
Abstract
Background
Pseudogenes provide a record of the molecular evolution of genes. As glycolysis is such a highly conserved and fundamental metabolic pathway, the pseudogenes of glycolytic enzymes comprise a standardized genomic measuring stick and an ideal platform for studying molecular evolution. One of the glycolytic enzymes, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), has already been noted to have one of the largest numbers of associated pseudogenes, among all proteins.
Results
We assembled the first comprehensive catalog of the processed and duplicated pseudogenes of glycolytic enzymes in many vertebrate model-organism genomes, including human, chimpanzee, mouse, rat, chicken, zebrafish, pufferfish, fruitfly, and worm (available at http://pseudogene.org/glycolysis/). We found that glycolytic pseudogenes are predominantly processed, i.e. retrotransposed from the mRNA of their parent genes. Although each glycolytic enzyme plays a unique role, GAPDH has by far the most pseudogenes, perhaps reflecting its large number of non-glycolytic functions or its possession of a particularly retrotranspositionally active sub-sequence. Furthermore, the number of GAPDH pseudogenes varies significantly among the genomes we studied: none in zebrafish, pufferfish, fruitfly, and worm, 1 in chicken, 50 in chimpanzee, 62 in human, 331 in mouse, and 364 in rat. Next, we developed a simple method of identifying conserved syntenic blocks (consistently applicable to the wide range of organisms in the study) by using orthologous genes as anchors delimiting a conserved block between a pair of genomes. This approach showed that few glycolytic pseudogenes are shared between primate and rodent lineages. Finally, by estimating pseudogene ages using Kimura's two-parameter model of nucleotide substitution, we found evidence for bursts of retrotranspositional activity approximately 42, 36, and 26 million years ago in the human, mouse, and rat lineages, respectively.
Conclusion
Overall, we performed a consistent analysis of one group of pseudogenes across multiple genomes, finding evidence that most of them were created within the last 50 million years, subsequent to the divergence of rodent and primate lineages.
Similar content being viewed by others


Background
Pseudogenes are inheritable genomic sequences sharing large amounts of sequence similarity to genes but exhibit limited or altered functionality because of disablements. They occur in many prokaryotic and eukaryotic genomes [1–11], but the abundance of pseudogenes is specific to each species. Pseudogenes comprise a significant portion of mammalian genomes and can be found primarily in non-coding regions such as intergenic regions and introns. Because of the high level of sequence similarity shared with the parent genes, the genes from which they were mostly likely generated, it has been a difficult task to biochemically and computationally distinguish pseudogenes from genes. Resolving the functional differences between genes and pseudogenes in spite of their sequence similarity would increase our understanding of regulatory mechanisms that determine gene expression [12, 13].
Pseudogenes can be classified into two main types, processed and duplicated [6]. Processed pseudogenes are generated via retrotransposition of the mRNA of their parent genes. After mRNAs of the parent genes are transcribed in the usual fashion by RNA polymerases, they are reverse transcribed and integrated into genomic DNA by reverse transcriptases and endonucleases encoded by long interspersed nuclear elements (LINEs) in primates and humans [14, 15, 5, 16, 17]. Because these pseudogenes are generated through mRNA intermediates, they are notable for their lack of introns, spliced out during mRNA maturation. On the other hand, duplicated pseudogenes are generated via direct DNA-to-DNA duplication followed by integration into genomic DNA and eventual disablement [18]. They retain most of the exon-intron arrangements with possible duplication of upstream and downstream regions.
We have developed computational methods for cataloguing processed and duplicated pseudogenes [19, 3, 4, 20, 2]. First we identify pseudogene candidates by aligning the genome in all six frames of the translated amino acid sequences to the known proteins in the organism [21]. Then we distinguish pseudogenes from their parent genes by identifying disablements such as insertions, deletions, and nonsense mutations, as these would interfere with the potential transcription and translation of the pseudogenes into a fully functional protein.
Because pseudogenes are released from the pressures of natural selection, they capture the sequences of genes at points in time and are subsequently subject to mutations at a neutral rate [22]. Understanding the subtleties of pseudogenes that effect their inactivation would aid in predicting genes de novo from genome sequences [23–25]. In addition to their passive role as genetic fossils, the functional roles of pseudogenes are still being characterized. Pseudogenes have been found to interact with the mRNA of their parent gene [26–28]. Some pseudogenes have also been implicated in chromosomal recombination and gene conversion events leading to diseases because of high sequence homology to their parent genes [7, 29]. Others have been reactivated and become fully expressed variants of their parent genes [30].
In order to characterize the factors influencing the generation of pseudogenes, it is useful to study a selected set of genes that are common to multiple species and have many associated pseudogenes [22]. We identified such a set that encodes the enzymes in glycolysis, a fundamental metabolic pathway conserved since ancient anaerobic prokaryotes. Using our pseudogene pipeline, we assembled the first detailed catalog of the processed and duplicated pseudogenes of glycolytic enzymes in the well-annotated eukaryotic genomes: human, chimpanzee, mouse, rat, chicken, zebrafish, pufferfish, fruitfly, and worm genomes [20, 31–39]. By comparing pseudogenes of orthologous genes in multiple genomes, we are able to identify general characteristics as well as species-specific characteristics. The dates of species divergence can be used as landmarks in the temporal evolution of the glycolytic pseudogenes.
