
Abstract
Background
The globe artichoke (Cynara cardunculus var. scolymus L.) is a significant crop in the Mediterranean basin. Despite its commercial importance and its both dietary and pharmaceutical value, knowledge of its genetics and genomics remains scant. Microsatellite markers have become a key tool in genetic and genomic analysis, and we have exploited recently acquired EST (expressed sequence tag) sequence data (Composite Genome Project - CGP) to develop an extensive set of microsatellite markers.
Results
A unigene assembly was created from over 36,000 globe artichoke EST sequences, containing 6,621 contigs and 12,434 singletons. Over 12,000 of these unigenes were functionally assigned on the basis of homology with Arabidopsis thaliana reference proteins. A total of 4,219 perfect repeats, located within 3,308 unigenes was identified and the gene ontology (GO) analysis highlighted some GO term's enrichments among different classes of microsatellites with respect to their position. Sufficient flanking sequence was available to enable the design of primers to amplify 2,311 of these microsatellites, and a set of 300 was tested against a DNA panel derived from 28 C. cardunculus genotypes. Consistent amplification and polymorphism was obtained from 236 of these assays. Their polymorphic information content (PIC) ranged from 0.04 to 0.90 (mean 0.66). Between 176 and 198 of the assays were informative in at least one of the three available mapping populations.
Conclusion
EST-based microsatellites have provided a large set of de novo genetic markers, which show significant amounts of polymorphism both between and within the three taxa of C. cardunculus. They are thus well suited as assays for phylogenetic analysis, the construction of genetic maps, marker-assisted breeding, transcript mapping and other genomic applications in the species.
Similar content being viewed by others

Background
The globe artichoke Cynara cardunculus is a member of the Asteraceae (Compositae) family, and originates from the Mediterranean basin [1]. The species is subdivided into three taxa - the globe artichoke (var. scolymus L.), the cultivated cardoon (var. altilis DC), and their progenitor, the wild cardoon [var. sylvestris (Lamk) Fiori]. The edible part of the globe artichoke plant is provided by its immature inflorescence, referred as a capitulum or head [2], and represents a significant component of the Mediterranean diet. The cultivated cardoon is grown for its fleshy stems, which are used in traditional cuisine. Leaf extracts of the species contain a number of bioactive compounds (e.g., quercetin, rutin, luteolin, gallic acid, di-caffeoylchinic acid, and sesquiterpene lactones) which have been shown to have anti-oxidative and anti-carcinogenic activity, to inhibit cholesterol biosynthesis, and to enhance lipid metabolism [3–8]. The antioxidant content per serving of globe artichoke ranks very highly among vegetables [9], while the roots provide a source of inulin, a proven enhancer of human intestinal flora [10, 11]. In spite of its economic importance, however, little breeding effort has been applied to date in the globe artichoke.
Progress has been made in the development of DNA marker based genetic maps in globe artichoke [12–14]. The most saturated map has been recently developed from a set of F1 progeny of a cross between a globe artichoke and a cultivated cardoon genotypes [14]. This map consisted of 20 linkage groups comprising 326 loci and spanned ~1500 cM with a mean inter-marker distance of ~4.5 cM. A set of loci common to this map and a previously developed one [12] allowed for map alignment and the definition of 17 homologous linkage groups, corresponding to the haploid chromosome number of the species.
It was long assumed that SSRs were primarily associated with non-coding DNA, but it has now become clear that they are more frequent in transcribed than non-transcribed sequences and equally frequent in the transcriptomes of plants with dramatically different nuclear DNA contents [15]. EST databases therefore represent a convenient resource for the identification of microsatellites, some of which, as a result of their presence within coding DNA, have the potential to deliver informative within gene markers, exploitable as COS (conserved orthologous set) for genomic comparative analysis.
Here, we report: i) the unigene assembly based on the globe artichoke EST database deposited in GenBank by the Compositae Genome Project (CGP), ii) the identification of a wide set of EST-based microsatellite markers and iii) the evaluation of the informativeness of a subset of these markers using a panel of C. cardunculus genotypes. Furthermore, we performed a comprehensive functional annotation, inferred from sequence alignment (ISA), as well as a gene ontology categorisation inferred from sequence orthology (ISO) of the SSR-containing unigenes. At last we assessed whether motif type and relative position within CDSs (Coding DNA Sequences)/UTRs (Untraslated Regions) may be preferentially associated with a particular gene ontology term.
