those of other species we considered aaRS sequences from several organisms representing three domains of life (see methods section). As expected, we found variable number of aaRS s in different species. M. jannaschii (archaebacteria) and M. tuberculosis (bacteria) have the lowest aaRS s count amongst other organisms like E. coli, S. cerevisiae, D. discoidium, P. falciparum, O. sativa, R. norvegicus, D. melanogaster, and H. sapiens. Human bears the highest number of aaRS s in this analysis (Figure 1a). Our analysis also shows that P. falciparum has the highest aaRS fraction (relative to its proteome size) when compared with bacteria, yeast and human counterparts (Figure 1b). The number of individual aaRS varies in different species. For example, when individual aaRS s from human and P. falciparum were compared it was evident that AlaRS and ThrRS were higher in number in humans (Figure 2). Presence of more than one copy of each aaRS in an organism may indicate additional biological, temporal or spatial roles for these enzymes as several aaRS s also perform non-canonical functions [33]. In this work we describe in detail the 37 Pf-aaRS s.

(a) Predictied number of aaRS s present in Plasmodium falciparum (Pf), Rattus norvegicus (Rn), Saccharomyces cerevisiae (Sc), Drosophila melanogaster (Dm), Homo sapiens (Hs), Oryza sativa (Os), Dictyostelium discoidium (Dd), Mycobacterium tuberculosis (Mtb), Escherichia coli (Ec) , and Methanocalclococcus jannaschii (Mj). (b) Diagram representing fraction of proteome (in percentage) dedicated to the aaRS proteins in various organisms.

Bar graph showing number of different aaRSs in Plasmodium falciparum and Homo sapiens. The number of alanyl- and threonyl- tRNA synthetases is higher in humans whereas P. falciparum seems richer in phe tRNA synthetases.
Indirect pathways of aminoacylation
It was earlier believed that 20 aaRS s were necessary for the incorporation of 20 amino acids in proteins. But surprisingly, some archaea, bacteria and chloroplasts lack GlnRS and AsnRS enzymes [34–38]. Interestingly, these organisms use an alternate pathway based on tRNA dependent amino acid transformation. A non-discriminating GluRS charges tRNAGln with glutamic amino acid and then a second enzyme called tRNA-dependent amidotransferase (AdT) amidates glutamate to make glutamine. A corresponding reaction occurs in case of asparagine residues. In case of P. falciparum, occurrence of glutamate-tRNA synthetase (PF13_0257, MAL13P1.281) and amidotransferase subunit A (PFD0780w) & subunit B (PFF1395c) together indicates presence of both direct and indirect pathways for aminoacylation [39, 40]. Both subunits of amidotransferase have apicoplast targeting signals suggesting an indirect pathway for aminoacylation in P. falciparum apicoplast. The expression of Pf-AdT subunit A is predicted in all life cycle stages of parasite based on proteomic and microarray data. We therefore feel that this pathway must also be active in the parasite apicoplast. We could not find sequence homologues of enzymes involved in indirect aminoacylation of cysteine residues [41–43] in the proteome of P. falciparum.
The multi-synthetase complex (MSC)
In mammalian cells, some aaRS s are present as a larger multi-aaRS complex (MSC). A constituent of the MSC - protein p43 - has sequence homologue (PF14_0401 - EMAP-II-like cytokine) in P. falciparum although there is no evidence for presence of MSC in malaria parasites. Interestingly, p43 is not only required for stability of the MSC complex but also functions as a proinflammatory cytokine [44–46]. Role of p43 homolog in P. falciparum is unknown, but evidence from other organisms indicates that MSC functions in protein stability, efficient protein translation and protein elongation [47]. Sequence identity between P. falciparum p43 and its human homolog is ~24% and based on microarray data p43 seems to be expressed at asexual life cycle stages of P. falciparum. A mitochondrial targeting signal was also predicted for parasite p43 but the role of p43 in parasite remains to be explored experimentally.
Targeting of aaRSs in the parasite
aaRS s are not only involved in protein synthesis but also in various other cellular activities including intron splicing, translational regulation and tRNA channeling. Diversified roles for aaRS s necessitate their presence (transit) into various cellular compartments. We therefore analyzed P. falciparum aaRS sequences for presence of putative signal sequences predicted by MITOPROT, PredictNLS and PATS for mitochondria, nucleus and apicoplast respectively. We found that 23 P. falciparum aaRS s have signal peptides, possibly for directing them to different cellular organelles. Another 14 aaRSs from P. falciparum may be resident in the parasite cytoplasm (Figure 3a). Apicoplast is known to have protein synthesis machinery which may use aaRS s [48]. Trafficking of nuclear encoded aaRS s to the apicoplast may explain why ~20 out of 37 Pf-aaRSs have apicoplast targeting signals. Our data indicate that out of total ~20 Pf-aaRSs bearing apicoplast targeting signals, ~12 aaRS s may be exclusive to this organelle. Others are predicted to be shared between apicoplast, nucleus and mitochondria (Figure 3b). It has been earlier shown that some tRNAs need to be aminoacylated in the nucleus before they can be exported to the cytoplasm, an observation indicating occurrence of aminoacylation reaction (mediated by aaRSs) inside the nucleus [49]. In P. falciparum, we found 10 aaRS s with nuclear localization signals but only one is predicted to be exclusively resident in the nucleus (PFA0480w- PheRS). Interestingly, we found no Pf-aaRS sequences with specific PEXEL (Plasmodium export element) motifs. This motif is found in parasite proteins that are exported beyond the parasitophorous vacuole membrane [50, 51].

(a) Percentage predicted distribution of Pf - aaRSs in different organelles within the parasite. (b) A schematic of all Pf-aaRS s and their predicted cellular localization. Detailed information regarding gene IDs can be found in additional file 1. Pf-aaRS s predicted to be common between apicoplast & mitochondria, mitochondria & nucleus and apicoplast & nucleus are marked with diamond, triangle and square shapes respectively.