From this analysis, we found that the number of processed and duplicated pseudogenes of GAPDH, as well as its spermatogenic isozyme, far exceeded the numbers of other glycolytic pseudogenes, and for this reason, most of the present work focuses on GAPDH specifically. In order to look for an evolutionary explanation for the large number of GAPDH pseudogenes, we matched orthologous regions by extensive synteny analysis, using genomes that had sufficiently complete and intact annotations and significant numbers of GAPDH pseudogenes, namely the human, mouse, and rat genomes. After considering various methods that aligned large genomic segments by nucleotide sequences [40], we decided to align the genomes using orthologous genes as anchors. Then, after applying Kimura's two-parameter model for neutral evolution [41], we calculated a burst in retrotranspositional activity dating to about 26 million years ago. This relative recentness is consistent with the low numbers of GAPDH pseudgenes syntenic between the primate and rodent lineages. Our study documents a careful analysis of a group of pseudogenes in multiple organisms, contrasting against recent studies devoted to draft pseudogene annotation of individual genomes and attempting to date the burst in retrotransposition [28, 42].
Methods
Genomic sequences and annotated genes
The human (Homo sapiens) NCBI 35 assembly, the chimpanzee (Pan troglodytes) 4× shotgun assembly released on November 13th 2003 from the Chimpanzee Sequencing Consortium, the mouse (Mus musculus) NCBI m34 assembly, the rat (Rattus norvegicus) assembly version 3.4 November 2004 update from the Rat Genome Project, and the chicken (Gallus gallus) first draft assembly were downloaded from ENSEMBL release 33. The zebrafish (Danio rerio) assembly version 7 (Zv7) released on 13 July 2007, the pufferfish (Tetraodon nigroviridis) assembly version 7, the fruitfly (Drosophila melanogaster) BDGP assembly release 5, and worm (Caenorhabditis elegans) WormBase 180 frozen database were downloaded from ENSEMBL release 49. Gene annotations, their intron and exon positions, and their protein sequences were also obtained from ENSEMBL. The segmental duplications for the human NCBI 35 assembly were obtained from http://eichlerlab.gs.washington.edu/database.html.
Computer programs were written in Perl and GNU Bash to collect and process data. The Perl API provided by ENSEMBL was used to query releases 33, 36, and 49 of its genome databases.
Pseudogene pipeline
We used a pseudogene pipeline containing separate routines to identify processed and duplicated pseudogenes. The pipeline had been tested on large parts of the human genome [3, 4, 28, 20, 43]. On one hand, protein sequences were used to query each genome for processed pseudogenes. Minimal thresholds for identifying processed pseudogenes were optimized at 40% sequence identity and 70% alignment without an insertion longer than 60 nucleotides. Pseudogene candidates that did not meet the second criterion were considered pseudogene fragments. On the other hand, nucleotide sequences spanning a parent gene's exons with 50-nucleotide extensions in both 5' and 3' directions were used to query each genome for duplicated pseudogenes. Repetitive sequences and exons were masked in all candidate matches for processed and duplicated pseudgenes. Please see the methods section of Zheng and Gerstein (2006) for thorough specifications of the pseudogene pipeline [43].
To examine the sensitivity of the pseudogene pipeline, we varied both the percent identity and e-value threshold used for the identification of the pseudogenes in the mouse genome. The total number of pseudogenes varied from 16,963 to 15,884 while the degree of similarity to the parent protein was incremented from 25% to 50%, which constituted a dramatic range. This showed that the number of pseudogenes did not change significantly with the sequence identity parameter, about 40 pseudogenes per 1% increase in sequence similarity. We used an identity threshold of 40%, which yielded 16,730 pseudogenes. We performed similar sensitivity analyses for other parameters and present those results in Additional File 1.
Synteny
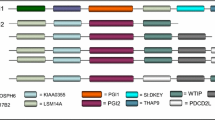
Syntenic analysis was conducted between two genomes using orthologous genes as anchors (Figure 1). A pair of GAPDH pseudogenes found in two genomes was considered a syntenic pair if it was flanked by the same two anchors. Gene orthology was assigned according to the annotations in ENSEMBL release 33. The human, mouse, and rat genomes were used for this analysis because they offered the most complete genomic annotations. We considered including the chimpanzee genome, but with its draft status and because it had only recently diverged from the human genome 5.4 million years ago, the chimpanzee genome would not have contributed significantly to the analysis. In contrast, the mouse-rat divergence occurred 41 million years ago and the human-murine divergence occurred 91 million years ago [44].

Syntenic analysis. Syntenic alignment using orthologous genes as anchors. In the example at top right, a orthologous pair of TASP1 genes is used as an anchor to determine that there is no syntenic mouse pseudogene corresponding to a human GAPDH pseudogene located in an intron of TASP1. In the example at bottom right, two orthologous pairs of CST genes are used as anchors to identify a syntenic pair of intergenic regions, in which we found a syntenic pair GAPDH pseudogenes. Solid and open bars indicate exons and introns, respectively.
Pseudogene ages
At the nucleotide level, we aligned pairs of orthologous GAPDH genes to each other and pairs of syntentic GAPDH pseudogenes to each other [45–47]. As shown in Table 1, nucleotide differences (P = fraction of transitions and Q = fraction of transversions) were used to calibrate Kimura's two-parameter model with the assumption that they began to accumulate T million years ago at the times of species divergence [41]. The divergence times between each species pair were 91 million years ago for the human-mouse divergence, 91 million years ago for the human-rat divergence, and 41 million years ago for the mouse-rat divergence [44]. The rates of transition and transversion mutations, α and β, respectively, were calculated by Equations 8-9 in Kimura (1980) as follows.