Results and Discussion
EST microsatellite discovery, frequency and primer design
Globe artichoke ESTs were trimmed, assembled, and annotated using a customized bioinformatic pipeline (Figure 1) into 19,055 unigenes (6,621 contigs and 12,434 singletons) spanning 15 Mbp. The transcript assembly and unigene consensus sequences are supplied as electronic supplementary materials (See Additional file 1, 2, 3: EST assembly, 19,055 unigenes, ACE assembly file). The unigenes had a mean length of 786 ± 1.7 bp, with a mean GC content of 43.9 ± 0.03% (range 24.7 - 67.9%, Figure 2) and a mean ambiguity code ratio of 0.51 ± 0.01. Within the unigene set, 3,308 contained 4,219 uninterrupted tracts of (perfect) di-, tri-, tetra-, penta-, and hexa-nucleotide repeats, delivering a mean microsatellite density of one per 3.6 kb. Comparable microsatellite frequencies and densities have been discovered in the transcriptomes of other Compositae species [16–18]. Only perfect repeats were selected, as these appear to be the more prone to strand slippage and, consequently, tend to be more polymorphic than interrupted ones [19]. Sufficient flanking sequence (in terms of both length and read quality) was present in 1,974 of the unigenes, containing 2,311 perfect microsatellites. The resulting PCR primers designed for these loci are given in Additional File 4 (primer pairs designed).

The schema used for EST assembly, annotation, primer design and amplicon screening.

Output of the EST assembly. Distribution of unigene (A) length, and (B) GC-content.
Allelic diversity with the EST microsatellites
A subset of 300 microsatellites (ranging in length from 10 to 84 bp, and representative of a broad spectrum of the repeat types) was surveyed for their informativeness. The targeted amplicon length ranged from 98 to 456 bp and the set was selected to optimize the possibility of multiplexing on the capillary genotyping platform employed. The test germplasm panel consisted of twelve genotypes of globe artichoke, nine of cultivated cardoon, and seven of wild cardoon (Table 1). In all, 238 (79.3%) of the assays were successful; of these, 236 were informative among the taxa, while 215, 216 and 223 were polymorphic within, respectively, globe artichoke, cultivated cardoon and wild cardoon (Table 2 and Additional file 5: full statistics on 300 SSR-containing loci). A total of 1,546 alleles was generated from the 238 successful assays, giving a mean of 3.8 (range 1-15) alleles per locus. The largest range in amplicon length observed was 196 - 252 bp, observed for a TCAn microsatellite (CyEM-171). In 85% of the loci, the assay generated the predicted length of amplicon, in 12.2% the amplicon was longer than expected, and in 2.8% it was shorter. The allelic range (in terms of amplicon length) was greater for the wild cardoon (17.1 ± 1.0 bp) than for globe artichoke (13.6 ± 0.8 bp) or cultivated cardoon (13.7 ± 0.7 bp).
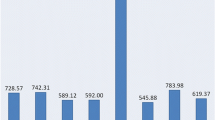
Allelic diversity was, as expected given the breeding history of these taxa [2, 20, 21], greater in the wild than in the two cultivated forms (Figure 3A). The frequency of taxon-specific alleles was two fold more in the wild cardoon, and the polymorphic information content (PIC) was higher in the wild cardoon (0.576 ± 0.015) than in either the globe artichoke (0.484 ± 0.013) or the cultivated cardoon (0.466 ± 0.015). The observed heterozygosity level (Ho) was significantly less in the cultivated cardoon than in globe artichoke, presumably because the globe artichoke is primarily a vegetatively propagated plant, and thus able to maintain a high level of heterozygosity over time [20, 21]; whereas cultivated cardoon is seed propagated and has been subjected to purifying selection aimed at increasing genetic uniformity for stabilizing its production. We previously developed three mapping population for the development of C. cardunculus genetic maps by crossing one globe artichoke non spiny genotype (common female parent) with a spiny genotype of globe artichoke or cultivated cardoon or wild cardoon. When the parents of the three mapping populations were tested with the set of microsatellites, 214 were informative in at least one combination, while 153 across all the three combinations, thus supplying landmarks for comparative mapping of phenotypic and quantitative trait loci (QTLs). As expected, the most polymorphic combination (198 microsatellites) was the one involving the cross between the most genetically divergent taxa: globe artichoke and wild cardoon (Figure 3B).

Allelic diversity revealed by the set of EST microsatellite markers. (A) Allelic patterns and the level of heterozygosity within each taxon. Observed (no), effective (ne) and the number of taxon-specific alleles per marker are represented by bars. PIC and Ho are indicated by points. (B) Markers showing segregation within the three mapping populations. GxG: within scolymus, GxC: scolymus × altilis, GxW: scolymus × sylvestris.
Diversity analysis
The informativeness of the newly developed EST microsatellites was comparable with that described for microsatellite markers in globe artichoke [13, 22], sunflower [23, 24] and lettuce [25]. A set of five EST microsatellites was sufficient to discriminate between each of the 28 members of the germplasm panel (e.g. CyEM-10, -37, -54, -105, -254). The inferred genetic relationships were in good concordance with those derived from AFLP profiling [20, 21, 26, 27]. Thus, the globe artichoke accessions clustered with one another (Figure 4A, cluster A), but two sub-clusters, corresponding to the contrasting capitulum types (i.e. non spiny vs. violet, spiny types), could be recognised. The clade most closely related to the globe artichokes contained the cultivated cardoons (Figure 4A, cluster B), and among these, the most well differentiated accession was 'Bianco Pieno Migliorato', as previously observed [21]. The Spanish cultivated cardoon accessions were genetically very similar to one another. The wild cardoon accessions formed a discrete, but rather loose group (Figure 4A, cluster C). A principal co-ordinate analysis further illustrated the genetic relationships between members of the germplasm panel (Figure 4B). Axes 1 and 2 accounted for ~ 73% of the genetic variation, the former contributing ~ 47%, and the latter ~ 26%. Axis 1 distinguished the globe artichokes from the cultivated and wild cardoons, while Axis 2 separated the two cardoon taxa. As expected, F1 progenies mapped to intermediate positions with respect to those of their parents (Figure 4B).

