
Abstract
Background
There are thousands of very diverse ciliate species from which only a handful mitochondrial genomes have been studied so far. These genomes are rather similar because the ciliates analysed (Tetrahymena spp. and Paramecium aurelia) are closely related. Here we study the mitochondrial genomes of the hypotrichous ciliates Euplotes minuta and Euplotes crassus. These ciliates are only distantly related to Tetrahymena spp. and Paramecium aurelia, but more closely related to Nyctotherus ovalis, which possesses a hydrogenosomal (mitochondrial) genome.
Results
The linear mitochondrial genomes of the hypotrichous ciliates Euplotes minuta and Euplotes crassus were sequenced and compared with the mitochondrial genomes of several Tetrahymena species, Paramecium aurelia and the partially sequenced mitochondrial genome of the anaerobic ciliate Nyctotherus ovalis. This study reports new features such as long 5'gene extensions of several mitochondrial genes, extremely long cox1 and cox2 open reading frames and a large repeat in the middle of the linear mitochondrial genome. The repeat separates the open reading frames into two blocks, each having a single direction of transcription, from the repeat towards the ends of the chromosome. Although the Euplotes mitochondrial gene content is almost identical to that of Paramecium and Tetrahymena, the order of the genes is completely different. In contrast, the 33273 bp (excluding the repeat region) piece of the mitochondrial genome that has been sequenced in both Euplotes species exhibits no difference in gene order. Unexpectedly, many of the mitochondrial genes of E. minuta encoding ribosomal proteins possess N-terminal extensions that are similar to mitochondrial targeting signals.
Conclusion
The mitochondrial genomes of the hypotrichous ciliates Euplotes minuta and Euplotes crassus are rather different from the previously studied genomes. Many genes are extended in size compared to mitochondrial genes from other sources.
Similar content being viewed by others


Background
Ciliates, unicellular eukaryotes, are extremely species-rich and colonize a very broad spectrum of ecological niches. They are characterized by complexes of cilia, used for swimming and food capturing and by a nuclear dimorphism that is unique for ciliates. All members possess a micronuclear genome, which is active in sexual reproduction, and a macronuclear genome that is transcriptionally active during somatic development and maintenance. In addition to the macronuclear and micronuclear genomes, aerobic ciliates also possess a mitochondrial genome. Although there are thousands of different ciliate species, only six mitochondrial genomes have been completely sequenced and analyzed thus far: P. aurelia [1] and five Tetrahymena species: T. pyriformis, T. thermophila, T. pigmentosa, T. paravorax and T. malaccensis [2–4]. Only minor differences between the mitochondrial genomes of the Tetrahymena species were found. The mitochondrial genomes of P. aurelia and Tetrahymena species are also very similar; only two large blocks of genes are shifted between them but within these blocks the gene order is conserved. A third smaller block, containing the split mitochondrial ribosomal rnl gene, is duplicated in Tetrahymena but not in Paramecium. Although most of the sequenced mitochondrial genomes are circular mapping, the mitochondrial genomes of Paramecium and Tetrahymena are monomeric linear and capped with telomeres. No mitochondrial genomes have been sequenced in the order of hypotrichous ciliates that contain intensively studied species such as Oxytricha and Stylonichia as well as Euplotes, a genus widely distributed in freshwater and seawater environments. The two Euplotes species studied here (E. crassus and E. minuta) are both marine ciliates that were collected in the Mediterranean sea.
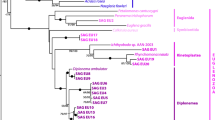
We investigated the mitochondrial genome organization of Euplotes for three reasons: firstly, because Euplotes is only distantly related to P. aurelia and the various Tetrahymena species, the only species from which mitochondrial genomes have been studied so far (Figure 1). Secondly, because Euplotes contains two morphologically different types of mitochondria, which might possess different genomes [5, 6] and thirdly, because we assumed that Euplotes is more closely related to Nyctotherus ovalis than Tetrahymena or Paramecium. Phylogenetic analysis, however, did not support this assumption because of lacking statistical support (Figure 1). Nevertheless, it is likely that its organellar genome is closely related to the hydrogenosomal genome of Nyctotherus ovalis, which exhibits characteristics of a ciliate mitochondrial genome and significant sequence similarity to certain Euplotes genes. Nyctotherus ovalis, which thrives in the hindgut of various cockroach species, has been investigated extensively, but only 14 kb of its hydrogenosomal genome have been sequenced so far [7–10]. Here, we show that the mitochondrial genomes of E. crassus and E. minuta are linear with a large repeat region in the middle that is potentially involved in transcription initiation. The gene content of the Euplotes genome is almost identical to that of Paramecium and Tetrahymena, but the order of the genes is completely different. We discuss the observation that Euplotes contains extremely large cox genes and several other mitochondrial genes with large extensions. It is shown that several N-terminal extensions of the mitochondrial genes have the potential to function as mitochondrial import signals.

Taxonomy of ciliates. A maximum likelihood phylogeny from selected 18S rRNA genes. Only bootstrap values equal or larger than 90/100 are indicated.