The parameters {(α i , β i )|i ∈ {human-mouse, human-rat, mouse-rat}} were calculated for GAPDH genes and pseudogenes for each pairwise comparison among human, mouse, and rat. We solved for the species-specific rates of transitions as follows.

The same equations are used, substituting β's for α's, to solve for species-specific rates of transversions. αmouse-rat-ancestor and βmouse-rat-ancestor were also calculated for the common ancestor of mouse and rat, in order to account for the time lapse of 50 million years between the human-murine divergence and mouse-rat divergence (Figure 2). The resultant values of αhuman, βhuman, αmouse, βmouse, αrat, βrat, αmouse-rat-ancestor, and βmouse-rat-ancestor are shown in Table 2. These parameters were then used to calculate the age of each GAPDH pseudogene from the nucleotide differences between it and its parent gene in the same species by solving for T in Equation 10 in Kimura (1980) as follows

Human-mouse-rat divergences. Rates of transitions and transversions were calculated for the human, mouse, and rat genomes as well as the presumed mouse-rat ancestral genome, in order to compensate for the 50 million years between the human-murine divergence and mouse-rat divergence.

where α is taken to be the averaged transition rate for genes and pseudogenes and β is taken to be the averaged transversion rate for genes and pseudogenes.
For mouse and rat pseudogenes older than 41 million years, α and β in the previous equation are replaced with

and

in order to accomodate the nucelotide substitution rates in the common ancestor of mouse and rat.
In these calculations, we derive different rates of nucleotide substitution in genes and pseudogenes because genes are subject to pressures of natural selection whereas pseudogenes are not. Although Kimura's model assumes neutral rates of nucleotide substitutions, we use it as an approximation of the mutation rates of the GAPDH genes for the sake of consistency, perhaps yielding conservative estimates or upper bounds on the ages of pseudogenes.
Results
Pseudogene abundances
We assembled a comprehensive catalogue of the processed and duplicated pseudogenes of genes encoding glycolytic enzymes in the human, chimpanzee, mouse, rat, chicken, zebrafish, pufferfish, fruitfly, and worm genomes (Table 3, http://pseudogene.org/glycolysis/). The chicken, zebrafish, pufferfish, fruitfly, and worm genomes contain the least number of GAPDH pseudogenes, none or almost none for each enzyme. The human and chimpanzee genomes both contain moderate numbers. The mouse and rat genomes contain the most, approximately five times as many as the primate genomes. The relative abundances for both processed and duplicated pseudogenes among the vertebrate genomes shows a consistent trend for each glycolytic enzyme: chicken/zebrafish/pufferfish/fruitfly/worm << primates << rodents. However, as previously reported, GAPDH surpasses the other glycolytic enzymes by far in pseudogene abundance (p = 0.0023 by Kolmogorov-Smirnov test), followed at a distant second by LDH. Processed pseudogenes outnumber duplicated pseudogenes in all the genomes except chicken, zebrafish, pufferfish, fruitfly, and worm.
Overall distribution
We mapped the chromosomal locations of the GAPDH pseudogenes in each genome. Figure 3 shows that GAPDH pseudogenes are distributed throughout the human, chimpanzee, mouse, and rat genomes, occuring on all or almost all chromosomes. While clusters of pseudogenes occur at some locations, the overall distribution appears to be uniform and shows no bias towards or against the locations of the parent genes. The other genomes we studied are not shown here because of their scarcity of processed and duplicated pseudogenes.

Pseudogene locations. Chromosomal distribution of GAPDH pseudogenes in the human genome. Pseudogenes are marked by triangles. The parent genes are marked by solid circles, occuring on human chromosomes 12 and 19 (spermatogenic), chimpanzee chromosomes 12 and 19 (spermatogenic), mouse chromosomes 6 and 7 (spermatogenic), and rat chromosome 1 (spermatogenic) and 4. The tick marks along the right edge of a chromosome mark 20 million base pair intervals, starting from the top.
Evolutionary analysis with synteny and mutation
To investigate the evolution of GAPDH pseudogenes, we attempted to identify syntenic relationships among them. As demonstrated by Figure 1, orthologous genes were used as anchors to delimit regions syntenic between two genomes. Table 4 shows the number of syntenic pseudogenes in each species pair. There were many pairs of pseudogenes syntenic between human and chimpanzee and between mouse and rat while there were very few pairs syntenic between the primate and rodent genomes, suggesting either recent pseudogene production occurring after the primate-rodent divergence or degradation beyond recognizability of pseudogenes older than 75-100 million years (Figure 4).

Phylogeny and numbers of syntenic pseudogenes. Phylogenic tree relating human, chimpanzee, mouse, rat, and chicken. Branch points are labeled with the number of syntenic GAPDH pseudogenes between the two branches and the approximate date of divergence. Branch lengths are not drawn in proportion to elapsed time.