Diversity analysis. (A) A UPGMA dendogram based on 1,546 EST microsatellite alleles. The parentheses indicate the globe artichoke, cultivated cardoon and wild cardoon clusters defined by [20, 21, 26]. (B) Principal co-ordinate analysis based on the genetic distance matrix of 46 individuals, including the parents (red circles) and progeny of the three mapping populations GxG: within scolymus, GxC: scolymus × altilis, GxW: scolymus × sylvestris.
Distribution of microsatellite
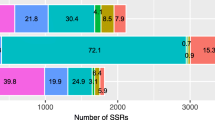
Of the 4,219 microsatellites, trinucleotide motifs accounted for 49%, dinucleotides for 33%, hexanucleotides for 13%, tetranucleotides for 3% and pentanucleotides for 2% (Figure 5). Only ESTs (2,498 of the 3,308) having a non-ambiguous ortholog in Arabidopsis thaliana were taken forward for the purpose of annotation. The position of the microsatellite tract (5'-UTR or 3'-UTR or CDS) was derived from the BlastX result in conjunction with the ORF (Open Reading Frame)-Predictor algorithm [28]. About 33% of the microsatellites were present in the 5'-UTRs, ~ 20% in the 3'-UTRs and ~ 47% in the CDSs (Figure 5), similar to the relative frequencies in both the A. thaliana and rice genomes [15]. Most of the CDS microsatellites consisted of trinucleotides, while dinucleotides were the most abundant in the 5'-UTRs, and di- and trinucleotides were equally represented in the 3'-UTRs. Tetra- and pentanucleotide motifs were more frequent in the 3'-UTRs than in either the CDSs or the 5'-UTRs (data not shown). Trinucleotide (and hexanucleotide) motifs are expected to predominate in the population of CDS microsatellites, as variation in their repeat number is not associated with a frame shift event [29]. The most abundant dinucleotide repeat was AG/CT, followed by AC/GT, although AT/TA predominated in the 3'-UTRs. Among the trinucleotides, the most frequent was AAG/CTT, followed by ATC/GAT and CAC/GTG (Figure 6). This distribution is consistent with the situation in A. thaliana and Brassica spp. orthologs, in which a preference for AG/CT and AAG/CTT motifs has been identified in the 5'-UTRs, thought to be associated with the cis-acting regulation of transcription [30]. In the globe artichoke 5' UTRs, dinucleotide motifs were over-represented, with AG/CT being the most abundant (Figure 6), similar to the situation in the 5'-UTRs of many plant (both mono- and dicotyledonous species) genes [31, 32], which has been reported to play a role in post-transcriptional gene regulation at the RNA level [33, 34]. Dinucleotide motifs were also frequent in the 3'-UTRs, possibly because AT-rich elements are able to act as cis mediators of mRNA turnover [33]. Overall, present data confirm that homopurine/homopyrimidine repeats contribute markedly in 5'-UTR and CDS, as previously reported by Morgante et al [15].

Position and motifs of EST microsatellites. (A) Distribution within specific regions of the unigenes. (B) Frequencies of repeat motifs within the unigene set.

Distribution of microsatellite classes. Di- and trinucleotide classes belonging to each unigene region (5'-UTR, coding sequence, 3-UTR).
The function of genes containing microsatellites
Microsatellites within coding sequences can have a major effect on gene activity, since the expansion/contraction of triplets within the coding sequence alters the gene product, thereby sometimes causing a significant phenotypic change. In humans, the effects on phenotype due to the presence of SSRs in coding regions of genes playing key roles in neuronal disorders and cancer have been extensively studied [35]. Among the microsatellites in the globe artichoke transcriptome, the six most frequent amino acid stretches present in the CDS were poly-serine (94 unigenes), poly-aspartic acid (75 unigenes), poly-glutamic acid (57 unigenes), poly-lysine (46 unigenes), poly-glycine (45 unigenes) and poly-threonine (35 unigenes). It has been reported that particular amino acid repeats tend to be associated with specific classes of proteins [36]. Acidic and polar amino acid repeats have generally been associated with transcription factors and protein kinases, whereas serine repeats are common within membrane transporter proteins [37]. In the globe artichoke, poly-serine and poly-glycine stretches are particularly frequent in the CDS. Poly-serine linkers are common in eukaryotic genomes, and are thought to provide a flexible interdomain. They are frequently associated with modular proteins, and are involved in complex carbohydrate degradation [38] and the binding of proteins with extracellular matrix components, such as the laminin binding protein. Poly-glycine (also poly-asparagine and poly-proline) microsatellites may provide a domain for DNA binding or protein-protein interactions, and has been found to be necessary for chloroplast envelope targeting. Poly-glutamic and poly-aspartic acid tracts feature in many NLS (nuclear localisation signal) proteins [39], and it has been suggested that both basic karyophilic and acidic clusters can enhance their selective binding to transport machinery components [40]. Poly-glutamic acid stretches have also been implicated in transcription activation/de-activation [41–45], and microsatellite allelic variants of these genes have been identified as the genetic basis of a number of human diseases [46].