Results and Discussion
Structure of the mitochondrial genomes
The linear mitochondrial DNA of E. minuta has been completely sequenced with exclusion of the telomeres and a repeat region of more then 1000 base pairs that is located almost in the middle of the mitochondrial genome. The length of the sequenced mitochondrial genome of E. minuta clearly exceeds 41600 bp, while 33273 bp (excluding the repeat region) of the mitochondrial genome of E. crassus have been sequenced (Figure 2). The length of the telomeres can only be estimated roughly since it is known from investigation of 5 different Tetrahymena species that the composition and length of mitochondrial telomeres can differ enormously [11, 12]. Also, in three Tetrahymena species the terminal repeats at both ends of the mitochondrial DNA are completely different. Moreover, analysis of the mitochondrial genome of T. malaccensis has shown that telomeric lengths can vary between 700 and 4200 bp with an average size of 2600 bp. [12]. Since pulsed field gel electrophoresis of E. minuta DNA has indicated that the total length of the mitochondrial genome is clearly less than 48 Kb (Figure 3), it is likely that we have sequenced the total mitochondrial genome with exception of the telomeres. This interpretation is supported by the observation that chromosome walking using organelle DNA failed to provide evidence for the presence of additional DNA at the ends of the sequenced mitochondrial genome.

Mitochondrial gene map of Euplotes minuta and Euplotes crassus. Red: Complex I genes, blue: rRNA genes, green: ribosomal proteins, yellow: Complex III and IV genes, grey: unidentified open reading frames, pink: repeat region, dark grey: atp9 gene, white: intergenic spacers. Capital letters indicate the various tRNA genes. Arrows: direction of transcription.

Pulsed field gel electrophoresis of genomic DNA of Euplotes minuta. Lanes 1 and 11 contain lambda concatamer (marker). Lanes 2-10 contain genomic DNA of Euplotes minuta. The mitochondrial band (arrow) is located just below the one lambda band (48 Kb).
The central repeat region is made up from 18-bp units that are palindromic except for the positions 3-4/15-16. The repeat units are identical in E. crassus and E. minuta (Figure 4).

Structure of the central repeat unit. The repeat unit is palindromic except for the positions 3-4/15-16. It is identical in E. crassus and E. minuta.
Because the direction of transcription of all mitochondrion encoded genes is away from this repeat region (Figure 2), we tested whether the palindrome exhibits significant sequence similarity to any known transcription factor binding site using the online motif comparison tool Tomtom [13]. As expected, no significant levels of sequence similarity were found (E-values > 0.1). Notably, it has been observed that unrelated A-T rich repeat units serve as autonomously replicating sequences in the mitochondrial DNA of Paramecium and Tetrahymena; these units are located at one end of the mitochondrial chromosome, close to the telomeric repeats [14–16]. Other examples of repeat regions in mitochondrial genomes of protists are found in the cryptophyte algae Rhodomonas salina [17] and Hemiselmis andersenii [18]. Both mitochondrial genomes contain a large complex repeat region that seems to play a role in transcription. However, the mitochondrial genomes of these unicellular cryptophyte algae are not linear but circular mapping.
The overall A+T content of the mitochondrial genome of E. minuta, (64.0%), is much lower than in T. pyriformis (78.7%) but significantly higher than in P. aurelia (59.0%) [1]. Genes are tightly packed and the intergenic regions (4.1% of the genome) are generally short (ranging from 1 to 385 nucleotides, with an average size of 66 nucleotides). These intergenic regions have an overall A+T content of 68.9% which is hardly higher than in the coding areas. We found eight cases where the orf s overlap (9-96 bp.) and no gene duplication. One gene (nad 1) was split into two parts that are located on different positions of the genome.
The overall A+T content of the sequenced part of the mitochondrial genome of E. crassus is 65.3%. The genes in the mitochondrial genome of E. crassus are also tightly packed and intergenic spacers (4.2%) have a length of 2 to 413 nucleotides with an average size of 77 nucleotides and an A+T content of 68.4%. Overlapping orf s were identified in 12 cases with overlaps varying in size from 3 to 100 base pairs.
The mitochondrial genes of Euplotes minuta and Euplotes crassus
The mitochondrial DNA of E. minuta includes 12 protein coding genes involved in the electron transport chain, 7 ribosomal protein coding genes, 2 ribosomal RNA genes, 7 transfer RNA genes, and one gene that encodes a cytochrome c assembly protein (ccmF/jeyR) (Table 1). Finally, 15 orf s were found with no detectable sequence similarity to known genes (Table 2). The sequenced part of the mitochondrial genome of E. crassus contains 10 genes of the electron transport chain, 6 ribosomal protein coding genes, 2 ribosomal RNA genes, 5 transfer RNA genes, the ccmF/jeyR gene and 11 orf s with significant sequence similarity to E. minuta genes, but no detectable sequence similarity to other known genes (Table 1, 2).
There are no differences in gene order between the closely related E. crassus and E. minuta, but their gene order is completely different than that of the Tetrahymena species and P. aurelia (Figure 2). Only four genes could be found that have a conserved order in all these ciliate species: rpl2-orf-nad10-rps12.