We applied Kimura's two-parameter model of nucleotide substitution to the orthologous GAPDH genes in human, mouse, and rat to estimate their rates of transitions and transversions in each species. We also applied this model on the pairs of syntenic pseudogenes between primates and rodents to estimate the rates of transitions and transversions in the GAPDH pseudogenes of each species (Table 2). Then we aligned each GAPDH pseudogene to its parent gene in the same genome and calculated the nucleotide difference in terms of transitions and transversions. By estimating nucleotide substitution rates for the GAPDH genes, our calculations compensated for mutations occurring after they diverged from a common ancestral gene and the ages of the pseudogenes were adjusted accordingly. From the nucleotide differences and the above estimated rates of transitions and transversions in genes and pseudogenes, we estimated the ages of the non-syntenic GAPDH pseudogenes, as shown in Figure 5. The ages of the non-spermatogenic GAPDH pseudogenes were not included, as they appeared to have become more severely degraded. These dating calculations are particularly sensitive to the quality of the underlying genome sequence and annotation. Consequently, we only report data for the three most completely finished and annotated genomes in our set: human, mouse, and rat. Because the chimpanzee genome diverged from the human genome so recently, we would not expect chimpanzee to have very different numbers for the comparison.

Pseudogene ages. Top three panels: Distributions of GAPDH pseudogenes by age in the human, mouse, and rat genomes. There appear to be three distinct bursts in retrotransposition which gave rise to GAPDH pseudogenes centered around medians (middle 50%) of 42.0 million years ago (26.4-49.3 million years) in human, 36.3 million years ago (17.4-52.8 million years) in mouse, and 25.9 million years ago (17.6-40.9 million years) in rat. Pairwise Kolmogorov-Smirnov testing shows that the age distributions among these three genomes are statistically different, with p-values of 0.01 (human-mouse), 7 × 10-7 (human-rat), and 7 × 10-10 (mouse-rat). Bottom two panels: Distributions of GAPDH pseudogenes syntenic between mouse and rat. Although the majority did occur before the mouse-rat divergence 41 million years ago, there is some noise or variation in nucleotide substitutions.
Discussion
As a central pathway in metabolism, glycolysis has been highly conserved across multiple species from archaea to humans. The omnipresence of the glycolytic enzymes makes for a crude but standardized genomic measuring stick, comprising an ideal platform for studying pseudogenes.
Despite the high degree of conservation in the glycolytic enzymes, there is much more variation in their pseudogene abundances. Some genomes, like chicken, zebrafish, pufferfish, fruitfly, and worm, have very few or none, while others, like mouse and rat, have hundreds. The differences in pseudogene abundances alone suggests significant differences in the processes of gene expression, duplication, and retrotransposition in the different genomes. Previous studies have suggested that the difference lies in the prolonged lampbrush stage of oogenesis in mammalians as compared to non-mammalian organisms [48, 49].
Most glycolytic pseudogenes are processed and can be assumed to be retrotransposed from an mRNA intermediate. It is possible that certain sequences intrinsic to the GAPDH and LDH genes may predispose them to be preferentially retrotranscribed, inserted, and preserved in the genome. These pseudogenes are classified as processed and not duplicated indicating their formation was the result of a retrotransposition event of the parent gene, rather than a duplication event. However, we must consider the possibility of formation of a processed pseudogene through a retrotransposition event and its subsequent duplication giving rise to so called "duplicated-processed" pseudogenes. Thus, while duplicated pseudogenes result from the duplication of parent gene, duplicated-processed pseudogenes result from the duplication of a processed pseudogene [50, 51]. One way to differentiate processed pseudogenes from duplicated-processed pseudogenes is to check if the segments of the genome surrounding a pair of processed pseudogenes are also similar. Hence, we checked for the presence of 60 processed pseudogenes of human GAPDH in duplicated regions of the genome called segmental duplications [52]. A pair of processed pseudogenes located in segmental duplication pairs indicates that one of the pseudogenes was likely formed by the duplication of the other one and hence is a duplicated-processed pseudogene (Figure 6). We identified eight duplicated-processed pseudogenes by this analysis, listed in Additional File 1. However, six of those eight pseudogenes occupy > 77% of the segments that are duplicated and could be the result of independent retrotransposition events. In this scenario perhaps the high sequence similarity of these segments led to their annotation as segmental duplications.

Aetiology of a duplicated-processed pseudogene. Alternative aetiology of a processed pseudogene. A parent gene is first retrotransposed into a processed pseudogene. Then the processed pseudogene undergoes segmental duplication to produce a duplicated-processed pseudogene.
As a coincident finding, GAPDH has many more biological roles outside glycolysis as compared to the other glycolytic enzymes. For example, GAPDH functions in DNA repair, telomeric DNA binding, transcriptional regulation, nuclear RNA export, apoptosis, membrane fusion, phosphorylation, tubulin bundling, and sperm motility [53–59]. Because the molecular processes of retrotransposition are separate from the enzymatic functionalities, we can only speculate that the preponderance of non-glycolytic roles may be correlated to the enrichment of GAPDH pseudogenes.
In an intergenomic analysis, GAPDH pseudogenes have about five- to six-fold greater abundance in the rodent genomes as in the primate genomes even though overall the mouse genome was found to have about half as many pseudogenes as the human genome [3]. The mouse genome has higher rates of nucleotide substitution, insertion, and deletion [33] than the human genome, leading to a higher rate of pseudogene decay. However, the higher rate of pseudogene decay seems to have preferentially spared the GAPDH pseudogenes.
To further characterize the molecular history of pseudogenes in the human, chimpanzee, mouse, and rat genomes, it was necessary to identify the pseudogenes that were most likely present prior to the primate-rodent ancestral divergence. We used orthologous genes to identify regions of synteny between primate-rodent genome pairs. This approach is based on the assumption that gene-coding regions are much less variable than intergenic regions because of functional constraints and are therefore more reliably matched between genome pairs.