Expression profiles of P. falciparum aaRSs
In order to study expression of aaRS during life cycle of the malaria parasite, we took advantage of available transcriptomics and proteomics data from PLASMODB. Firstly, we analyzed proteomic data from several independent experiments and compared them with transcriptomics data by Le Roch [52]. The latter sets of data were obtained using the affimetrix technology and hence provide a quantitative measure of mRNA levels in the parasite. Our results are provided in Table 2. Interestingly, we found that mRNA levels of potential apicoplast proteins (AP in the table) are lower on average (mean1 = 44.6; mean2 = 41.5; gam = 91.3; spor = 58.1) than those of potential cytoplasmic proteins (mean1 = 259; mean2 = 264.8; gam = 174.8; spor = 73.8). Proteomic data confirmed that while the cytoplasmic aaRS are found in almost all stages, the apicoplast aaRS are rarely found in the parasite. This could be in part due to experimental limits in the identification of apicoplast proteins by mass spectrometry. Indeed, when we carried out a chi-quadro test we found that proteins predicted to be targeted to apicoplast are significantly less represented (p < 10-4) in the sample of proteins identified by mass spectrometry. For these reasons we limited analysis of gene expression profiles only for putative cytoplasmic proteins. We considered trascriptomics data for sexual stages and asexual stages [52, 53]. We considered a reduced set of the time course gene expression data (22 time points instead of 48) and normalized data by Le Roch (see methods for details). This allowed us to analyse the expression of aaRS genes along all the intra-erythrocytic life cycle of the parasite (Table 2). Further observations of the protein expression profiles indicated that some aaRSs were exclusively detected at specific stages like, LeuRS (PF08_0011) and AspRS (PFE0715w) in sporozoites; IleRS (PFL1210w), SerRS (PF07_0073), GlnRS (PF13_0170), HisRS (PF14_0428) and PheRS (PFA0480w) in merozoites; AsnRS (PFE0475w), PheRS (PF11_0051) and HisRS (PFI1645c) in trophozoites and TrpRS (PF13_0205) in gametocyte stages (Figure 4).

Diagrammatic representation of Pf-aaRS protein expression which are specifically expressed in different life stages of the parasite based on mass spectrometry data [82].
Domain architecture of P. falciparum aaRSs
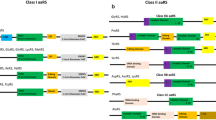
aaRS s are multi-domain proteins typically consisting of a conserved catalytic domain and an anti-codon binding domain. In addition, some aaRS s have RNA binding and editing domains that cleave incorrectly aminoacylated tRNA molecules [54]. Additional functional domains may be appended to aaRS s in the course of biological evolution [55, 56]. Careful examination of 37 identified P. falciparum aaRS s using Pfam database showed that most of them have a generic modular architecture that adheres to prototypical aaRSs (Figure 5). The remaining P. falciparum aaRS s or related proteins like PF14_0423 (protein having serine-threonine kinase domain in fusion with an anti-codon binding module) have complex domain architectures. In several, concatenation of unusual domains such as Ybak, GST, Ser-Thr kinase and DNA binding domains is evident (Figure 5). The functional relevance of these additional domains fused to typical aaRS in P. falciparum needs to be experimentally addressed. Intriguingly, two of the four Pf-PheRS subunits contain DNA binding domains (PF11_0051, PFA0480w). It is likely that the PheRS, in addition to its aminoacylation function, influences other cellular processes via DNA binding [57]. Consistent with its potential DNA binding property, the P. falciparum PheRS (PFA0480w) has a nuclear localization signal. The CysRS of B. subtilis (which also contains a DNA binding domain) is believed to play a role in initiating chromosomal replication [58]. Therefore, functional roles for P. falciparum PheRSs may extend from aminoacylation to DNA recognition and replication - a suggestion that requires experimental investigation. Similarly, it has been shown that GST or GST homology domains can help in complex formation of aaRS s with multifunctional factors (p38, p18) [56, 57]. Additional data show that deletion of GST homology domain from the C-terminal region of p38 results in the dissociation of EPRS (Glutamyl-prolyl-tRNA synthetase) and MetRS from the MSC complex [59]. Mammalian ValRS associated with elongation factor subunits also contain the GST homology domain [60–62]. Thus, the presence of GST domains might be a crucial feature of aaRS s. P. falciparum proteome has two such proteins with GST domains appended to MetRS (PF10_0340) and GluRS (PF13_0257). We also found a most interesting fusion of anticodon binding domain with a serine-threonine kinase (PF14_0423) in P. falciparum. This unusual kinase seems to be expressed throughout the life cycle of parasite (microarray data) and interestingly is predicted to be localized to the parasite nucleus. Clearly, the presence of unusual domain fusions in P. falciparum aaRS s suggests multiple functional roles for many of these P. falciparum enzymes as has been shown in other organisms.

Representation of unusual domain architectures in Pf-aaRS s and related proteins. A generic aaRS is also shown on top. Domain name abbreviations are YB, Ybak associating domain; TS-II, class II tRNA synthetase; AC, anticodon binding site; ED, editing domain; GST, glutathione-Stransferase C-terminal region; RBD, S4 RNA binding domain; TS, tRNA synthetase core domain; STK, serine-threonine kinase; FTS, phenylalanine-tRNA synthetase; PTS, prolinetRNA synthetase; VTS, valine-tRNA synthetase; MTS, methionine-tRNA synthetase; YTS, tyrosine-tRNA synthetase; ETS, glutamate-tRNA synthetase.