To support the occurrence of certain repeated motifs in the assembled unigenes we have exploited sequence alignment and gene ontology in order to annotate their functions and assess whether their motif type and position are preferentially associated with a particular gene ontology term.
In preparation, the set of globe artichoke unigenes was filtered to include only those with a BlastX E-value of < 1e-29 when matched with the A. thaliana reference protein set. In all, 12,783 queries satisfied this criterion (Additional file 6: 12,783 globe artichoke unigenes annotation). The A. thaliana gene accession numbers were used to categorize the unigenes using TAIR gene ontology (data not shown). The GoStat2 web interface was then used to identify gene ontology categories which were over-represented. By comparing either the set of microsatellite-containing unigenes, or subsets of it (e.g.: genes including di- or trinucleotide motifs in their CDS or UTRs) with the complete set of annotated unigenes, it was possible to identify over-representation in gene ontology (GO) categories (Table 3). Microsatellites appeared to be over-represented in loci involved in certain biological processes and functions, while no significant association was found with GO cell components (data not shown).
Most of the unigenes containing trinucleotide motifs in their CDS were associated with nucleic acid metabolic processes (GO:0006139), transcription (GO:0006350) and the regulation of transcription (GO:0006355), consistent with the encoding by the GAT trinucleotide of aspartic acid, since stretches of this residue are characteristic of 'karyophilic' acidic clusters in NLS (nuclear localization signal) proteins. Similarly, the AAG/TTC motif, which occurred frequently in the CDS, encodes poly-glutamate, which is thought to be involved in both protein-DNA complex assembly (GO:0065004) and heterocyclic metabolic processes (GO:0046483). Unigenes carrying dinucleotide motifs in their CDS were found to be specifically associated with the response to stimulus (GO:0050896). The AG/CT repeats in the CDSs were over-represented among genes responding to stress (GO:0006950), involved in DNA repair (GO:0006281) and in nucleic acid binding (GO:0003676). This is consistent with the presence of domains involved in protein-RNA/protein-protein sticky interactions.
The commonest microsatellite motifs occurring in 5'-UTR of unigenes were dinucleotide repeats (mostly AG/CT). These unigenes were associated with nucleic acid metabolism (GO:0006139), the regulation of gene expression (GO:0010468), transcription (GO:0006350) and the regulation of transcription (GO:0006355). AG/CT repeats were also over-represented in genes involved in the response to ABA (GO:0009737 and GO:0009738). Moreover, trans-acting elements (GO:0003700: "transcription factor activity"), which show an over-representation of trinucleotidic (ATC/GAT) in their CDSs, were also frequently enriched in their 5'UTRs by AG/CT motifs, suggesting a cascade of signal transmission. Trinucleotide motifs were not common in the 5'-UTRs, except in genes involved in the response to auxin stimulus (GO:0009733).
Conclusion
We have demonstrated here the utility of a set of de novo globe artichoke EST-based microsatellite markers for the definition of genetic diversity, phylogeny and genetic mapping. Since EST microsatellites lie within expressed sequences, they have the potential to represent perfect markers for genes underlying phenotypic variation. Most of these assays are fully transferable to other C. cardunculus taxa, providing anchor points for the integration of taxon-specific genetic maps. The functional annotation of these EST sequences increases their utility as a source of gene-based markers for the study of synteny and other applications.
Methods
EST microsatellites discovery and primer design
A collection of 36,321 EST, generated from the 'Green Globe' variety of C. cardunculus var. scolymus, as part of the output of the Compositae Genome Project http://compgenomics.ucdavis.edu, was downloaded from the NCBI database http://www.ncbi.nlm.nih.gov. To generate a set of unique assemblies, the sequences were first trimmed to remove any remaining vector fragments and polyA tails, using the perl script SeqCleaner, and assembled adopting a second perl script, TGICL, employing the following parameters: p = 95 (identity percentage), l = 40 (minimum overlap length), v = 10 (maximum length of unmatched overhangs); the maximum mismatch overhang was set to 10 bp, since the sequences had already been purged of vector stretches and polyA tails. The two scripts are available at http://compbio.dfci.harvard.edu/tgi/software. The unigene set was then searched for perfect microsatellite sequences using a modified SSRIT perl script [47], with the minimum number of dinucleotides set as five, of tri-, tetra- and penta-nucleotides set as four, and of hexanucleotides as three. A sample of 300 non redundant microsatellite-containing sequences, selected to include the longer microsatellite motifs, was taken forward for PCR screening. Primer design was carried out using BatchPrimer3 http://probes.pw.usda.gov/batchprimer with an optimal GC content of 50%, a maximum melting temperature difference of 3°C, variable amplicon size (to allow multiplexing), and all other parameters set to default values. The de novo microsatellite markers were prefixed with 'CyEM' (Cynara Expressed Microsatellite) and numbered sequentially.