Genes encoding components of the electron transport chain
As shown in Table 1 all mitochondrion-encoded Complex I genes that were found in T. pyriformis and P. aurelia [4], were also found in E. minuta with the exception of nad6/ymf62 that was identified as nad6 in T. pyriformis [3]. The mitochondrial genomes of all sequenced Tetrahymena species possess nad6/ymf62, which exhibits a significant sequence similarity with orf 265 in P. aurelia.
In all Tetrahymena species and in P. aurelia the Complex I gene nad1 is split into a larger A and a smaller B part, which is located on the opposite strand. In E. minuta this gene is also split but located on the same strand. In E. crassus the corresponding part of the mitochondrial genome has not been sequenced (Figure 2).
The length of the nad2 gene of T. pyriformis, P. aurelia and N. ovalis (var. Bla. Ams) is almost the same but about 180 amino acids smaller than the nad2 gene of Bos taurus (cow). In contrast, the nad2 genes of both Euplotes species have large N terminal extensions. The nad2 gene of E. crassus has an extension of about 250 amino acids, and the homologous gene of E. minuta is around 500 amino acids longer. These extensions have no detectable sequence similarity to each other or to other known genes.
The Complex I gene nad4L has been identified by Brunk et al. in T. thermophila [3] (named ymf 58 in the other mitochondrial genomes of Tetrahymena species), in the hydrogenosomal genome of the anaerobic ciliate N. ovalis and in both Euplotes species (Table 1). It has not been annotated in Paramecium, but alignments of orf113 in P. aurelia with the Tetrahymena species and with the nad4L gene of N. ovalis shows that this orf113 is homologous to nad4L [10]. The nad7 genes of both Euplotes species are highly conserved; both have a small N-terminal extension (19 and 36 amino acids, respectively). These extensions are not similar to each other and are not found in other ciliates.
The only Complex III gene that is found in the mitochondrial genomes of the Tetrahymena species and P. aurelia is cytochrome-b (cob), which has also been identified in both Euplotes species (Table 1; Figure 2). The cob genes of both Euplotes species possess small N-terminal extensions that are not conserved while the remaining part of the gene is very well conserved. The Complex IV genes cytochrome oxidase 1 and 2 (cox1 and cox2), are found in all Tetrahymena species, in P. aurelia and in both Euplotes species (Table 1; Figure 2). As shown earlier in T. pyriformis and P. aurelia both genes contain large (in frame) upstream open reading frames [4]. In Euplotes the cox2 frames reach extreme lengths, 1021 amino acids in E. crassus and 1017 amino acids in E. minuta (Figure 5a, b). The insert does not show significant similarity to any known gene, precluding the inference of function and functional constraints by sequence similarity. The sequencing of two Euplotes species, however, allows us to assess whether there is any selection on the protein coding sequence by calculating the ratio of non-synonymous over synonymous substitutions (dn/ds) and test for protein sequence conservation. Figure 6a shows the ClustalW alignment [19] between the Cox2 proteins in Euplotes and several other ciliate species. There are only two regions that could be unequivocally aligned among all the ciliates and of which the alignment did not depend on the program used [20, 21]. These regions are indicated by the high conservation and quality bars in Figure 6. They are also highly conserved between the two Euplotes sequences (dn = 0), and overlap with the regions for which we detected likely sequence similarity with the PFAM Cox2 domain, albeit with an insignificant E-value for the N-terminal part [22]. In contrast, there appear to be less constraints on the primary sequence of the ~700 amino acid in-frame cox2 insert (65% identity between the two Euplotes sequences). For cox1, we found a similar situation (see Additional file 1: Fig. S1 and Additional file 2:Fig. S2) When the Cox1 protein sequences of both Euplotes species are compared with the Cox1 protein sequence of Bos taurus a large insert in frame of 380 amino acids was identified between positions 119-120. T. pyriformis and P. aurelia possess an insert of 271 amino acids in exactly the same position. Furthermore, it seems that the cox1 genes of T. pyriformis and both Euplotes species contain N-terminal extensions of about 40 amino acids. The N-terminal extension in P. aurelia is a bit longer, about 83 amino acids. The N-terminal extensions of the cox1 gene in both Euplotes species and in P. aurelia harbour a potential mitochondrial import signal that has been identified by the program Mitoprot [23]. In a recent publication [24] it was observed that latent mitochondrial targeting signals are present on the mitochondrial genomes of Arabidopsis thaliana and Oryza sativa. It is possible that some of the N-terminal extensions we find in Euplotes spp. play a role as latent mitochondrial targeting signals. Alternatively, they could function as an internal localization signal, resulting from a bias in nucleotide alteration, or even hint at the possibility of back-transfer of genes from the nucleus to the organelle [25]. Furthermore, the cox1 gene of E. minuta possesses a C-terminal extension (267 amino acids) that has not been found in the other ciliates, including E. crassus.

Length of cox1 and cox2 open reading frames (in amino acids) from E. crassus and E. minuta compared to other organisms.

Alignment analysis of Euplotes cox2. a) ClustalW alignment of cox2 from other ciliates and Euplotes; b) conservation, quality and consensus scores of the multiple alignment in (a) according to Jalview; c) Pfam search result including an insignificant hit to the Cox2 Pfam domain in the N-terminal conserved region of the gene; d) number of non-synonymous (ds) and synonymous (ds) base substitutions observed between E. minuta and E. crassus per 69 nt sliding window; e) dn/ds ratio based on (d).