Phylogenetics
Overall the percentage identity between matching human and P. falciparum aaRS domains varies from 17 to 51. Clearly, Pf-aaRSs which have low sequence identity with human counterparts might serve as good drug targets. In order to study evolutionary relationships of P. falciparum aaRS s with other species, phylogenetic trees were developed in PHYML using maximum likelihood method. For each type of P. falciparum aaRS a separate tree was constructed (see additional file 2). aaRS sequences from 102 different species were used for multiple sequence alignments. As an example, phylogenetic tree of TyrRS from various species (including two sequences from P. falciparum) was constructed. Interestingly, one Pf-TyrRS (MAL8P1.125) clustered with human TyrRS whereas the second Pf-TyrRS (PF11_0181) clustered with bacterial TyrRS indicating different evolutionary origins (Figure 6a). Based on distance matrices, several P. f alciparum aaRS sequences clustered as being closer to plants (A. thaliana) or to bacteria (E. coli) (Figure 6b). It is already known that apicomplexan parasites like P. falciparum house a secondary endosymbiotic plastid, possibly hijacked by lateral genetic transfer from an alga. Therefore, the P. falciparum aaRS sequences which are evolutionary close to bacteria and plants are likely to be the outcome of horizontal gene transfer from the plastid. P. falciparum contains ~12 such aaRS sequences which cluster with bacterial or plant sequences. Functional and structural characterization of these bacterial/plant-like aaRS may be relevant in focusing efforts at using aaRS as drug targets.

(a) Evolutionary tree was constructed using the PHYML based on maximum likelihood method. P. falciparum TyrRSs (PlasmoDB id -MAL8p1.125 and PF11_0181) are labeled as green triangles. One of the TryRSs (MAL8p1.125) is evolutionarily closer to H. sapiens whereas the other TyrRS (PF11_0181) is closer to E. coli. Total of 102 species were considered for the evolutionary analysis and were taken from three domains of life. (b) List of Pf-aaRS sequences evolutionarily closer to their E. coli and A. Thaliana counterparts.
Homology modeling and structure comparisons
To date, no crystal structures have been obtained for any aaRS from P. falciparum. Hence, we performed homology modeling of several P. falciparum aaRSs using homologous structures available in PDB. Known structural templates (≥ 40% identity) were used for molecular modeling of several P. falciparum aaRSs including the two TyrRSs (PF11_0181, MAL8P1.125), the PheRS (PFA0480w), ThrRS (PF11_0270), LysRS (PF13_0262), MetRS (PF10_0340) and TrpRS (PF13_0205). The program Align2D (sequence alignment module in Modeller) was used to perform dynamic programming-based global alignments of the target and template sequences. This program uses variable gap penalty for structural loops and core regions using information derived from template structures. We found key differences in the conserved motifs in various aaRS s. For example, the class I motif 'KYSKS' in P. falciparum TyrRS (PF11_0181) and 'KMSKS' in MAL8P1.125 differs from 'KLGKS' of human mitochondrial TyrRS (2PID) and 'KMSSS' of human cytoplasmic (1N3L) respectively. Similarly, class I motif 'HIGH' has subtle sequences variations between P. falciparum and H. sapiens TyrRSs (Figure 7a, Table 3). Using the above procedures, we could generate structural models for several Pf-aaRSs. Stereo-chemical qualities of the generated protein models were assessed using PROCHECK (85-90% residues are in allowed regions of Ramachandran plot). The overall superimposed three-dimension models were visualized in CHIMERA and PYMOL (Figure 7b). Many sequence insertions were observed for P. falciparum enzymes when compared to their homologous [63]. Location of insertions in P. falciparum TyrRS between well-conserved secondary structures suggests ability of TyrRS anticodon binding core to accommodate larger sequence inserts with minimum disruption to the catalytic domain. Direct comparison of modeled P. falciparum aaRS s with human aaRS s revealed several other important structural differences. For example, numerous insertions are present in the loop regions linking various α-helices (α10 to α13) in anticodon binding domain of P. falciparum TyrRSs (PF11_0181 and MAL8p1.125) when compared to its human homologous (2PID and 1N3L) respectively. Structural differences between TyrRS (from P. falciparum) and human counterparts are summarized in Table 3 and shown in Figure 7c. These subtle structural changes that manifest as partial conservation of important motifs in P. falciparum aaRSs reflect evolutionary divergence, and may be useful for exploitation of parasite-specific features as drug targets.

Left and right panels of the figure represent sequence and structural comparison of bacterial type Plasmodium TyrRS (PF11_0181) with human mitochondrial TyrRS (2PID) and the cytosolic Plasmodium TyrRS (Mal8p1.125) with human cytosolic TyrRS (1N3L). a) A structure-based sequence alignment of the catalytic domain of Plasmodium TyrRSs with human TyrRSs. Insertions in Pf and human sequences are colored in light blue and orange respectively. Class I synthetase conserved motifs are colored red. Residues involved in tRNA recognition and catalysis are indicated in green (same residues in Pf and Hs) and violet & boxed (different in Pf and Hs). The secondary structural elements are shown above the sequence alignments. Conserved residues are indicated by asterisk below the sequence alignment. (b) Superposition of Pf-TyrRS and Hs-TyrRS depicting the structural differences. Pf-Tyr is colored grey and Hs-TyrRS is colored tan. Insertions in Pf-TyrRSs are highlighted in blue whereas Hs-TyrRS insertions are in orange. Motif 1 in Pf (PF11_0181 - HLGN and Mal8p1.125 - HIAQ) and Hs (2PID - HVGH and 1N3L - HVAY) TyrRSs has been encircled red whereas Motif 2 in Pf (PF11_0181 - KLGKS and Mal8p1.125 - KMSKS) and Hs (2PID - KYSKS and 1N3L - KMSSS) is encircled green. (c) Snapshot of the active sites of Pf and Hs TyrRS s (superimposed) structures. Non-conserved active site residues colored violet are encircled.