Plant materials and genomic DNA isolation
DNA was extracted from young leaves following a modified CTAB method [48]. The primers were used to amplify genomic DNA template extracted from a germplasm panel consisting of twelve globe artichoke genotypes, representative of crops grown in the Mediterranean Basin [20]; nine cultivated cardoon genotypes, representative of both the Spanish and Italian gene pools [21]; and seven wild cardoon genotypes sampled from both Sicily and Sardinia [26]. Full genotypes details are reported in Table 1. The set also included DNA of the four parents of three established mapping populations, i.e. two globe artichoke accessions ['Romanesco C3' (C3) and 'Spinoso di Palermo' (SP)], one cultivated cardoon ('Altilis 41') and one wild cardoon ('Creta 4'); furthermore six F1 individuals from each of the segregating populations (C3 × SP, C3 × Altilis 41 and C3 × Creta 4) were included in the analyses.
Genotyping and diversity analysis
Primer pairs CyEM-001 to CyEM-300 (Additional file 4: primer pairs designed) were tested for their informativeness on the germplasm panel. Amplification was carried out in 10 μl reactions containing 7 ng template DNA, 1× PCR Buffer (Qiagen Inc., Venlo, Netherlands), 1.0 mM MgCl2, 0.5 U Taq DNA polymerase (Qiagen), 40 nM 5'-labelled (FAM, HEX or TAMRA) forward primer, 40 nM unlabelled reverse primer and 0.2 mM dNTPs. A touchdown cycling regime was applied, consisting of 1 cycle at 94°C for 150 sec, 9 cycles at 94°C for 30 sec, 63°C for 30 sec (-0,7°C/cycle) and 72°C for 60 sec, then 30 cycles at 94°C for 30 sec, 57°C for 30 sec and 72°C for 60 sec, followed by a final extension at 72°C for 5 min.
Weakly amplified reactions were re-run using 1.5 mM MgCl2 and applying a final annealing temperature of 55°C. Amplicons were separated on an ABI3730 capillary DNA sequencer (Applied Biosystem Inc., Foster City, CA, USA). Internal ROX-labelled GS500 size standards were included in each capillary. Fragment data were analysed using GeneMapper v3.5 software (Applied Biosystems). The genotypic data were analysed using the GenAlex Excel package [49]. Genetic diversity was calculated separately for the globe artichoke, cultivated cardoon and wild cardoon genotypes on the basis of (1) the mean number of alleles observed per locus (no), (2) the effective number of alleles per locus (ne) as predicted by 1/Σpi2 where pi is the frequency of the ith allele at the locus, (3) the mean observed heterozygosity (Ho), and (4) the polymorphic information content (PIC), estimated following [49]. A co-phenetic distance matrix for co-dominant markers was generated as described by Smouse and Peakall [50] and used to construct a UPGMA-based dendrogram [51] by means of NTSYS software package v2.10 [52]. Principal co-ordinate analysis was based on the distance matrix, with data standardization provided by the GenAlex package.
Annotation of the unigene set
The unigene set was aligned by a BlastX [53] search against the A. thaliana reference proteins database (NCBI), applying an E-value threshold of e-29. The location within the gene sequence of the microsatellite (5'-UTR, CDS or 3'-UTR) was inferred from this alignment, while ORF-Predictor [28] was used to predict the position of the stop codon. The frequencies of peptide repeat tracts within the CDS were used to identify any over-representation of particular triplets. For this purpose, the unigenes were divided into ten subgroups on the basis of the identity of the trinucleotide motif present in the CDS. Each subgroup was then subjected to an analysis based on the ORF-Predictor algorithm, considering only the positive reading frames (+1, +2, +3), since the sequenced transcripts were originally directionally cloned. All the unigenes were assigned a function based on the GeneOntology tool http://www.arabidopsis.org, using the A. thaliana orthologs (from BlastX output) as input (using AGI codes from TAIR). Enrichment within the GO hierarchical levels by mean of different subset of unigenes was estimated using the GoStat2 interface http://gostat.wehi.edu.au/cgi-bin/goStat2.pl based on Fisher's exact test [54], adopting a threshold p-value of 0.01 and considering terms starting from the 3rd hierarchical level of the DAG (directed acyclic graph; Table 3).
References
Rottenberg A, Zohary D: The wild ancestry of the cultivated artichoke. Genet Resour Crop Ev. 1996, 43 (1): 53-58. 10.1007/BF00126940.
Lanteri S, Portis E: Globe Artichoke and Cardoon. Vegetables I. 2008, Edited by Springer, New York: Springer, 1: 49-74. full_text.