The scarcity of GAPDH pseudogenes syntenic between the primate and rodent genomes suggests an increase in retrotranspositional activity after the primate-rodent divergence 91 million years ago, which is consistent with the findings of previous investigators [6]. In order to achieve more detail in the timeline and provide further corroboration, we used Kimura's two-parameter model of nucleotide substitution to estimate the rates of change in the GAPDH genes and pseudogenes and thereby calculate the insertion date of each pseudogene. The creation dates formed three distinct distributions centered at 42.0, 36.3, and 25.9 million years ago in the human, mouse, and rat genomes, respectively, signifying a burst in retrotranspositional activity around those times. Kimura's model assumes neutrally evolving sequences, as in many pseudogenes [42], but some may initially be subject to natural selection [12] and the ages of these pseudogenes may be underestimated. In the human genome, the bursts in retrotranspositional activity may coincide with the "Alu burst" that occurred about 40 million years ago in primate genomes [60, 1, 5, 61]. By examining the sensitivity of our pseudogene pipeline, as decribed under Methods, we found that the number of pseudogenes does not vary significantly with the threshold for sequence identity or BLAST score when compared to the parent gene. Thus, we believe this dating method accurately reflects all GAPDH pseudogenes and is not significantly biased towards longer and therefore younger pseudogenes.
Conclusion
The ubiquitous nature of glycolytic enzymes rendered their pseudogenes most appropriate for comparing retrotransposition among multiple genomes. There was no evidence for preferential distribution of GAPDH pseudogenes in relation to individual chromosomes and to the location of the parent genes. We were able to calculate synteny using orthologous genes as anchors between two genomes. Whereas retrotransposition and gene annotation have been previously characterized on an individual genome basis, our syntenic method allowed us to perform a careful analysis of one pseudogene family across multiple genomes. This and a molecular clock analysis indicated that three distinct bursts in the insertion of GAPDH pseudogenes occurred at approximately 42, 36, and 26 million years ago in the human, mouse, and rat genomes, respectively, with evidence that most were created within the last 50 million years, subsequent to the divergence of rodent and primate lineages.
References
Zhang Z, Harrison P, Gerstein M: Identification and analysis of over 2000 ribosomal protein pseudogenes in the human genome. Genome Res. 2002, 12 (10): 1466-82. 10.1101/gr.331902.
Zhang Z, Harrison PM, Liu Y, Gerstein M: Millions of years of evolution preserved: a comprehensive catalog of the processed pseudogenes in the human genome. Genome Res. 2003, 13 (12): 2541-58. 10.1101/gr.1429003.
Zhang Z, Carriero N, Gerstein M: Comparative analysis of processed pseudogenes in the mouse and human genomes. Trends Genet. 2004, 20 (2): 62-7. 10.1016/j.tig.2003.12.005.
Zhang Z, Gerstein M: Large-scale analysis of pseudogenes in the human genome. Curr Opin Genet Dev. 2004, 14 (4): 328-35. 10.1016/j.gde.2004.06.003.
Ohshima K, Hattori M, Yada T, Gojobori T, Sakaki Y, Okada N: Whole-genome screening indicates a possible burst of formation of processed pseudogenes and Alu repeats by particular L1 subfamilies in ancestral primates. Genome Biol. 2003, 4 (11): R74-10.1186/gb-2003-4-11-r74.
Torrents D, Suyama M, Zdobnov E, Bork P: A genome-wide survey of human pseudogenes. Genome Res. 2003, 13 (12): 2559-67. 10.1101/gr.1455503.
Bischof JM, Chiang AP, Scheetz TE, Stone EM, Casavant TL, Sheffield VC, Braun TA: Genome-wide identification of pseudogenes capable of disease-causing gene conversion. Hum Mutat. 2006, 27 (6): 545-52. 10.1002/humu.20335.
Lerat E, Ochman H: Psi-Phi: exploring the outer limits of bacterial pseudogenes. Genome Res. 2004, 14 (11): 2273-8. 10.1101/gr.2925604.
Lerat E, Ochman H: Recognizing the pseudogenes in bacterial genomes. Nucleic Acids Res. 2005, 33 (10): 3125-32. 10.1093/nar/gki631.
Ochman H, Davalos LM: The nature and dynamics of bacterial genomes. Science. 2006, 311 (5768): 1730-3. 10.1126/science.1119966.
Andersson JO, Andersson SG: Pseudogenes, junk DNA, and the dynamics of Rickettsia genomes. Mol Biol Evol. 2001, 18 (5): 829-39.
Balakirev ES, Ayala FJ: Pseudogenes: are they "junk" or functional DNA?. Annu Rev Genet. 2003, 37: 123-51. 10.1146/annurev.genet.37.040103.103949.
van Baren MJ, Brent MR: Iterative gene prediction and pseudogene removal improves genome annotation. Genome Res. 2006, 16 (5): 678-85. 10.1101/gr.4766206.
Feng Q, Moran JV, Kazazian J, H H, Boeke JD: Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 1996, 87 (5): 905-16. 10.1016/S0092-8674(00)81997-2.
Jurka J: Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc Natl Acad Sci USA. 1997, 94 (5): 1872-7. 10.1073/pnas.94.5.1872.