Conclusion
Aminoacyl-tRNA synthetases (aaRS s) link RNA with protein translation. Besides their key role in protein synthesis, aaRS s are also integral to various other cellular processes. aaRS enzymes have been the focus for antimicrobial drug discovery [64, 65]. An example of clinical application of an aaRS inhibitor is provided by the antibiotic mupirocin (marketed as Bactroban), which selectively inactivates bacterial isoleucyl-tRNA synthetase [66]. Similarly, it has been shown that the broad-spectrum antifungal 5-fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole (AN2690) inhibits yeast cytoplasmic leucyl-tRNA synthetase by blocking editing site of the enzyme [67, 68]. Therefore, presence of distinct or tinkered P. falciparum aaRS lends an opportunity for their exploitation as new drug targets against malaria. In this study, we have extensively analyzed aaRS sequences from Plasmodium species in terms of their mRNA/protein expression profiles, their cellular localization, their organelle targeting and their unique sequence/domain attributes. We have discovered several distinct aaRS s in P. falciparum with no clear human counterparts in terms of their overall domain structures. We have also highlighted deviations of some highly conserved sequence motifs and active site sequence clusters. Our analyses clearly show that a larger fraction of P. falciparum proteome is devoted to aaRS when compared with many other organisms. The phylogenetic data hint at evolutionary closeness of some Pf-aaRSs to bacteria and plants - this further supports the fact of secondary endosymbiosis in this apicomplexan. We hope that our in-depth phylogenetic, protein targeting, domain architecture, protein expression profiling and homology modeling data on Pf-aaRSs can be used as a platform for experimental studies of this important protein family in malaria parasites.
Methods
Sequence extraction
The P. falciparum genome database PlasmoDB Release 5.4 was used for the present analyses. Sequence sets of all the aaRS s from other organisms includes P. berghei, P. chabaudi, P. falciparum, P. knowlesi, P. yoelii, P. vivax, H. sapiens, M. tuberculosis, D. discoidium, M. jannaschii, R. norvegicus, C. parvum, B. bovis, S. cerevisiae, D. melanogaster, Y. pestis, T. aquaticus, S. pneumoniae, S. entrica, E. coli, A. thaliana, A. pisum, A. salmonicida, B. cereus, B. thuringiensis, B. afzelii, B. burgdorferi, B. garinii, B. valaisiana, Bradyrhizobium, B. pennsylvanicus, C. acidaminovorans, H. defensa, C. taiwanensis, E. fergusonii, F. bacterium, F. novicida, F. tularensis, F. alni, G. tenuistipitata, H. arsenicoxydans, A. cellulolyticus, A. chlorophenolicus, A. ferrooxidans, Algoriphagus, A. muciniphila, Anaeromyxobacter, A. thermophilum, B. ambifaria, B. indica, B. mycoides, B. taurus, B. tribocorum, C. atlanticus, Caulobacter, C. aurantiacus, C. cellulolyticum, Citrobacter, C. pinensis, C. Ruthia, Cyanothece, D. desulfuricans, D. hafniense, Diaphorobacter, D. shibae, D. turgidum, E. cuniculi, E. lenta, E. ruminantium, Exiguobacterium, G. diazotrophicus, Geobacillus, M. maris, N. multipartita, Nocardioides, O. terrae, P. abelii, P. atlantica, P. denitrificans, P. ingrahamii, P. lavamentivorans, R. castenholzii, S. arenicola, S. fumaroxidans, X. autotrophicus, V. vadensis, V. paradoxus, T. whipplei, T. auensis, S. stellata, Ch. parvum, S. heliotrinireducens, Silicibacter, S. putrefaciens, S. usitatus, Thauera, X. laevis, Theileria annulata, Vibrio fischeri, W. succinogenes, X. tropicalis, Zeamays. Additional sequences were obtained based on sequence similarity via NCBI BLAST [69] and ENSEMBL [70] databases. Known sequence motifs of aaRS s have been used as templates to retrieve sequences of aaRS from other organisms. Some aaRS sequences were manually annotated based on the presence of signature motifs. Protein domains and motifs in the predicted aaRS s were identified using following programs - Superfamily [71], SMART [72] and MotifScan available at expasy web server. The following databases - Pfam [73], TIGR, PIR, EBI and PlasmoDB were also extensively used. Hidden Markov Model (HMM) for each of the 20 aaRS were constructed by the software package Sequence Alignment and Modeling System version 2.2.1 (SAM) [74] exploiting sequences in the aaRS database [75]. HMM profiles were then used to carry out database search vs P. falciparum proteins. A score was assigned to each protein by calculating the probability that the corresponding sequence is generated by the HMM model, hence for each database search a score distribution was obtained. The score distributions were normalized and 4 ranges of values were considered to identify aaRS (c > 5, 10 < c < 20, 20 < c < 50, c < 50).
Expression and Localization
The prediction of signal sequences for cellular localization in P. falciparum was performed using various available online web-servers - MITOPROT [76], PredictNLS [77] and PATS [78] for mitochondria, nucleus and apicoplast respectively. PEXEL motif prediction was been carried out by querying PlasmoDB. To identify specific gene expression profiles, we have combined information from different data sets. For the spotted oligonucleotide array data, only half of the 48 time points of the intra-erythrocytic cycle are shown for simplicity, and ratios (versus a common reference) were log2-transformed prior to cluster analysis. For the photolithography data, CEL files were downloaded from website and transferred into Bioconductor package for analysis using a robust multi-array averaging algorithm (RMA) for background adjustment and quantiles normalization [79]. Genes whose expression level was less than 10 (too close to background) or the logP was greater than -0.5 (too few probes per gene) were removed from dataset. Total intensity values for each time point were converted to mean-centered ratios by dividing the total intensity by the average intensity for that gene across all experimental conditions and were then log2-transformed prior to clustering. These data manipulations were necessary because the oligo-nucleotide array data was collected as the intensity ratio between the experimental sample and a common reference, while the photolithography data was collected as the total signal intensity at each spot. Gene expression patterns where the minimum percentage of existing values was less than 80% were eliminated from rest of the analysis. The remaining missing values were replaced by using the KNN-imputation method [80].
Phylogenetic analysis
To explore the evolutionary relationships amongst aaRSs phylogenetic analyses were performed for each P. falciparum aaRS on an expanded set of 102 sequences. Multiple sequence alignments of these sequences were obtained from CLUSTALW with default parameters (performed locally) in PHYLIP format [81]. These MSAs were used as seed sequences to run PHYML_v2.4.4 using Jones-Taylor-Thornton (JTT) model [82]. The resulting file was further used in MEGA4.2 for visualization of trees [83].