Kraft K: Artichoke leaf extract - Recent findings reflecting effects on lipid metabolism, liver and gastrointestinal tracts. Phytomedicine. 1997, 4 (4): 369-378.
Gebhardt R: Antioxidative and protective properties of extracts from leaves of the artichoke (Cynara scolymus L) against hydroperoxide-induced oxidative stress in cultured rat hepatocytes. Toxicol Appl Pharm. 1997, 144 (2): 279-286. 10.1006/taap.1997.8130.
Gebhardt R: Inhibition of cholesterol biosynthesis in primary cultured rat hepatocytes by artichoke (Cynara scolymus L.) extracts. J Pharmacol Exp Ther. 1998, 286 (3): 1122-1128.
Llorach R, Espin J, Tomas-Barberan F, Ferreres F: Artichoke (Cynara scolymus L.) byproducts as a potential source of health-promoting antioxidant phenolics. J Agr Food Chem. 2002, 50 (12): 3458-3464. 10.1021/jf0200570.
Wang M, Simon J, Aviles I, He K, Zheng Q, Tadmor Y: Analysis of antioxidative phenolic compounds in artichoke (Cynara scolymus L.). J Agr Food Chem. 2003, 51 (3): 601-608. 10.1021/jf020792b.
McDougall B, King P, Wu B, Hostomsky Z, Reinecke M, Robinson W: Dicaffeoylquinic and dicaffeoyltartaric acids are selective inhibitors of human immunodeficiency virus type 1 integrase. Antimicrob Agents Ch. 1998, 42 (1): 140-146.
Halvorsen B, Carlsen M, Phillips K, Bohn S, Holte K, Jacobs D, Blomhoff R: Content of redox-active compounds (ie, antioxidants) in foods consumed in the United States. Am J Clin Nutr. 2006, 84 (1): 95-135.
Lopez-Molina D, Navarro-Martinez M, Melgarejo F, Hiner A, Chazarra S, Rodriguez-Lopez J: Molecular properties and prebiotic effect of inulin obtained from artichoke (Cynara scolymus L.). Phytochemistry. 2005, 66 (12): 1476-1484. 10.1016/j.phytochem.2005.04.003.
Raccuia S, Melilli M: Cynara cardunculus L., a potential source of inulin in the Mediterranean environment: screening of genetic variability. Aust J Agr Res. 2004, 55 (6): 693-698. 10.1071/AR03038.
Lanteri S, Acquadro A, Comino C, Mauro R, Mauromicale G, Portis E: A first linkage map of globe artichoke (Cynara cardunculus var. scolymus L.) based on AFLP, S-SAP, M-AFLP and microsatellite markers. Theor Appl Genet. 2006, 112 (8): 1532-1542. 10.1007/s00122-006-0256-8.
Acquadro A, Lanteri S, Scaglione D, Arens P, Vosman B, Portis E: Genetic mapping and annotation of genomic microsatellites isolated from globe artichoke. Theor Appl Genet. 2009, 118 (8): 1573-1587. 10.1007/s00122-009-1005-6.
Portis E, Mauromicale G, Mauro R, Acquadro A, Scaglione D, Lanteri S: Construction of a reference molecular linkage map of globe artichoke (Cynara cardunculus var. scolymus). Theor Appl Genet. 2009,
Morgante M, Hanafey M, Powell W: Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat Genet. 2002, 30 (2): 194-200. 10.1038/ng822.
Kumpatla SP, Mukhopadhyay S: Mining and survey of simple sequence repeats in expressed sequence tags of dicotyledonous species. Genome. 2005, 48 (6): 985-998. 10.1139/g05-060.
Heesacker A, Kishore V, Gao W, Tang S, Kolkman J, Gingle A, Matvienko M, Kozik A, Michelmore R, Lai Z, et al: SSRs and INDELs mined from the sunflower EST database: abundance, polymorphisms, and cross-taxa utility. Theor Appl Genet. 2008, 117 (7): 1021-1029. 10.1007/s00122-008-0841-0.
Simko I, Hu J: Population structure in cultivated lettuce and its impact on association mapping. J Am Soc Hortic Sci. 2008, 133 (1): 61-68.
Ellegren H: Microsatellites: Simple sequences with complex evolution. Nat Rev Genet. 2004, 5 (6): 435-445. 10.1038/nrg1348.
Lanteri S, Saba E, Cadinu M, Mallica G, Baghino L, Portis E: Amplified fragment length polymorphism for genetic diversity assessment in globe artichoke. Theor Appl Genet. 2004, 108 (8): 1534-1544. 10.1007/s00122-003-1576-6.
Portis E, Barchi L, Acquadro A, Macua J, Lanteri S: Genetic diversity assessment in cultivated cardoon by AFLP (amplified fragment length polymorphism) and microsatellite markers. Plant breeding. 2005, 124 (3): 299-304. 10.1111/j.1439-0523.2005.01098.x.