Another cytochrome c related gene, ccmF/yejR, is also found in both Euplotes species (Table 1). It is a cytochrome c assembly protein that encodes the potential catalytic subunit of cytochrome c lyase. There is a large difference in the lengths of the ccmF/yejR genes between these ciliates. T. pyriformis (513 amino acids) and E. minuta (461 amino acids) have a large C-terminal extension. The corresponding extensions in P. aurelia (256 amino acids) and E. crassus (243 amino acids) are significantly smaller.
Only one Complex V gene, ATPase 9, has been identified in E. minuta. It is also located on the mitochondrial genomes of T. pyriformis and P. aurelia (Table 1). The corresponding region in E. crassus was not sequenced.
Ribosomal proteins
Mitochondrial genes encoding mitochondrial ribosomal proteins are common in plants and protists but have never been found in the mitochondrial genomes of animals. Ciliates possess a limited number of ribosomal proteins on their mitochondrial genomes. So far 7 ribosomal proteins have been identified in E. minuta (Table 1). Another ribosomal protein, rpl14 from E. crassus, which is present in all other ciliate mitochondrial genomes, has an N-terminal extension (33 amino acid) that has no significant similarity with other known proteins. Similar extensions were observed for the rpl16, rps4 and rps12 genes in both ciliates E. crassus and E. minuta (Table 3). When the mitochondrial genes were examined with the mitochondrial import signal prediction program Mitoprot [23] we found high scoring hits for all ribosomal proteins in E. minuta and all ribosomal proteins in E. crassus except one. All these ribosomal proteins also contained a predicted cleavage site. Analyses of the mitochondrial ribosomal proteins of T. pyriformis and P. aurelia indicated that also some of these proteins possessed a potential import signal. An analysis based on the signal prediction programs Predotar [26] and TargetP [27] gave less hits but still identified a significant number of potential mitochondrial import signals (Table 3).
tRNA genes
Among eukaryotes the number of mitochondrial-encoded tRNA genes varies from 26 tRNAs in Reclinomonas americana to zero in apicomplexa [28]. Seven different tRNA genes were identified in the mitochondrial genome of E. minuta (trnE, trnF, trnH, trnM, trnY, trnQ and trnW) in contrast to only four such tRNA genes in P. aurelia (trnF, trnM, trnW and trnY) (Table 1) [1]. In E. crassus only 5 tRNA genes were identified. Also, in T. pyriformis a set of seven tRNA genes were identified i.e. trnE, trnF, trnH, trnL, trnM, trnW and trnY [4]. The mitochondrial-encoded tRNA for Glutamine (trnQ) seems to be unique for Euplotes, since it was not identified in either T. pyriformis or in P. aurelia; trnL is duplicated in T. pyriformis. Two different programs (tRNAscan-SE and ARAGORN) did not detect a tRNA for tryptophan (W); instead, this tRNA was identified as a tRNA for selenocystein. Recently however, the presence of trnW in the mitochondrial genome of E. crassus was experimentally confirmed by Turanov et al. [29].
Open reading frames
Additional 17 orf s have been identified in E. minuta and 13 orf s in E. crassus (Table 2). One orf (rps3) of E. minuta and E. crassus has, after BlastX and BlastN searches, detectable sequence similarity with orf s from T. pyriformis and P. aurelia (ymf64/orf234). In T. thermophila the gene ymf64 has been identified as a putative ribosomal protein, based on physicochemical parameters of the predicted protein [3]. Comparison of an alignment of the ymf64 homologs in the ciliates with the Hidden Markov Models (HMMs) in PFAM, using the sensitive profile-profile based homology detection tool HHsearch [30] indicates that ymf64 exhibits significant sequence similarity with the C-terminal domain of the ribosomal protein S3 (P < 2.1 E-5, Additional file 3: Fig. S3). An HMM of the genes that are currently annotated as rps3 in the Tetrahymena species and in P. aurelia indicated that they are homologous to the N-terminal domain of the ribosomal protein S3 (P < 2.1 E-5). The gene length of ymf64 in T. thermophila is 330 amino acids; in P. aurelia (orf234) it has a length of 234 amino acids. The orthologous Euplotes genes are much larger (767 and 768 amino acids, respectively). We could not detect significant sequence similarity of the S3 N-terminal domain to any of the Euplotes sequences.
For the remaining 16 orfs in E. minuta and 12 in E. crassus no homologous genes were found using BlastX and BlastN searches. However, one of these, orf267(orf 297 in E. crassus), which is part of the conserved region of four genes in Euplotes spp., is weakly conserved when compared to orf161/ymf74 in T. pyriformis and orf178-2/ymf84 in P. aurelia (Table 2)
Mitochondrial ribosomal RNA genes
The mitochondrial large and small subunit ribosomal RNA genes in five Tetrahymena species and in P. aurelia are split into two pieces [4]. In all these Tetrahymena species the rnl gene is duplicated. Analysis of the mitochondrial genomes of E. minuta and E. crassus by BlastN identified the regions were the rnl and rns genes are situated. The rnl gene in Euplotes species is not duplicated as in the Tetrahymena species. Even by sensitive Smith-Waterman queries [31] with selected parts of the rnl and rns sequences from other ciliate species, we did not find any indication that these genes were split in Euplotes (not shown). Both, the region containing the putative rnl and the region containing the putative rns, have significant sequence similarity to the rnl and the rns of the published mitochondrial ciliate ribosomal RNAs. Nevertheless, the regions of significant sequence similarity do not cover the complete published ribosomal RNAs, prohibiting complete sequence alignment and therewith assessment as to whether these RNAs are complete or interrupted. As expected, a 5S rRNA gene could not be identified.
