
Abstract
Background
In response to pathogen attack, grapevine synthesizes phytoalexins belonging to the family of stilbenes. Grapevine cell cultures represent a good model system for studying the basic mechanisms of plant response to biotic and abiotic elicitors. Among these, modified β-cyclodextrins seem to act as true elicitors inducing strong production of the stilbene resveratrol.
Results
The transcriptome changes of Vitis riparia × Vitis berlandieri grapevine cells in response to the modified β-cyclodextrin, DIMEB, were analyzed 2 and 6 h after treatment using a suppression subtractive hybridization experiment and a microarray analysis respectively. At both time points, we identified a specific set of induced genes belonging to the general phenylpropanoid metabolism, including stilbenes and hydroxycinnamates, and to defence proteins such as PR proteins and chitinases. At 6 h we also observed a down-regulation of the genes involved in cell division and cell-wall loosening.
Conclusions
We report the first large-scale study of the molecular effects of DIMEB, a resveratrol inducer, on grapevine cell cultures. This molecule seems to mimic a defence elicitor which enhances the physical barriers of the cell, stops cell division and induces phytoalexin synthesis.
Similar content being viewed by others

Background
Plants respond to pathogens through constitutive and inducible mechanisms [1]. Structural barriers represent preformed constitutive defences, while the accumulation of pathogenesis-related proteins (PR), phytoalexins and reactive oxygen species is part of an active mechanism stimulated by the pathogen [2]. Grapevine also responds to fungal infection via PR-protein synthesis and phytoalexin accumulation [3]. Plant phytoalexins are low-molecular-weight secondary metabolites with antimicrobial properties and they show wide chemical diversity among different plant species [4]. In grapevine they mainly belong to the stilbene family and consist of trans-resveratrol (3,5,4'-trihydroxystilbene) its oligomers, called viniferins [5–7] and pterostilbene, a dimethylated derivative of resveratrol [8]. Stilbene synthesis in berries [9] and leaves can be elicited by fungal infection [5, 10], but also by treatment with UV-irradiation [11], ozone [12] and heavy metals [13].
Plant cell cultures are a useful tool for studying plant cell defence response to biotic and abiotic elicitors [14]. Stilbene accumulation has been reported in grapevine cells treated with different elicitors: fungal cell wall fragments [15], Na-orthovanadate, jasmonic acid and methyljasmonate [16, 17] and laminarin, a β-glucan polysaccharide from brown algae [18]. In addition, special attention has been given to the β-cyclodextrin molecular class. These are cyclic oligosaccharides consisting of seven α-D-glucopyranose residues linked by α 1 → 4 glucosidic bonds forming a structure with a hydrophobic central cavity and a hydrophilic external surface [19]. Among β-cyclodextrins, heptakis(2,6-di-O-methyl)-β-cyclodextrin (DIMEB), was reported to be the most effective resveratrol elicitor in different Vitis vinifera cultivars [19, 20]. The ability of the modified β-cyclodextrins to act as elicitors probably resides in their chemical similarity to the alkyl-derivatized pectic oligosaccharides released from the cell walls during fungal infection [20]. Along with stilbene accumulation these experiments highlighted a more general response involving peroxidase activity as well as inhibition of Botrytis cinerea growth [19, 20].
Zamboni et al. [21] further investigated DIMEB activity on additional Vitis genotypes and observed that its effect was more pronounced when tested on Vitis riparia × Vitis berlandieri cell cultures. The kinetics of resveratrol synthesis showed that trans-resveratrol, the induced form, started to accumulate from 6 h after treatment and reached its maximum at 24 h. Moreover, this metabolite was much more localized in the medium than within the cell.
With these results [21] as our starting point, we report here the first large-scale transcriptional characterization of the early response of Vitis riparia × Vitis berlandieri cells to DIMEB treatment.
After 2 h, 127 positively modulated genes were identified by suppression subtractive hybridization (SSH), whereas after 6 h, 371 genes turned out to be differentially expressed when control and treated cells on the Vitis vinifera GeneChip® Genome Array (Affymetrix) were compared. These results showed that DIMEB specifically modulates the expression of a small number of genes involved in resveratrol and lignin biosynthesis, PR synthesis, cell division and cell wall modification.
Results and discussion
The ability of DIMEB to elicit defence responses in grapevine cell culture was suggested by previous results showing stilbene accumulation, changes in peroxidase activity, as well as inhibition of Botrytis cinerea growth [19, 20]. Considerable stilbene accumulation in response to DIMEB treatment was also observed by our group using non-vinifera (Vitis riparia × Vitis berlandieri) liquid cell cultures [21]. In this study we analyzed the changes in gene expression of these cells elicited with DIMEB after 2 h and 6 h using SSH and microarray experiments, respectively.