Weiner AM: Do all SINEs lead to LINEs?. Nat Genet. 2000, 24 (4): 332-3. 10.1038/74135.
Esnault C, Maestre J, Heidmann T: Human LINE retrotransposons generate processed pseudogenes. Nat Genet. 2000, 24 (4): 363-7. 10.1038/74184.
Glusman G, Yanai I, Rubin I, Lancet D: The complete human olfactory subgenome. Genome Res. 2001, 11 (5): 685-702. 10.1101/gr.171001.
Harrison PM, Hegyi H, Balasubramanian S, Luscombe NM, Bertone P, Echols N, Johnson T, Gerstein M: Molecular fossils in the human genome: identification and analysis of the pseudogenes in chromosomes 21 and 22. Genome Res. 2002, 12 (2): 272-80. 10.1101/gr.207102.
Zhang Z, Carriero N, Zheng D, Karro J, Harrison PM, Gerstein M: PseudoPipe: an automated pseudogene identification pipeline. Bioinformatics. 2006, 22 (12): 1437-9. 10.1093/bioinformatics/btl116.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25 (17): 3389-402. 10.1093/nar/25.17.3389.
Hardison RC, Gelinas RE: Assignment of orthologous relationships among mammalian alpha-globin genes by examining flanking regions reveals a rapid rate of evolution. Mol Biol Evol. 1986, 3 (3): 243-61.
Brent MR, Guigo R: Recent advances in gene structure prediction. Curr Opin Struct Biol. 2004, 14 (3): 264-72. 10.1016/j.sbi.2004.05.007.
Khelifi A, Duret L, Mouchiroud D: HOPPSIGEN: a database of human and mouse processed pseudogenes. Nucleic Acids Res. 2005, D59-66. 33 Database
Mighell AJ, Smith NR, Robinson PA, Markham AF: Vertebrate pseudogenes. FEBS Lett. 2000, 468 (2-3): 109-14. 10.1016/S0014-5793(00)01199-6.
Hirotsune S, Yoshida N, Chen A, Garrett L, Sugiyama F, Takahashi S, Yagami K, Wynshaw-Boris A, Yoshiki A: An expressed pseudogene regulates the messenger-RNA stability of its homologous coding gene. Nature. 2003, 423 (6935): 91-6. 10.1038/nature01535.
Korneev SA, Park JH, O'Shea M: Neuronal expression of neural nitric oxide synthase (nNOS) protein is suppressed by an antisense RNA transcribed from an NOS pseudogene. J Neurosci. 1999, 19 (18): 7711-20.
Zheng D, Zhang Z, Harrison PM, Karro J, Carriero N, Gerstein M: Integrated pseudogene annotation for human chromosome 22: evidence for transcription. J Mol Biol. 2005, 349: 27-45. 10.1016/j.jmb.2005.02.072.
Druker R, Whitelaw E: Retrotransposon-derived elements in the mammalian genome: a potential source of disease. J Inherit Metab Dis. 2004, 27 (3): 319-30. 10.1023/B:BOLI.0000031096.81518.66.
Cheng JF, Krane DE, Hardison RC: Nucleotide sequence and expression of rabbit globin genes zeta 1, zeta 2, and zeta 3. Pseudogenes generated by block duplications are transcriptionally competent. J Biol Chem. 1988, 263 (20): 9981-93.
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M: Initial sequencing and analysis of the human genome. Nature. 2001, 409 (6822): 860-921. 10.1038/35057062.
Chimpanzee Sequencing and Analysis Consortium: Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005, 437 (7055): 69-87. 10.1038/nature04072.
Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigo R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK: Initial sequencing and comparative analysis of the mouse genome. Nature. 2002, 420 (6915): 520-62. 10.1038/nature01262.
Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, Okwuonu G, Hines S, Lewis L, DeRamo C, Delgado O, Dugan-Rocha S, Miner G, Morgan M, Hawes A, Gill R, Celera , Holt RA, Adams MD, Amanatides PG, Baden-Tillson H, Barnstead M, Chin S, Evans CA, Ferriera S, Fosler C, Glodek A, Gu Z, Jennings D, Kraft CL, Nguyen T, Pfannkoch CM, Sitter C, Sutton GG, Venter JC, Woodage T, Smith D, Lee HM, Gustafson E, Cahill P, Kana A, Doucette-Stamm L, Weinstock K, Fechtel K, Weiss RB, Dunn DM, Green ED, Blakesley RW, Bouffard GG, De Jong PJ, Osoegawa K, Zhu B, Marra M, Schein J, Bosdet I, Fjell C, Jones S, Krzywinski M, Mathewson C, Siddiqui A, Wye N, McPherson J, Zhao S, Fraser CM, Shetty J, Shatsman S, Geer K, Chen Y, Abramzon S, Nierman WC, Havlak PH, Chen R, Durbin KJ, Egan A, Ren Y, Song XZ, Li B, Liu Y, Qin X, Cawley S, Cooney AJ, D'Souza LM, Martin K, Wu JQ, Gonzalez-Garay ML, Jackson AR, Kalafus KJ, McLeod MP, Milosavljevic A, Virk D, Volkov A, Wheeler DA, Zhang Z, Bailey JA, Eichler EE, Tuzun E: Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004, 428 (6982): 493-521. 10.1038/nature02426.