Genetic code
Analysis of the codon usage as described in the Methods section confirmed that both Euplotes species use the protozoan mitochondrial code, with TGA encoding tryptophan. There are a few spurious predictions (TCG, ATG and ACC in E. crassus and TAG, ATT, ATG and ACC in E. minuta), but for all these cases we find the correct translation at an almost equal score. The prediction that TAG would code for a serine in E. minuta is only based on a single aligned occurrence of the codon, caused possibly by a sequence error or a misalignment (not shown).
Conclusion
When the mitochondrial genome of T. pyriformis was published and compared with that of P. aurelia, it seemed that the mitochondrial gene order in ciliates was very well conserved [4]. With the determination of the mitochondrial genome of a third ciliate genus, belonging to a completely different taxon, we have shown that the gene order in mitochondrial genomes of ciliates can be very different while a similar set of genes is conserved. Also the linearity of the mitochondrial chromosomes is conserved. This might suggest that monomeric linear mitochondrial chromosomes, which are relatively rare among protozoa and animals [32], are characteristic for ciliates. This possibility is corroborated by the observation that also species belonging to the sister taxon apicomplexa possess linear mitochondrial chromosomes [33]. However, it should be noted that among yeasts even the mitochondrial genomes of closely related species differ with respect to their linearity/circularity [32].
From the 17 unidentified open reading frames in the mitochondrial genome of Euplotes minuta two could be found with significant sequence similarity to T. pyriformis and P. aurelia (Table 2). This contrasts with the situation in T. pyriformis and P. aurelia where 13 out of the 22 unidentified open reading frames in T. pyriformis were also found in P. aurelia [4]. One of these orfs, ymf64, has now, with the aid of the Euplotes sequences and profile based homology searches been shown to be significantly similar to a known protein domain, the C-terminal part of the Rps3 protein. This suggests that with the sequencing of more mitochondrial genomes of the ciliates also for other orfs sequence similarity might be detected with known mitochondrial genes.
One of the rare mitochondrial features present in the mitochondrial genomes of T. pyriformis and P. aurelia is a split nad1 gene. This split gene has also been identified in Euplotes and thus seems to be specific for a large group of (maybe all) ciliates.
One of the most striking differences between the mitochondrial genomes of Euplotes and those of Tetrahymena species and P. aurelia is the presence of a large repeat region in the middle of the mitochondrial genome of both Euplotes species that seems to be used as a bi-directional transcription start. No such repeat was found in Tetrahymena species and P. aurelia and, in contrast to Euplotes, the transcription direction changes several times.
Another striking feature of the genes in the mitochondrial genome of Euplotes species is the presence of very large open reading frames. Most of these large orf s contain N-terminal extensions, but in some cases, like the cox1 and cox2 genes, large inserts in frame cause this effect. Such inserts in frame were also detected in Tetrahymena sp. and P. aurelia. Surprisingly, all of the N-terminal extensions of genes encoding ribosomal proteins of Euplotes minuta contain a potential targeting signal for import into mitochondria. This is the first report identifying such import signals in mitochondrial-encoded genes in organisms other than plants.
Sequencing and analyzing the mitochondrial genomes of E. crassus and E. minuta shows that the mitochondrial genomes of ciliates are rearranged more extensively than previously thought. Sequencing of the mitochondrial genome of E. minuta also did not provide any evidence for the presence of a slightly deviating, alternative genome that might be expected for the two morphs of mitochondria observed in this species. Studying these mitochondrial genomes has provided additional information about the evolution of mitochondria in general and in particular about the evolution of the elusive hydrogenosomal genome of Nyctotherus ovalis [10], which appeared to be more related to the mitochondrial genome of Euplotes than to those of Paramecium and Tetrahymena.
Methods
E. minuta cells were collected in 2005 in the Mediterranean sea near Stareso, Corsica, France (Em. S1, E. minuta Stareso1), cultured in the laboratory in artificial sea water obtained from the Botanical and Zoological Garden Stuttgart (Wilhelma) and fed with Klebsiella minuta grown on nutrient agar. For the isolation of DNA, a concentrated sample of living cells was mixed with 8 M guanidiniumchloride. A 10:1 mixture with 1 M phosphate buffer pH 7.0 was made, adsorbed on a hydroxyapatite (Biorad, bio-gel HTP) column (1 cm × 0.4 cm) and washed with 4 M guanidiniumchloride, 100 mM phosphate buffer pH = 7.0, followed by washing with 4 M guanidiniumchloride, 200 mM phosphate buffer pH = 7.0. Subsequently, the bulk of DNA was eluted with 4 M guanidiniumchloride 500 mM phosphate buffer pH = 7.0. The DNA was diluted with 1 volume water and precipitated with 10 v/v% 3 M sodiumactetate pH = 5.2 and 50 v/v % propanol-2 for 10 minutes at room temperature. After precipitation and washing the pellet was air dried. Finally, the DNA pellet was dissolved in DEPC treated water (Invitrogen).