Model Building and Validation
We used Sali's Modeller8v2 [84] tool for building various P. falciparum aaRS s models. The stereo-chemical quality of modeled proteins was verified by PROCHECK [85]. Structural mapping of active site residues and other motifs was performed using CHIMERA [86] and PYMOL [87].
References
Ibba M, Soll D: Aminoacyl-tRNA synthesis. Annu Rev Biochem. 2000, 69: 617-650. 10.1146/annurev.biochem.69.1.617.
Ibba M, Soll D: The renaissance of aminoacyl-tRNA synthesis. EMBO Rep. 2001, 2: 382-387.
Eriani G, Delarue M, Poch O, Gangloff J, Moras D: Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature. 1990, 347: 203-206. 10.1038/347203a0.
Burbaum JJ, Schimmel P: Structural relationships and the classification of aminoacyl- tRNA synthetases. J Biol Chem. 1991, 266: 16965-16968.
Wolf YI, Aravind L, Grishin NV, Koonin EV: Evolution of aminoacyl-tRNA synthetases-analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999, 9 (8): 689-710.
Mirande M: Aminoacyl tRNA synthetase family from prokaryotes and eukaryotes: structural domains and their implications. Prog Nucleic Acid Res Mol Biol. 1991, 40: 95-142. full_text.
Cahuzac B, Berthonneau E, Birlirakis N, Guittet E, Mirande M: A recurrent RNA binding domain is appended to eukaryotic aminoacyl-tRNA synthetases. EMBO J. 2000, 19: 445-452. 10.1093/emboj/19.3.445.
Robinson JC, Kerjan P, Mirande M: Macromolecular assemblage of aminoacyl-tRNA synthetases: quantitative analysis of protein-protein interactions and mechanism of complex assembly. J Mol Biol. 2000, 304: 983-994. 10.1006/jmbi.2000.4242.
Guigou L, Shalak V, Mirande M: The tRNA-interacting factor p43 associates with mammalian arginyl-tRNA synthetase but does not modify its tRNA aminoacylation properties. Biochemistry. 2004, 43: 4592-4600. 10.1021/bi036150e.
Hausmann CD, Ibba M: Aminoacyl-tRNA synthetase complexes: molecular multitasking revealed. FEMS Microbiology Reviews. 2008, 32: 705-721. 10.1111/j.1574-6976.2008.00119.x.
Bandyopadhyay AK, Deutscher MP: Complex of aminoacyl-transfer RNA synthetases. J Mol Biol. 1971, 60: 113-122. 10.1016/0022-2836(71)90451-7.
Kerjan P, Cerini C, Semeriva M, Mirande M: The multienzyme complex containing nine aminoacyl-tRNA synthetases is ubiquitous from Drosophila to mammals. Biochem Biophys Acta. 1994, 1199: 293-297.
Wolfson A, Knight R: Occurrence of the aminoacyl-tRNA synthetases in high-molecular weight complexes correlates with the size of substrate amino acids. FEBS Lett. 2005, 579: 3467-3472. 10.1016/j.febslet.2005.05.038.
Schimmel P, Schmidt E: Residues in a class I tRNA synthetase which determine selectivity of amino acid recognition in the context of tRNA. Biochemistry. 1995, 34: 11204-11210. 10.1021/bi00035a028.
Lin L, Schimmel P: Mutational analysis suggests the same design for editing activities of two tRNA synthetases. Biochemistry. 1996, 35: 5596-5601. 10.1021/bi960011y.
Sokabe M, Okada A, Yao M, Nakashima T, Tanaka I: Molecular basis of alanine discrimination in editing site. Proc Natl Acad Sci USA. 2005, 102: 11669-11674. 10.1073/pnas.0502119102.
Sokabe M, Ose T, Nakamura A, Tokunaga K, Nureki O, Yao M, Tanaka I: The structure of alanyl-tRNA synthetase with editing domain. Proc Natl Acad Sci U S A. 2009, 106 (27): 11028-11033.
Tardif KD, Liu M, Vitseva O, Hou YM, Horowitz J: Misacylation and editing by Escherichia coli valyl-tRNA synthetase: evidence for two tRNA binding sites. Biochemistry. 2001, 40: 8118-8125. 10.1021/bi0103213.
Betha AK, Williams AM, Martinis SA: Isolated CP1 domain of Escherichia coli leucyl- tRNA synthetase is dependent on flanking hinge motifs for amino acid editing activity. Biochemistry. 2007, 46: 6258-6267. 10.1021/bi061965j.
Zhao MW, Zhu B, Hao R, Xu MG, Eriani G, Wang ED: Leucyl-tRNA synthetase from the ancestral bacterium Aquifex aeolicus contains relics of synthetase evolution. Embo J. 2005, 24: 1430-1439. 10.1038/sj.emboj.7600618.
Ambrogelly A, Ahel I, Polycarpo C: Methanocaldococcus jannaschii prolyl-tRNA synthetase charges tRNAPro with cysteine. J Biol Chem. 2002, 277: 34749-34754. 10.1074/jbc.M206929200.
Ruan B, Söll D: The bacterial YbaK protein is a Cys-tRNAPro and Cys-tRNACys deacylase. J Biol Chem. 2005, 280: 25887-25891. 10.1074/jbc.M502174200.
Chong YE, Yang XL, Schimmel P: Natural homolog of tRNA synthetase editing domain rescues conditional lethality caused by mistranslation. J Biol Chem. 2008, 283: 30073-30078. 10.1074/jbc.M805943200.
An S, Musier-Forsyth K: Trans-editing of Cys-tRNAPro by Haemophilus influenzae YbaK protein. J Biol Chem. 2004, 279: 42359-42362. 10.1074/jbc.C400304200.
Dwivedi S, Kruparani SP, Sankaranarayanan R: A D-amino acid editing module coupled to the translational apparatus in archaea. Nat Struct Mol Biol. 2005, 12: 556-7. 10.1038/nsmb943.
Ko YG, Kang YS, Kim EK, Park SG, Kim S: Nucleolar localization of human methionyl-tRNA synthetase and its role in ribosomal RNA synthesis. J Cell Biol. 2000, 149: 567-574. 10.1083/jcb.149.3.567.