The rationale behind the two approaches was that after 2 h of treatment, a small number of genes are expected to be modulated, and only to a limited extent, whereas after 6 h an increase in the number of genes and in their expression level is envisaged. The SSH technique appeared then the right choice for identifying the low abundance differential transcripts at 2 h, while the Affymetrix GeneChip® microarray was used to measure the expression of a larger number of genes (~14,500 unigenes) after 6 h of treatment [22].
Starting with 384 clones from the constructed cDNA subtractive library and then performing a hybridization screening to eliminate clones which were not really differentially expressed (false positives), we obtained 168 high-quality sequences which clustered in 127 tentative consensuses (Additional File 1). The microarray experiments instead identified 371 (223 upregulated and 148 downregulated) significantly modulated probe sets in the treated cells compared with the control ones (Additional File 2). Sequence annotation and classification according to Gene Ontology categories [23], revealed that at both time points primary (mainly signal transduction related genes) and secondary metabolisms, together with response to the stimulus, were the most affected categories (Additional Files 3 and 4). At 6 h, the analysis also highlighted downregulation of the cellular component organization and the biogenesis category (Additional file 4).
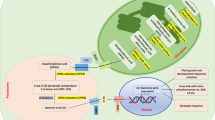
In general, the two experiments showed modulation of specific mechanisms had already occurred at 2 h and continued more extensively at 6 h after DIMEB treatment. The data summarized in Table 1 suggest that the grapevine cell responds to the elicitor by the activation of a signal transduction cascade which leads to the induction of specific classes of transcription factors. The downstream effect of this process is, on the one hand, the induction of some branches of the secondary metabolism and defence response, and, on the other hand, the blockage of cell duplication (Figure 1).

Molecular events triggered by DIMEB as deduced by transcriptional profiling.
At 2 h the treatment caused positive transcriptional regulation of a grapevine gene (CLU090) encoding a protein with homology to an Arabidopsis kinase-associated protein phosphatase (KAPP) (Table 1). KAPP protein may function as a signalling component in the pathway involving the serine-threonine receptor-like kinase, RLK5 of Arabidopsis [24]. In rice the RLK XA21 confers resistance to bacterial blight disease [25]. Other genes possibly involved in signal transduction showed overexpression at 6 h: a gene (1620080_at) with homology to a putative receptor-like protein kinase ARK1 of Oryza sativa and a gene (1611172_at) homologous to a Glycine max Salt Overly Sensitive gene encoding a SOS2-like protein kinase (Table 1). In Arabidopsis thaliana ARK genes seem to be involved in plant defence response to wounding and to bacterial infections [26], while SOS2 is a signalling kinase involved in salt tolerance response [27]. Phospholipid-derived molecules are emerging as novel second messengers in plant defence signalling and phospholipases are key enzymes for their synthesis [14, 28]. In the array experiment we observed the overexpression of a putative phospholipase gene (1608981_at), which may generate lipid messengers for the signalling response (Table 1).
The activation of a signal cascade generally induces the expression of genes encoding for specific transcription factors, which in turn regulate downstream effector genes.
Two genes, upregulated at 6 h, showed homology to a hot pepper WRKY-b (1610775_s_at) and Arabidopsis WRKY11 (1611285_s_at) respectively (Table 1). WRKY proteins are plant-specific transcription factors whose expression is modulated in response to wounding, pathogen infection and abiotic stress [29]. Other classes of transcription factors appeared to take part in regulation of the response of grapevine cells to DIMEB treatment. The grape homologue (1619311_at) of a tomato pathogenesis-related gene transcriptional activator PTI5 was upregulated at 6 h (Table 1). This transcription factor binds to the GCC-box cis element present in the promoter region of many plant PR genes [30] and its upregulation could explain the observed induction of many PR proteins in this experiment. Another sequence (CLU059), induced at 2 h, which might modulate the expression of PR genes is the homologue of the tobacco bZIP TGA10 factor (Table 1). It has been reported that this protein can bind to the regulatory activation sequence-1 (as-1) [31] identified in the promoter of Arabidopsis PR-1 gene [32].
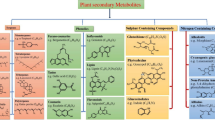
Our results indicated that one of the final grapevine cell responses to the DIMEB-elicited signal consists in the modulation of phenolic metabolism, especially stilbene and monolignol biosynthesis (Figure 2).