The dissolved DNA was loaded on a pulsed field agarose gel (1% agarose type II medium EEO, Sigma) and run at 170 V (145 mA) ramping from 2.5 s - 25 s for 16 hours PFGE with a LKB 2015 Pulsaphor plus control unit.
The band just below the first band of the lambda marker (Figure 3) was cut out and the DNA extracted. The position of the mitochondrial band on pulsed field gel is a clear indication of a linear mitochondrial genome. Circular mitochondrial genomes of this size should run much faster in the gel. The DNA of the band was digested with Sau 3A and then size fractionated on an agarose gel. The DNA from these fractions was isolated from the gel, ligated in pUC-18 digested with BamH1 and transformed in E. coli DH101B cells. The titre of the library was 1.12 × 105. From this library, plasmid DNA from 288 different colonies was sequenced with an ABI prism 3730 online capillary sequencing machine and the mitochondrial genome was assembled as described below. The gene library was constructed by Genterprise, Mainz, Germany.
E. crassus was collected from shallow coastal waters of the sandy beach of Porto Recanati (43° 26' N, 13° 40' E) on the Italian Adriatic Coast, 50 km south of Ancona, July 1984 and cultured in the laboratory in artificial sea water (NaCl 465 mM, KCl 10 mM, MgCl2.6H2O 24.8 mM, MgSO4.7H2O 28.1 mM, CaCl2 10.4 mM, NaHCO3 2.4 mM pH 8.0).
Initially, a culture was kept in artificial seawater in an Erlenmeyer flask and fed with a small piece of raw beef. Alternatively, a set of 200 ml tissue flasks was first siliconized, filled with approximately 50 ml of artificial seawater, and inoculated with E. crassus cells. These cultures were fed with HB101 E. coli cells.
Total DNA of E. crassus was isolated by dissolving cells in 8 M guanidiniumchloride and purification by hydroxyapatite as described above for E. minuta.Four fragments of different mitochondrial genes were obtained by PCR with degenerated primers on this DNA, i.e. primers directed against the ribosomal genes rnl (5'-GTCAAGAGAGAAACAGC-3', 5'-GCATAGGGTCTTCCCGTC-3'), rns (5'-TGTGCCAGCAGCCGCGGTAA-3', 5'-TCCCMTACCRGTACCTTGTGT-3') and the complex I genes nad7 (5'-TTCGGWCCHCARCAYCCHGC-3', 5'-CTRTCRACYTCWCCRAARAC-3') and nad10 (5'-TTYGGHYTNGCHTGHTG-3', 5'-ARDGCYTCDSWDGTDGGDGGDCA-3') On these gene fragments primers for long range PCR were developed and long range PCR with LA-Taq-polymerase (5 U/μl) (Takara bio inc.) was performed. The long range PCR products were digested with different restriction enzymes, subcloned in pUC-18 (Sigma) or in pGEM-T easy (Promega) and sequenced. Sequencing was performed at the DNA diagnostics centre of the Nijmegen University Medical Center using M13 forward and reverse primers.
All sequences have been submitted to NCBI GenBank. The GenBank accession-numbers are for E. minuta GQ903130 and for E. crassus GQ903131. The protein identifiers are displayed in additional file 4.
Analysis of the sequence data
Sequences were edited using chromas Lite 2.01 http://www.technelysium.com.au The edited sequences were assembled using BioEdit version 7.0.9.0 [34]. Open reading frames were identified with orf Finder http://www.ncbi.nlm.nih.gov/gorf/gorf.html. tRNAs were identified with tRNAscan-SE http://lowelab.ucsc.edu/tRNAscan-SE/ and ARAGORN [35]. Sequence similarity searches of deduced amino acid sequences were performed with BLASTX and BLAST2 [36]. The nucleotide sequence similarity searches were conducted with BLASTN (NCBI) and FASTA (EMBL-EBI). Import signal prediction was done with Mitoprot [23], Predotar [26] and TargetP [27]. Alignments were made with ClustalX2 and ClustalW [37]. The program Nucleic Acid Dot Plots http://www.vivo.colostate.edu/molkit/dnadot/index.html was used for identifying repeat structures.
The sequences for the 18s rRNA phylogeny were aligned using the SINA Webaligner http://www.arb-silva.de/aligner, which aligns them in accordance with the ARB/SILVA rRNA alignment [38] which is based on a secondary structure model [39]. Subsequently we used Gblocks [40] to identify reliably aligned parts, using the default settings except that we did not require the coverage for every position to be 100%, but rather 80%. We then used PhyML v3.0.1 (HKY85 model, optimised equilibrium frequencies, estimated ts/tv ratio, estimated proportion of invariable sites, 4 substitution rate categories, estimated gamma distribution parameter, NNI tree topology search, 100 bootstrap iterations [41]) to obtain the phylogeny.