Martinis SA, Plateau P, Cavarelli J, Florentz C: Aminoacyl-tRNA synthetases: a family of expanding functions. EMBO J. 1999, 18: 4591-4596. 10.1093/emboj/18.17.4591.
Cherniack AD, Garriga G, Kittle JD, Akins RA, Lambowitz AM: Function of Neurospora mitochondrial tyrosyl-tRNA synthetase in RNA splicing requires an idiosyncratic domain not found in other synthetases. Cell. 1990, 62: 745-755. 10.1016/0092-8674(90)90119-Y.
Sampath P, Mazumder B, Seshadri V, Gerber CA, Chavatte L, Kinter M, Ting SM, Dignam JD, Kim S, Driscoll DM, Fox PL: Noncanonical function of glutamyl-prolyl-tRNA synthetase: gene-specific silencing of translation. Cell. 2004, 119: 147-148. 10.1016/j.cell.2004.09.030.
Herzog W, Muller K, Huisken J, Stainier DYR: Genetic evidence for a noncanonical function of seryl-tRNA synthetase in vascular development. Circulation Research. 2009, 104: 1260-1266. 10.1161/CIRCRESAHA.108.191718.
Ramya TNC, Karmodiya K, Surolia A, Surolia N: 15-Deoxyspergualin Primarily Targets the Trafficking of Apicoplast Proteins in Plasmodium falciparum. J Biol Chem. 2007, 282: 6388-6397. 10.1074/jbc.M610251200.
Stoeckert CJ, Fischer S, Kissinger JC, Heiges M, Aurrecoechea C, Gajria B, Roos DS: PlasmoDB v5: new looks, new genomes. Trends Parasitol. 2006, 22: 543-546. 10.1016/j.pt.2006.09.005.
Francklyn C, Perona JJ, Puetz J, Hou YM: Aminoacyl-tRNA synthetases: Versatile players in the changing theater of translation. RNA. 2002, 8: 1363-1372. 10.1017/S1355838202021180.
Feng L, Sheppard K, Tumbula-Hansen D, Söll D: Gln-tRNAGln formation from Glu-tRNAGln requires cooperation of an asparaginase and a Glu-tRNAGln kinase. J Biol Chem. 2005, 280: 8150-8155. 10.1074/jbc.M411098200.
Tumbula DL, Becker HD, Chang WZ, Söll D: Domain-specific recruitment of amide amino acids for protein synthesis. Nature. 2000, 407: 106-110. 10.1038/35024120.
Sheppard K, Akochy PM, Salazar JC, Söll D: The Helicobacter pylori amidotransferase GatCAB is equally efficient in glutamine-dependent transamidation of Asp-tRNAAsn and Glu-tRNAGln. J Biol Chem. 2007, 282: 11866-11873. 10.1074/jbc.M700398200.
Schön A, Kannangara CG, Gough S, Söll D: Protein biosynthesis in organelles requires misaminoacylation of tRNA. Nature. 1988, 331: 187-190. 10.1038/331187a0.
Jahn D, Kim YC, Ishino Y, Chen MW, Söll D: Purification and functional characterization of the Glu-tRNA(Gln) amidotransferase from Chlamydomonas reinhardtii. J Biol Chem. 1990, 265: 8059-8064.
Kim SI, Stange-Thomann N, Martins O, Hong KW, Söll D, Fox TD: A nuclear genetic lesion affecting Saccharomyces cerevisiae mitochondrial translation is complemented by a homologous Bacillus gene. J Bact. 1997, 179: 5625-5627.
Sheppard K, Söll D: On the evolution of the tRNA-dependent amidotransferases, GatCAB and GatDE. J Mol Biol. 2008, 377: 831-844. 10.1016/j.jmb.2008.01.016.
Hauenstein SI, Perona JJ: Redundant synthesis of cysteinyl-tRNACys in Methanosarcina mazei. J Biol Chem. 2008, 283: 22007-22017. 10.1074/jbc.M801839200.
Sauerwald A, Zhu W, Major TA, Roy H, Palioura S, Jahn D, Whitman WB, Yates JR, Ibba M, Söll D: RNA-dependent cysteine biosynthesis in archaea. Science. 2005, 307: 1969-1972. 10.1126/science.1108329.
Fukunaga R, Yokoyama S: Structural insights into the second step of RNA-dependent cysteine biosynthesis in Archaea: crystal structure of Sep-tRNA:Cys-tRNA synthase from Archaeoglobus fulgidus. J Mol Biol. 2007, 370: 128-141. 10.1016/j.jmb.2007.04.050.
Quevillon S, Agou F, Robinson JC, Mirande M: The p43 component of the mammalian multi-synthetase complex is likely to be the precursor of the endothelial monocyte-activating polypeptide II cytokine. J Biol Chem. 1997, 272: 32573-32579. 10.1074/jbc.272.51.32573.
Behrensdorf HA, van de Craen M, Knies UE, Vandenabeele P, Clauss M: The endothelial monocyte-activating polypeptide II (EMAP II) is a substrate for caspase-7. FEBS Lett. 2000, 466: 143-147. 10.1016/S0014-5793(99)01777-9.
Faisal W, Symonds P, Panjwani S, Heng Y, Murray JC: Cell-surface associated p43/endothelial-monocyte-activating-polypeptide-II in hepatocellular carcinoma cells induces apoptosis in T-lymphocytes. Asian J Surg. 2007, 30: 13-22.
Shalak V, Kaminska M, Mitnacht-Kraus R, Vandenabeele P, Clauss M, Mirande M: The EMAPII cytokine is released from the mammalian multisynthetase complex after cleavage of its p43/proEMAPII component. J Biol Chem. 2001, 276: 23769-23776. 10.1074/jbc.M100489200.
Hopkins J, Fowler R, Krishna S, Wilson I, Mitchell G, Bannister L: The plastid in Plasmodium falciparum asexual blood stages: a three-dimensional ultrastructural analysis. Protist. 1999, 150: 283-295. 10.1016/S1434-4610(99)70030-1.