Acquadro A, Portis E, Albertini E, Lanteri S: M-AFLP-based protocol for microsatellite loci isolation in Cynara cardunculus L. (Asteraceae). Mol Ecol Notes. 2005, 5 (2): 272-274. 10.1111/j.1471-8286.2005.00897.x.
Tang S, Knapp S: Microsatellites uncover extraordinary diversity in native American land races and wild populations of cultivated sunflower. Theor Appl Genet. 2003, 106 (6): 990-1003.
Paniego N, Echaide M, Munoz M, Fernandez L, Torales S, Faccio P, Fuxan I, Carrera M, Zandomeni R, Suarez E, et al: Microsatellite isolation and characterization in sunflower (Helianthus annuus L.). Genome. 2002, 45 (1): 34-43. 10.1139/g01-120.
Wiel van de C, Arens P, Vosman B: Microsatellite retrieval in lettuce (Lactuca sativa L.). Genome. 1999, 42 (1): 139-149. 10.1139/gen-42-1-139.
Portis E, Acquadro A, Comino C, Mauromicale G, Saba E, Lanteri S: Genetic structure of island populations of wild cardoon [Cynara cardunculus L. var. sylvestris (Lamk) Fiori] detected by AFLPs and SSRs. Plant Sci. 2005, 169 (1): 199-210. 10.1016/j.plantsci.2005.03.014.
Portis E, Mauromicale G, Barchi L, Mauro R, Lanteri S: Population structure and genetic variation in autochthonous globe artichoke germplasm from Sicily Island. Plant Sci. 2005, 168 (6): 1591-1598. 10.1016/j.plantsci.2005.02.009.
Min X, Butler G, Storms R, Tsang A: OrfPredictor: predicting protein-coding regions in EST-derived sequences. Nucleic Acids Res. 2005, 33: W677-W680. 10.1093/nar/gki394.
Metzgar D, Bytof J, Wills C: Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000, 10 (1): 72-80.
Zhang L, Zuo K, Zhang F, Cao Y, Wang J, Zhang Y, Sun X, Tang K: Conservation of noncoding microsatellites in plants: implication for gene regulation. BMC Genomics. 2006, 7: 323-10.1186/1471-2164-7-323.
Guo H, Moose S: Conserved noncoding sequences among cultivated cereal genomes identify candidate regulatory sequence elements and patterns of promoter evolution. Plant cell. 2003, 15 (5): 1143-1158. 10.1105/tpc.010181.
Yang Y, Lai K, Tai P, Li W: Rates of nucleotide substitution in angiosperm mitochondrial DNA sequences and dates of divergence between Brassica and other angiosperm lineages. J Mol Evol. 1999, 48 (5): 597-604. 10.1007/PL00006502.
Mignone F, Grillo G, Licciulli F, Iacono M, Liuni S, Kersey P, Duarte J, Saccone C, Pesole G: UTRdb and UTRsite: a collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 2005, 33: D141-D146. 10.1093/nar/gki021.
Martin P, Makepeace K, Hill S, Hood D, Moxon E: Microsatellite instability regulates transcription factor binding and gene expression. P Natl Acad Sci USA. 2005, 102 (10): 3800-3804. 10.1073/pnas.0406805102.
Li Y, Korol A, Fahima T, Nevo E: Microsatellites within genes: Structure, function, and evolution. Mol Biol Evol. 2004, 21 (6): 991-1007. 10.1093/molbev/msh073.
Richard G, Dujon B: Association of transcripts from a group-I intron-containing gene with high sedimentation coefficient particles. Curr Genet. 1997, 32 (3): 175-181. 10.1007/s002940050263.
Alba M, Santibanez-Koref M, Hancock J: Amino acid reiterations in yeast are overrepresented in particular classes of proteins and show evidence of a slippage-like mutational process. J Mol Evol. 1999, 49 (6): 789-797. 10.1007/PL00006601.
Shen H, Schmuck M, Pilz I, Gilkes N, Kilburn D, Miller R, Warren R: Deletion of the linker connecting the catalytic and cellulose-binding domains of endoglucanase-A (cena) of cellulomonas-fimi alters its conformation and catalytic activity. J Biol Chem. 1991, 266 (17): 11335-11340.
Pearson C, Edamura K, Cleary J: Repeat instability: Mechanisms of dynamic mutations. Nat Rev Genet. 2005, 6 (10): 729-742. 10.1038/nrg1689.
Vancurova I, Vancura A, Lou W, Paine P: Nucleoplasmins poly-glutamic acid tract is required for facilitated transport and enhances intranuclear binding. Mol Biol Cell. 1995, 6: 1826-1826.
Toth G, Gaspari Z, Jurka J: Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res. 2000, 10 (7): 967-981. 10.1101/gr.10.7.967.
Berger M, Sionov R, Levine A, Haupt Y: A role for the polyproline domain of p53 in its regulation by Mdm2. J Biol Chem. 2001, 276 (6): 3785-3790. 10.1074/jbc.M008879200.