The genetic code used for the translation of the Euplotes mitochondrial DNA was derived using the standard genetic code for translation of the complete DNA sequence in 6 frames, and searching the resulting protein sequences for conserved Pfam-fs protein domains [42] using HMMPFAM [30]. The amino acid frequencies provided by the Pfam HMM profiles were then used to predict the translation of each codon. Averaging over all aligned occurrences of the codon, the highest scoring (i.e. most often aligned) amino acid was predicted to be the translation of the codon in vivo.
References
Pritchard AE, Seilhamer JJ, Mahalingam R, Sable CL, Venuti SE, Cummings DJ: Nucleotide sequence of the mitochondrial genome of Paramecium. Nucleic Acids Res. 1990, 18 (1): 173-180. 10.1093/nar/18.1.173.
Moradian MM, Beglaryan D, Skozylas JM, Kerikorian V: Complete Mitochondrial Genome Sequence of Three Tetrahymena Species Reveals Mutation Hot Spots and Accelerated Nonsynonymous Substitutions in Ymf Genes. PLoS ONE. 2007, 2 (7):
Brunk CF, Lee LC, Tran AB, Li J: Complete sequence of the mitochondrial genome of Tetrahymena thermophila and comparative methods for identifying highly divergent genes. Nucleic Acids Res. 2003, 31 (6): 1673-1682. 10.1093/nar/gkg270.
Burger G, Zhu Y, Littlejohn TG, Greenwood SJ, Schnare MN, Lang BF, Gray MW: Complete sequence of the mitochondrial genome of Tetrahymena pyriformis and comparison with Paramecium aurelia mitochondrial DNA. J Mol Biol. 2000, 297 (2): 365-380. 10.1006/jmbi.2000.3529.
Jurland A, Lipps HJ: Two types of mitochondria in Euplotes minuta. Arch Protistenk Bd. 1973, 115, S: 133-136.
Görtz HD: Untersuchungen zur Feinstruktur von Euplotes minuta Yocum (Ciliata, Hypotrichida) unter besonderer Berücksichtigung von Cortexstrukturen. PhD thesis. 1975, Münster: University of Münster
Ricard G, de Graaf RM, Dutilh BE, Duarte I, van Alen TA, van Hoek AH, Boxma B, Staay van der GW, Staay Moon van der SY, Chang WJ, et al: Macronuclear genome structure of the ciliate Nyctotherus ovalis: Single-gene chromosomes and tiny introns. BMC Genomics. 2008, 9 (1): 587-10.1186/1471-2164-9-587.
Boxma B, Ricard G, van Hoek AH, Severing E, Staay Moon-van der SY, Staay van der GW, van Alen TA, de Graaf RM, Cremers G, Kwantes M, et al: The [FeFe] hydrogenase of Nyctotherus ovalis has a chimeric origin. BMC Evol Biol. 2007, 7: 230-10.1186/1471-2148-7-230.
Akhmanova A, Voncken F, van Alen T, van Hoek A, Boxma B, Vogels G, Veenhuis M, Hackstein JH: A hydrogenosome with a genome. Nature. 1998, 396 (6711): 527-528. 10.1038/25023.
Boxma B, de Graaf RM, Staay van der GW, van Alen TA, Ricard G, Gabaldon T, van Hoek AHAM, Staay Moon-van der SY, Koopman WJ, van Hellemond JJ, et al: An anaerobic mitochondrion that produces hydrogen. Nature. 2005, 434 (7029): 74-79. 10.1038/nature03343.
Morin GB, Cech TR: Mitochondrial telomeres: surprising diversity of repeated telomeric DNA sequences among six species of Tetrahymena. Cell. 1988, 52 (3): 367-374. 10.1016/S0092-8674(88)80029-1.
Morin GB, Cech TR: Telomeric repeats of Tetrahymena malaccensis mitochondrial DNA: a multimodal distribution that fluctuates erratically during growth. Mol Cell Biol. 1988, 8 (10): 4450-4458.
Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS: Quantifying similarity between motifs. Genome Biol. 2007, 8 (2): R24-10.1186/gb-2007-8-2-r24.
Lazdins I, Cummings D: Structural and functional analysis of the origin of replication of mitochondrial DNA from Paramecium aurelia. Curr Genet. 1984, 8: 483-487. 10.1007/BF00410433.
Kiss GB, Amin AA, Pearlman RE: Two separate regions of the extrachromosomal ribosomal deoxyribonucleic acid of Tetrahymena thermophila enable autonomous replication of plasmids in Saccharomyces cerevisiae. Mol Cell Biol. 1981, 1 (6): 535-543.
Pritchard AE, Laping JL, Seilhamer JJ, Cummings DJ: Inter-species sequence diversity in the replication initiation region of Paramecium mitochondrial DNA. J Mol Biol. 1983, 164 (1): 1-15. 10.1016/0022-2836(83)90084-0.
Hauth AM, Maier UG, Lang BF, Burger G: The Rhodomonas salina mitochondrial genome: bacteria-like operons, compact gene arrangement and complex repeat region. Nucleic Acids Res. 2005, 33 (14): 4433-4442. 10.1093/nar/gki757.