Lund E, Dahlberg JE: Proofreading and aminoacylation of tRNAs before export from the nucleus. Science. 1998, 282: 2082-2085. 10.1126/science.282.5396.2082.
Cooke BM, Lingelbach K, Bannister LH, Tilley L: Protein trafficking in Plasmodium falciparum -infected red blood cells. Trends Parasitol. 2004, 20: 581-589. 10.1016/j.pt.2004.09.008.
Hiller NL, Bhattacharjee S, van Ooij C, Liolios K, Harrison T: A host-targeting signal in virulence proteins reveals a secretome in malarial infection. Science. 2004, 306: 1934-1937. 10.1126/science.1102737.
Le Roch KG, Zhou Y, Blair PL, Grainger M, Moch JK, Haynes JD, De La, Vega P, Holder AA, Batalov S, Carucci DJ, Winzeler EA: Discovery of gene function by expression profiling of the malaria parasite life cycle. Science. 2003, 301: 1487-1488.
Bozdech Z, Llinas M, Pulliam BL, Wong ED, Zhu J, DeRisi JL: The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003, 1: e5-10.1371/journal.pbio.0000005.
O'Donoghue P, Luthey-Schulten Z: Evolution of Structure in Aminoacyl-tRNA synthetases. Microbiology and Molecular Biology Reviews. 2003, 67: 550-573. 10.1128/MMBR.67.4.550-573.2003.
Salazar JC, Ambrogelly A, Crain PF, McCloskey JA, Söll D: A truncated aminoacyl-tRNA synthetase modifies RNA. Proc Natl Acad Sci USA. 2004, 101: 7536-7541. 10.1073/pnas.0401982101.
An S, Musier-Forsyth K: Cys-tRNA(Pro) editing by Haemophilus influenzae YbaK via a novel synthetase.YbaK.tRNA ternary complex. J Biol Chem. 2005, 280: 34465-72. 10.1074/jbc.M507550200.
Dou X, Limmer S, Kreutzer R: DNA-binding of phenylalanyl-tRNA synthetase is accompanied by loop formation of the double-stranded DNA. Journal of Molecular Biology. 2001, 305: 451-458. 10.1006/jmbi.2000.4312.
Seror SJ, Casaregola S, Vannier F, Zoauri N, Dahl M, Boye E: A mutant cysteinyl-tRNA synthetase affecting timing of chromosomal replication initiation in B. subtilis and conferring resistance to a protein kinase C inhibitor. EMBO J. 1994, 13: 2472-2480.
Kim JY, Kang YS, Lee JW, Kim HJ, Ahn YH, Park H, Ko YG, Kim S: p38 is essential for the assembly and stability of macromolecular tRNA synthetase complex: implications for its physiological significance. Proc Natl Acad Sci USA. 2002, 99: 7912-7916. 10.1073/pnas.122110199.
Bec G, Kerjan P, Zha XD, Waller JP: Valyl-tRNA synthetase from rabbit liver. I. Purification as a heterotypic complex in association with elongation factor 1. J Biol Chem. 1989, 264: 21131-21137.
Bec G, Kerjan P, Waller JP: Reconstitution in vitro of the valyl-tRNA synthetase-elongation factor (EF) 1 beta gamma delta complex. Essential roles of the NH2- terminal extension of valyl-tRNA synthetase and of the EF-1 delta subunit in complex formation. J Biol Chem. 1994, 269: 2086-2092.
Brandsma M, Kerjan P, Dijik J, Janssen GM, Moller W: Valyl-tRNA synthetase from Artemia. Purification and association with elongation factor 1. Eur J Biochem. 1995, 233: 277-282. 10.1111/j.1432-1033.1995.277_1.x.
Kumar R, Musiyenko A, Oldenburg A, Adams B, Barik S: Post-translational generation of constitutively active cores from larger phosphatases in the malaria parasite, Plasmodium falciparum: implications for proteomics. BMC Mol Biol. 2004, 5: 6-10.1186/1471-2199-5-6.
He CY, Shaw MK, Pletcher CH, Striepen B, Tilney LG, Roos DS: A plastid segregation defect in the protozoan parasite Toxoplasma gondii. EMBO J. 2001, 20: 330-339. 10.1093/emboj/20.3.330.
Waller RF, McFadden GI: The apicoplast: a review of the derived plastid of apicomplexan parasites. Curr Issues Mol Biol. 2005, 7: 57-79.
Fichera ME, Roos DS: A plastid organelle as a drug target in apicomplexan parasites. Nature. 1997, 390: 407-409. 10.1038/37132.
Boyce JM: MRSA patients: proven methods to treat colonization and infection. J Hosp Infect. 2001, 48: S9-S14. 10.1016/S0195-6701(01)90005-2.
Rock FL, Mao W, Yaremchuk A, Tukalo M, Crépin T, Zhou H, Zhang YK, Hernandez V, Akama T, Baker SJ, Plattner JJ, Shapiro L, Martinis SA, Benkovic SJ, Cusack S, Alley MRK: An Antifungal Agent Inhibits an Aminoacyl-tRNA Synthetase by Trapping tRNA in the Editing Site. Science. 2007, 316: 1759-1761. 10.1126/science.1142189.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
Stalker J, Gibbins B, Meidl P, Smith J, Spooner W, Hotz HR, Cox AV: The Ensembl Web Site: Mechanics of a Genome Browser. Genome Res. 2004, 14: 951-955. 10.1101/gr.1863004.
Wilson D, Pethica R, Zhou Y, Talbot C, Vogel C, Madera M, Chothia C, Gough J: SUPERFAMILY - Comparative Genomics, Datamining and Sophisticated Visualisation. Nucleic Acids Res. 2009, 37: 380-386. 10.1093/nar/gkn762.
Letunic I, Doerks T, Bork P: SMART 6: Recent updates and new developments. Nucleic Acids Res. 2008, 37: 229-232. 10.1093/nar/gkn808.
Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL: The Pfam protein families database. Nucleic Acids Res. 2002, 30: 276-280. 10.1093/nar/30.1.276.
Hughey R, Krogh A: Hidden Markov models for sequences analysis: Extension and analysis of the basic method. Computer Applications in the Biosciences. 1996, 12: 95-107.
Szymanski M, Deniziak MA, Barciszewski J: Aminoacy-tRNA synthetases database. Nucleic Acids Res. 2001, 29: 288-290. 10.1093/nar/29.1.288.
Claros MG, Vincens P: Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem. 1996, 241: 779-786. 10.1111/j.1432-1033.1996.00779.x.
Cokol M, Nair R, Rost B: Finding nuclear localization signals. EMBO reports. 2000, 1: 411-415. 10.1093/embo-reports/kvd092.
Zuegge J, Ralph S, Schmuker M, McFadden GI, Schneider G: Deciphering apicoplast targeting signals - feature extraction from nuclear-encoded precursors of Plasmodium falciparum apicoplast proteins. Gene. 2001, 280: 19-26. 10.1016/S0378-1119(01)00776-4.
Irirzarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP: Exploration, normalization and summaries of high densities of oligonucleotide array probe level data. Biostatistics. 2003, 4: 249-264. 10.1093/biostatistics/4.2.249.
Troyanskaya O, Cantor M, Sherlock G: Missing value estimation method for DNA microarrays. Bioinformatics. 2001, 17: 520-525. 10.1093/bioinformatics/17.6.520.
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22: 4673-4680. 10.1093/nar/22.22.4673.
Stéphane G, Franck L, Patrice D, Olivier G: PHYML Online--a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 2005, W557-W559.
Kumar S, Dudley J, Nei M, Tamura K: MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Briefings in Bioinformatics. 2008, 9: 299-306. 10.1093/bib/bbn017.
Renom MA, Stuart A, Fiser A, Sánchez R, Melo F, Sali A: Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000, 29: 291-325. 10.1146/annurev.biophys.29.1.291.
Laskowski RA, MacArthur MW, Moss DS, Thornton JM: PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993, 26: 283-291. 10.1107/S0021889892009944.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE: UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J Comput Chem. 2004, 25: 1605-1612. 10.1002/jcc.20084.
DeLano WL: The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA . 2002, [http://www.pymol.org]
Lasonder E, Ishihama Y, Andersen JS, Vermunt AM, Pain A, Sauerwein RW, Eling WM, Hall N, Waters AP, Stunnenberg HG, Mann M: Analysis of the Plasmodium falciparum proteome by high-accuracy mass spectrometry. Nature. 2002, 419: 537-542. 10.1038/nature01111.
Acknowledgements
TKB, CK and AS are supported by grants from the Department of Biotechnology, Govt. Of India. SK is supported by MEPHITIS grant. This work has been conducted as part of MEPHITIS project and partially funded by the European Commission (Grant Agreement no: HEALTH-F3-2009-223024).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
TKB, CK and SK carried out the computational experiments and data analysis and wrote the paper; MAJ and VS contributed to the manuscript writing; DS and EP carried out HMM construction and database search by HMM; FS performed analysis of transcriptomic and proteomic data; AS designed the study and supervised the work. All authors have read and approved the final manuscript.
Electronic supplementary material
12864_2009_7087_MOESM1_ESM.PDF
Additional file 1:List of Pf-aaRSs categorized into class I, class II, and related proteins. Gene ID, gene location, description of product and its length are given. (PDF 44 KB)
12864_2009_7087_MOESM2_ESM.PDF
Additional file 2:Phylogenetic trees of aaRSs from P. falciparum. The evolutionary tree was constructed by the method PHYML using the MEGA 4.0. P. falciparum aaRSs are labeled green triangles. 102 species considered for the evolutionary analysis are taken from the three domains of life viz. P. berghei, P. chabaudi, P. falciparum, P. knowlesi, P. yoelii, P. vivax, H. sapiens, M. tuberculosis, D. discoidium, M. jannaschii, R. norvegicus, C. parvum, B. bovis, S. cerevisiae, D. melanogaster, Y. pestis, T. aquaticus, S. pneumoniae, S. entrica, E. coli, A. thaliana, A. pisum, A. salmonicida, B. cereus, B. thuringiensis, B. afzelii, B. burgdorferi, B. garinii, B. valaisiana, Bradyrhizobium, B. pennsylvanicus, C. acidaminovorans, H. defensa, C. taiwanensis, E. fergusonii, F. bacterium, F. novicida, F. tularensis, F. alni, G. tenuistipitata, H. arsenicoxydans, A. cellulolyticus, A. chlorophenolicus, A. ferrooxidans, Algoriphagus, A. muciniphila, Anaeromyxobacter, A. thermophilum, B. ambifaria, B. indica, B. mycoides, B. taurus, B. tribocorum, C. atlanticus, Caulobacter, C. aurantiacus, C. cellulolyticum, Citrobacter, C. pinensis, C. Ruthia, Cyanothece, D. desulfuricans, D. hafniense, Diaphorobacter, D. shibae, D. turgidum, E. cuniculi, E. lenta, E. ruminantium, Exiguobacterium, G. diazotrophicus, Geobacillus, M. maris, N. multipartita, Nocardioides, O. terrae, P. abelii, P. atlantica, P. denitrificans, P. ingrahamii, P. lavamentivorans, R. castenholzii, S. arenicola, S. fumaroxidans, X. autotrophicus, V. vadensis, V. paradoxus, T. whipplei, T. auensis, S. stellata, Ch. parvum, S. heliotrinireducens, Silicibacter, S. putrefaciens, S. usitatus, Thauera, X. laevis, Theileria annulata, Vibrio fischeri, W. succinogenes, X. tropicalis, Zeamays. (PDF 5 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Bhatt, T.K., Kapil, C., Khan, S. et al. A genomic glimpse of aminoacyl-tRNA synthetases in malaria parasite Plasmodium falciparum. BMC Genomics 10, 644 (2009). https://doi.org/10.1186/1471-2164-10-644
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-10-644