Modulation of secondary metabolism at 2 and 6 h after DIMEB treatment. Modulation (+ or -) of genes encoding enzymes of phenylalanine biosynthesis, general phenylpropanoid metabolism, monolignol, stilbene and anthocyanin pathways are reported within a simplified secondary metabolism scheme. Abbreviations: DHAP synthase, 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase; DHQ synthase, 3-dehydroquinate synthase; CM, chorismate mutase; PDT, prephenate dehydratase; PAL, phenylalanine ammonia-lyase; C4H, cinnamate 4-hydroxylase; 4CL, 4-coumarate-CoA ligase; CAD, cinnamyl alchol dehydrogenase; CCoAOMT, caffeoyl-CoA 3-O-methyltransferase; COMT, caffeic acid O-methyltransferase; CCR, cinnamoyl-CoA reductase; F5H, ferulate-5-hydroxylase; STS, stilbene synthase; CHI, chalcone isomerase; UFGT, flavonoid-3-O-glucosyltransferase.
Genes encoding enzymes involved in phenylalanine biosynthesis such as 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase (CLU083; 1611211_at; 1614440_at; 1619357_at; 1621405_at), 3-dehydroquinate synthase (1609646_at), prephenate dehydratase (1609932_at; 1621307_at) and chorismate mutase (1611895_at) were positively modulated both at 2 and 6 h after DIMEB treatment (Table 1). These enzymes participate in the synthesis of aromatic amino acids, particularly of phenylalanine, which is the link between primary and secondary metabolism, being a precursor of general phenylpropanoid metabolism. A recent report showed that cyclodextrins stimulates the expression of the structural genes of the general phenylpropanoids metabolism which sustains the synthesis of p-cumaroyl CoA, one of the two precursors of stilbenes [17].
Although we focused on the earlier cell response time, at both time points we also observed upregulation of this pathway's genes, namely phenylalanine ammonia lyase (1613113_at), cinnamic acid 4-hydroxylase (CLU024; 1610821_at; 1616191_s_at) and 4-coumarate-CoA ligase (1615801_at; 1619320_at) (Table 1). Similarly, several stilbene synthase genes were induced at 2 h and 6 h (CLU009, CLU022, CLU023, CLU049, CLU097, CLU103, 1606750_at, 1608009_s_at, 1609696_x_at, 1609697_at, 1610824_s_at, 1610850_at, 1611190_s_at, 1612804_at, 1614621_at, 1616575_at, 1620964_s_at, 1622638_x_at). According to the classification proposed by Richter et al. [33], they correspond to 7 different stilbene synthase genes plus one pseudogene (1606750_at). In particular, the probeset 1616575_at, encoding a stilbene synthase 2, appeared to be the most induced one, being 23 times higher in the DIMEB treated sample with respect to the control. In agreement, the chemical analysis proved stilbene accumulation in the medium already at 2 h and at higher levels after 6 h, as previously reported [21].
The accumulation of stilbenes in the growth medium requires, besides stilbene biosynthesis, the presence of export machinery. In fact, induction of genes encoding putative secondary metabolite transporters, such as those belonging to the ATP-binding cassette (ABC) transporter family, was found. Genes encoding for pleiotropic drug resistance (PDR)-like ABC transporters (CLU106; CLU119), ABC transporter-like proteins (1613763_at; 1618493_s_at) and a CjMDR transporter (1610363_at) were indeed induced (Table 1). The ABC transporters play an important role in some host-pathogen interactions [34]. In some pathogenic fungi they are involved in resistance to plant phytolexins and antifungal compounds, while in plants they seem to take part in plant defence response [34]. The induction of genes encoding glutathione S-transferase (1609330_at; 1611890_at) at 6 h correlates well with the ABC-mediated transport (Table 1). A glutathione moiety seems to function as a "recognition tag" for the transport of phenols [35]. Resveratrol translocation outside the cells has two main objectives: to mediate the defence response against pathogens and to avoid intracellular accumulation of this compound at cytotoxic levels.
Phenylpropanoid metabolism also produces the precursors (p-coumarate and p-coumaroyl-CoA) for the synthesis of monolignols, which are used to reinforce the cell wall during defence response [36]. DIMEB treatment caused a general induction of genes involved in their synthesis at 6 h: the genes for caffeic acid O-methyltransferase (1607475_s_at, 1619682_x_at, 1620342_at), caffeoyl-CoA O-methyltransferase (1611897_s_at; 1614643_at), cinnamyl alcohol dehydrogenase (1613900_at), ferulate 5-hydroxylase (1614045_at; 1614502_at) and cinnamoyl-CoA reductase (1619065_at) were overexpressed (Table 1, Figure 2). Genes coding for enzymes such as polyphenol oxidase and diphenol oxidase, probably responsible for the lignin polymerization process [36], were induced as well (1622651_at; 1610806_at) (Table 1).