Kim E, Lane CE, Curtis BA, Kozera C, Bowman S, Archibald JM: Complete sequence and analysis of the mitochondrial genome of Hemiselmis andersenii CCMP644 (Cryptophyceae). BMC Genomics. 2008, 9: 215-10.1186/1471-2164-9-215.
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22 (22): 4673-4680. 10.1093/nar/22.22.4673.
Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ: Jalview Version 2 - a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009, 25 (9): 1189-91. 10.1093/bioinformatics/btp033.
Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32 (5): 1792-1797. 10.1093/nar/gkh340.
Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL: The Pfam protein families database. Nucleic Acids Res. 2008, D281-288. 36 Database
Claros MG, Vincens P: Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem. 1996, 241 (3): 779-786. 10.1111/j.1432-1033.1996.00779.x.
Ueda M, Fujimoto M, Arimura S, Tsutsumi N, Kadowaki K: Presence of a latent mitochondrial targeting signal in gene on mitochondrial genome. Mol Biol Evol. 2008, 25 (9): 1791-1793. 10.1093/molbev/msn139.
Huynen MA: Presence of a latent mitochondrial targeting signal in gene on mitochondrial genome Faculty Comments & Author Responses. Faculty of 1000 Biology. 2009, [http://f1000biology.com/article/id/1158887/evaluation]
Small I, Peeters N, Legeai F, Lurin C: Predotar: A tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics. 2004, 4 (6): 1581-1590. 10.1002/pmic.200300776.
Emanuelsson O, Brunak S, von Heijne G, Nielsen H: Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007, 2 (4): 953-971. 10.1038/nprot.2007.131.
Gray MW, Lang BF, Cedergren R, Golding GB, Lemieux C, Sankoff D, Turmel M, Brossard N, Delage E, Littlejohn TG, et al: Genome structure and gene content in protist mitochondrial DNAs. Nucleic Acids Res. 1998, 26 (4): 865-878. 10.1093/nar/26.4.865.
Turanov AA, Lobanov AV, Fomenko DE, Morrison HG, Sogin ML, Klobutcher LA, Hatfield DL, Gladyshev VN: Genetic code supports targeted insertion of two amino acids by one codon. Science. 2009, 323 (5911): 259-261. 10.1126/science.1164748.
Soding J: Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005, 21 (7): 951-960. 10.1093/bioinformatics/bti125.
Smith TF, Waterman MS: Identification of common molecular subsequences. J Mol Biol. 1981, 147 (1): 195-197. 10.1016/0022-2836(81)90087-5.
Nosek J, Tomaska L, Fukuhara H, Suyama Y, Kovac L: Linear mitochondrial genomes: 30 years down the line. Trends Genet. 1998, 14 (5): 184-188. 10.1016/S0168-9525(98)01443-7.
Feagin JE: The extrachromosomal DNAs of apicomplexan parasites. Annu Rev Microbiol. 1994, 48: 81-104. 10.1146/annurev.mi.48.100194.000501.
Hall TA: BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999, 41: 95-98.
Laslett D, Canback B: ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32 (1): 11-16. 10.1093/nar/gkh152.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25 (17): 3389-3402. 10.1093/nar/25.17.3389.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al: Clustal W and Clustal X version 2.0. Bioinformatics. 2007, 23 (21): 2947-2948. 10.1093/bioinformatics/btm404.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO: SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35 (21): 7188-7196. 10.1093/nar/gkm864.
Gutell RR, Larsen N, Woese CR: Lessons from an evolving rRNA: 16S and 23S rRNA structures from a comparative perspective. Microbiol Rev. 1994, 58 (1): 10-26.
Castresana J: Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000, 17 (4): 540-552.
Guindon S, Gascuel O: A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, 52 (5): 696-704. 10.1080/10635150390235520.
Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL: The Pfam protein families database. Nucleic Acids Res. 2002, 30 (1): 276-280. 10.1093/nar/30.1.276.
Acknowledgements
This work was partially suported by the European Union 5th framework grant 'CIMES' (QLK3-2002-02151).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
RdG participated and coordinated cloning, sequencing and analyzing of both mitochondrial genomes and drafted the manuscript. BD analyzed the genetic code and performed the bioinformatic analysis. TvA participated in sequencing and analyzing the mitochondrial genomes. HvZ and MBH cloned and sequenced parts of the mitochondrial genome of Euplotes crassus. JK participated in cloning, sequencing and analyzing the mitochondrial genome of Euplotes minuta. HDG cultivated Euplotes minuta and provided cells for PFGE. MH supervised sequence analysis and participated in drafting the manuscript. JH initiated and coordinated the study and participated in drafting the manuscript. All authors read and approved the final version of the manuscript.
Electronic supplementary material
12864_2009_2398_MOESM3_ESM.pdf
Additional file 3: Figure S3. Multiple sequence alignment of the C-terminal part of the ribosomal protein S3. (PDF 296 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
de Graaf, R.M., van Alen, T.A., Dutilh, B.E. et al. The mitochondrial genomes of the ciliates Euplotes minuta and Euplotes crassus. BMC Genomics 10, 514 (2009). https://doi.org/10.1186/1471-2164-10-514
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-10-514