Gerber H, Seipel K, Georgiev O, Hofferer M, Hug M, Rusconi S, Schaffner W: Transcriptional activation modulated by homopolymeric glutamine and proline stretches. Science. 1994, 263 (5148): 808-811. 10.1126/science.8303297.
Kolaczkowska A, Kolaczkowski M, Delahodde A, Goffeau A: Functional dissection of Pdr1p, a regulator of multidrug resistance in Saccharomyces cerevisiae. Mol Genet Genomics. 2002, 267 (1): 96-106. 10.1007/s00438-002-0642-0.
Perutz M, Johnson T, Suzuki M, Finch J: Glutamine repeats as polar zippers - their possible role in inherited neurodegenerative diseases. P Natl Acad Sci. 1994, 91 (12): 5355-5358. 10.1073/pnas.91.12.5355.
Leroy X, Leon K, Branchard M: Plant genomic instability detected by microsatellite-primers. Electronic Journal of Biotechnology. 2000, 3 (2): 140-148.
Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch S: Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): Frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 2001, 11 (8): 1441-1452. 10.1101/gr.184001.
Lanteri S, Di Leo I, Ledda L, Mameli M, Portis E: RAPD variation within and among populations of globe artichoke cultivar 'Spinoso sardo'. Plant breeding. 2001, 120 (3): 243-246. 10.1046/j.1439-0523.2001.00605.x.
Peakall R, Smouse P: GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes. 2006, 6 (1): 288-295. 10.1111/j.1471-8286.2005.01155.x.
Smouse P, Peakall R: Spatial autocorrelation analysis of individual multiallele and multilocus genetic structure. Heredity. 1999, 82: 561-573. 10.1038/sj.hdy.6885180.
Sneath PHA, Sokal RR: Numerical taxonomy -- the principles and practice of numerical classification. 1973, W. H. Freeman: San Francisco
Rohlf FJ: NTSYSpc Version 2.0: User Guide. Applied Biostatistics Inc. 1998
Altschul S, Madden T, Schaffer A, Zhang J, Zhang Z, Miller W, Lipman D: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25 (17): 3389-3402. 10.1093/nar/25.17.3389.
Beissbarth T, Speed T: GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics. 2004, 20 (9): 1464-1465. 10.1093/bioinformatics/bth088.
Acknowledgements
Globe artichoke ESTs were produced by the Compositae Genome Project http://compgenomics.ucdavis.edu/. This research was supported by grants from: (i) the National Science Foundation Plant Genome Research Program (No. 0421630), (ii) the Georgia Research Alliance, (iii) the University of Georgia Research Foundation, and (iv) by MIPAAF (Ministero delle Politiche Agricole, Alimentari e Forestali - Italy) through the CAR-VARVI ("Valorizzazione di germoplasma di carciofo attraverso la costituzione varietale ed il risanamento da virus") project.
We are grateful to Prof. Giovanni Mauromicale and Dr. Rosario Mauro (D.A.C.P.A. Agronomical Sciences, University of Catania) for providing the C. cardunculus parental accessions and the F1 individuals.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
SL and SK planned and supervised the work. DS were responsible for the in silico analysis of the EST sequence data, primer design and amplification; AA and EP selected the constitution of the C. cardunculus gerplasm panel; DS, AA, EP and CT analysed the data. All the authors contributed to the final version of the manuscript.
Electronic supplementary material
12864_2009_2338_MOESM1_ESM.XLS
Additional file 1: EST assembly. The data provided represent the list of 6,621 assembled globe artichoke contigs derived from 23,871 ESTs. (XLS 1 MB)
12864_2009_2338_MOESM2_ESM.TXT
Additional file 2: 19,055 unigenes. The data provided represent the fasta sequences of the assembled unigenes (contigs and singletons). (TXT 15 MB)
12864_2009_2338_MOESM3_ESM.ZIP
Additional file 3: ACE assembly file (EagleView available at http://bioinformatics.bc.edu/marthlab/EagleView. Contig representation file parsed from TGICL output file by a customised PERL script. (ZIP 12 MB)
12864_2009_2338_MOESM4_ESM.XLS
Additional file 4: primer pairs designed. The data provided represent the list of the 2,311 designed primer pairs and loci/SSRs description. (XLS 497 KB)
12864_2009_2338_MOESM5_ESM.XLS
Additional file 5: full statistics on 300 SSR-containing loci. The data provided represent the list of 300 selected SSR-containing loci, their allele statistics, polymorphism information, repetitive motif position and gene annotation. (XLS 642 KB)
12864_2009_2338_MOESM6_ESM.XLS
Additional file 6: 12,783 globe artichoke unigenes annotation. The data provided represent the list of 12,783 globe artichoke unigenes annotation using BlastX (e-value threshold < 1e-29) on Arabidopsis reference protein database (June, 2008). (XLS 3 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Scaglione, D., Acquadro, A., Portis, E. et al. Ontology and diversity of transcript-associated microsatellites mined from a globe artichoke EST database. BMC Genomics 10, 454 (2009). https://doi.org/10.1186/1471-2164-10-454
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-10-454