Hillier LW, Miller W, Birney E, Warren W, Hardison RC, Ponting CP, Bork P, Burt DW, Groenen MA, Delany ME, Dodgson JB, Chinwalla AT, Cliften PF, Clifton SW, Delehaunty KD, Fronick C, Fulton RS, Graves TA, Kremitzki C, Layman D, Magrini V, McPherson JD, Miner TL, Minx P, Nash WE, Nhan MN, Nelson JO, Oddy LG, Pohl CS, Randall-Maher J, Smith SM, Wallis JW, Yang SP, Romanov MN, Rondelli CM, Paton B, Smith J, Morrice D, Daniels L, Tempest HG, Robertson L, Masabanda JS, Griffin DK, Vignal A, Fillon V, Jacobbson L, Kerje S, Andersson L, Crooijmans RP, Aerts J, Poel van der JJ, Ellegren H, Caldwell RB, Hubbard SJ, Grafham DV, Kierzek AM, McLaren SR, Overton IM, Arakawa H, Beattie KJ, Bezzubov Y, Boardman PE, Bonfield JK, Croning MD, Davies RM, Francis MD, Humphray SJ, Scott CE, Taylor RG, Tickle C, Brown WR, Rogers J, Buerstedde JM, Wilson SA, Stubbs L, Ovcharenko I, Gordon L, Lucas S, Miller MM, Inoko H, Shiina T, Kaufman J, Salomonsen J, Skjoedt K, Wong GK, Wang J, Liu B, Yu J, Yang H, Nefedov M, Koriabine M, Dejong PJ, Goodstadt L, Webber C, Dickens NJ, Letunic I, Suyama M, Torrents D, von Mering C, Zdobnov EM: Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004, 432 (7018): 695-716. 10.1038/nature03154.
Danio rerio Sequencing Project: (unpublished zebrafish genome) April 2008., [http://mar2008.archive.ensembl.org/Danio_rerio/index.html]
Jaillon O, Aury JM, Brunet F, Petit JL, Stange-Thomann N, Mauceli E, Bouneau L, Fischer C, Ozouf-Costaz C, Bernot A, Nicaud S, Jaffe D, Fisher S, Lutfalla G, Dossat C, Segurens B, Dasilva C, Salanoubat M, Levy M, Boudet N, Castellano S, Anthouard V, Jubin C, Castelli V, Katinka M, Vacherie B, Biemont C, Skalli Z, Cattolico L, Poulain J, De Berardinis V, Cruaud C, Duprat S, Brottier P, Coutanceau JP, Gouzy J, Parra G, Lardier G, Chapple C, McKernan KJ, McEwan P, Bosak S, Kellis M, Volff JN, Guigo R, Zody MC, Mesirov J, Lindblad-Toh K, Birren B, Nusbaum C, Kahn D, Robinson-Rechavi M, Laudet V, Schachter V, Quetier F, Saurin W, Scarpelli C, Wincker P, Lander ES, Weissenbach J, Roest Crollius H: Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature. 2004, 431 (7011): 946-57. 10.1038/nature03025.
Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, George RA, Lewis SE, Richards S, Ashburner M, Henderson SN, Sutton GG, Wortman JR, Yandell MD, Zhang Q, Chen LX, Brandon RC, Rogers YH, Blazej RG, Champe M, Pfeiffer BD, Wan KH, Doyle C, Baxter EG, Helt G, Nelson CR, Gabor GL, Abril JF, Agbayani A, An HJ, Andrews-Pfannkoch C, Baldwin D, Ballew RM, Basu A, Baxendale J, Bayraktaroglu L, Beasley EM, Beeson KY, Benos PV, Berman BP, Bhandari D, Bolshakov S, Borkova D, Botchan MR, Bouck J, Brokstein P, Brottier P, Burtis KC, Busam DA, Butler H, Cadieu E, Center A, Chandra I, Cherry JM, Cawley S, Dahlke C, Davenport LB, Davies P, de Pablos B, Delcher A, Deng Z, Mays AD, Dew I, Dietz SM, Dodson K, Doup LE, Downes M, Dugan-Rocha S, Dunkov BC, Dunn P, Durbin KJ, Evangelista CC, Ferraz C, Ferriera S, Fleischmann W, Fosler C, Gabrielian AE, Garg NS, Gelbart WM, Glasser K, Glodek A, Gong F, Gorrell JH, Gu Z, Guan P, Harris M, Harris NL, Harvey D, Heiman TJ, Hernandez JR, Houck J, Hostin D, Houston KA, Howland TJ, Wei MH, Ibegwam C: The genome sequence of Drosophila melanogaster. Science. 2000, 287 (5461): 2185-95. 10.1126/science.287.5461.2185.
C elegans Sequencing Consortium: Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998, 282 (5396): 2012-8. 10.1126/science.282.5396.2012.
Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D: Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc Natl Acad Sci USA. 2003, 100 (20): 11484-9. 10.1073/pnas.1932072100.
Kimura M: A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980, 16 (2): 111-20. 10.1007/BF01731581.
Zheng D, Frankish A, Baertsch R, Kapranov P, Reymond A, Choo SW, Lu Y, Denoeud F, Antonarakis SE, Snyder M, Ruan Y, Wei CL, Gingeras TR, Guigo R, Harrow J, Gerstein MB: Pseudogenes in the ENCODE regions: consensus annotation, analysis of transcription, and evolution. Genome Res. 2007, 17 (6): 839-51. 10.1101/gr.5586307.
Zheng D, Gerstein MB: A computational approach for identifying pseudogenes in the ENCODE regions. Genome Biol. 2006, 7 (Suppl 1): S13-10.1186/gb-2006-7-s1-s13. 1-10
Hedges SB: The origin and evolution of model organisms. Nat Rev Genet. 2002, 3 (11): 838-49. 10.1038/nrg929.
Li WH, Gojobori T, Nei M: Pseudogenes as a paradigm of neutral evolution. Nature. 1981, 292 (5820): 237-239. 10.1038/292237a0.
Miyata T, Yasunaga T: Rapidly evolving mouse alpha-globin-related pseudo gene and its evolutionary history. Proc Natl Acad Sci USA. 1981, 78: 450-453. 10.1073/pnas.78.1.450.
Ercolani L, Florence B, Denaro M, Alexander M: Isolation and complete sequence of a functional human glyceraldehyde-3-phosphate dehydrogenase gene. J Biol Chem. 1988, 263 (30): 15335-41.
Drouin G: Processed pseudogenes are more abundant in human and mouse X chromosomes than in autosomes. Mol Biol Evol. 2006, 23 (9): 1652-5. 10.1093/molbev/msl048.
Weiner AM, Deininger PL, Efstratiadis A: Nonviral retroposons: genes, pseudogenes, and transposable elements generated by the reverse flow of genetic information. Annu Rev Biochem. 1986, 55: 631-61. 10.1146/annurev.bi.55.070186.003215.
Hazkani-Covo E, Sorek R, Graur D: Evolutionary dynamics of large numts in the human genome: rarity of independent insertions and abundance of post-insertion duplications. J Mol Evol. 2003, 56 (2): 169-74. 10.1007/s00239-002-2390-5.
Garcia-Meunier P, Etienne-Julan M, Fort P, Piechaczyk M, Bonhomme F: Concerted evolution in the GAPDH family of retrotransposed pseudogenes. Mamm Genome. 1993, 4 (12): 695-703. 10.1007/BF00357792.
Bailey JA, Eichler EE: Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet. 2006, 7 (7): 552-64. 10.1038/nrg1895.
Kim JW, Dang CV: Multifaceted roles of glycolytic enzymes. Trends Biochem Sci. 2005, 30 (3): 142-50. 10.1016/j.tibs.2005.01.005.
Sundararaj KP, Wood RE, Ponnusamy S, Salas AM, Szulc Z, Bielawska A, Obeid LM, Hannun YA, Ogretmen B: Rapid shortening of telomere length in response to ceramide involves the inhibition of telomere binding activity of nuclear glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 2004, 279 (7): 6152-62. 10.1074/jbc.M310549200.
Zheng L, Roeder RG, Luo Y: S phase activation of the histone H2B promoter by OCA-S, a coactivator complex that contains GAPDH as a key component. Cell. 2003, 114 (2): 255-66. 10.1016/S0092-8674(03)00552-X.
Sirover MA: Minireview. Emerging new functions of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in mammalian cells. Life Sci. 1996, 58 (25): 2271-7. 10.1016/0024-3205(96)00123-3.
Sirover MA: Role of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in normal cell function and in cell pathology. J Cell Biochem. 1997, 66 (2): 133-40. 10.1002/(SICI)1097-4644(19970801)66:2<133::AID-JCB1>3.0.CO;2-R.
Sirover MA: New insights into an old protein: the functional diversity of mammalian glyceraldehyde-3-phosphate dehydrogenase. Biochim Biophys Acta. 1999, 1432 (2): 159-84.
Miki K, Qu W, Goulding EH, Willis WD, Bunch DO, Strader LF, Perreault SD, Eddy EM, O'Brien DA: Glyceraldehyde 3-phosphate dehydrogenase-S, a sperm-specific glycolytic enzyme, is required for sperm motility and male fertility. Proc Natl Acad Sci USA. 2004, 101 (47): 16501-6. 10.1073/pnas.0407708101.
Kapitonov V, Jurka J: The age of Alu subfamilies. J Mol Evol. 1996, 42: 59-65. 10.1007/BF00163212.
Marques AC, Dupanloup I, Vinckenbosch N, Reymond A, Kaessmann H: Emergence of young human genes after a burst of retroposition in primates. PLoS Biol. 2005, 3 (11): e357-10.1371/journal.pbio.0030357.
Acknowledgements
We would like to acknowledge financial support from grants from the NIH and from the Yale University School of Medicine Summer Research Grant. The authors would also like to acknowledge Rajkumar Sasidharan and Hugo Lam for helpful discussion.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
YJL carried out the tabulation of processed and duplicated pseudogenes of glycolytic enzymes, syntenic and evolutionary analysis, and calculation of pseudogene ages. DZ, SB, NC, RR, and MBG were involved in developing and calibrating our pseudogene pipeline. EK carried out the analysis of potential duplicated processed pseudogenes in sequence-duplicated regions of the human genome. MBG conceived of the study and participated in its design and coordination. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2009_2364_MOESM1_ESM.PDF
Additional file 1: Supplement. The sensitivity of our pseudogene pipeline is clarified and the sets of duplicated-processed pseudogenes are cataloged. (PDF 28 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Liu, YJ., Zheng, D., Balasubramanian, S. et al. Comprehensive analysis of the pseudogenes of glycolytic enzymes in vertebrates: the anomalously high number of GAPDH pseudogenes highlights a recent burst of retrotrans-positional activity. BMC Genomics 10, 480 (2009). https://doi.org/10.1186/1471-2164-10-480
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-10-480