The other branches of phenolic metabolism seemed not to be affected by DIMEB. Only two genes of the anthocyanin pathway (a chalcone-flavonone isomerase (CLU122) and a flavonol-3-O-glucosyltransferase (CLU048)) were induced at 2 h but not at 6 h (Table 1, Figure 2). Interestingly, selective induction of the early steps of phenylpropanoid metabolism and of the late steps leading to monolignol biosynthesis was also described in Arabidopsis in the early response to oligogalacturonide treatment [37].
The results strongly suggest that DIMEB acts as an elicitor modifying cell metabolism to promote the accumulation of phytoalexins and cell wall lignification. These two defence responses have been described as typical biochemical responses occurring in vegetal cells after elicitor exposure [14].
The transcriptional profiling results, however, show that the response to DIMEB seems to include other defence mechanisms. Overexpression of sequences for pathogenesis-related proteins such as chitinase (CLU088; 1613871_at; 1617192_at; 1617430_s_at), PR-10 (CLU001; 1610011_s_at; 1618568_s_at) and PR-4 (CLU021), but also for a prolin-rich protein (CLU036) and a protease inhibitor (1609875_at; 1611666_s_at) was observed in both experiments, while upregulation of two genes encoding the S-adenosyl-L-methyonine:salicylic acid carboxyl methyltransferase (1612552_at; 1620309_at) was recorded at 6 h (Table 1). Interestingly, this enzyme mediates the synthesis of gaseous methyl salicylate which was recently demonstrated to be a key mediator in plant systemic acquired resistance [38] in tobacco, as well as an inducer of the expression of PR-1 gene and TMV resistance [39]. This result strengthens the hypothesis that DIMEB acts as a true elicitor. The increase in the expression of a gene encoding for a 1-aminocyclopropane-1-carboxylate oxidase (1622147_at), would suggest the involvement of ethylene as well (Table 1). This hormone is a major regulator of the plant's reaction to pathogen attack [40] and via the action of a group of ethylene responsive factors it modulates the expression of plant defence-related genes such as, for example, phenylalanine ammonia-lyase, hydroxylproline-rich glycoprotein and acid class II chitinase [41, 42]. It appears from the finding that a gene (1616358_at) homologous to an MLO-like 11 of Arabidopsis was downregulated at 6 h (Table 1), that the similarities between the cell's responses upon DIMEB treatment and upon pathogen attack are even greater. In barley, downregulation of the Mlo gene is involved in response to powdery mildew caused by the fungus Blumeria graminis f.sp.hordei[43], and in the dicot Arabidopsis thaliana, resistance to powdery mildews also depends on loss-of-function mlo alleles [44].
Our data support another effect of DIMEB on grapevine cells: blockage of the cell-division process. Upon treatment, we measured a lower expression of the genes involved in modification of the cell wall structure, cell division and microtubule organization. At 6 h, downregulation of genes related to cell wall modification [45], such as those encoding expansins (1608074_s_at; 1620840_at), xyloglucan endotransglycosylase (1615995_at; 1620003_at), pectin methylesterases (1608799_at; 1619468_at), a β-galactosidase (1619522_at), a polygalacturonase (1608756_at) and endoglucanases (1606763_at; 1609506_at; 1610263_at), was observed (Table 1). The sequence 1609506_at corresponds to the VvCEL2 transcript which encodes a grapevine cellulase. Since in Arabidopsis the expression of the cel1 gene was related to growing tissues [46], downregulation of VvCEL2 could be related to repression of the cell growth. Microtubules play an essential role in cell division and cell elongation too. They set the cellular division planes and axes of elongation and influence the deposition and orientation of cellulose microfibrils [47]. The downregulation of genes coding for α- and β-tubulin (1612320_a_at; 1616815_at; 1618413_at; 1619167_at; 1621015_at; 1622466_at) is indication of a stop in cell expansion and cell division (Table 1). mRNA degradation of a β-tubulin isoform was observed in soybean cells elicited by Phytophthora sojae-derived glucan fragments suggesting re-routing of the cellular resources towards the defence-related metabolism and repression of the cellular growth [48].
Further indication of cell division reduction were the lower transcription of genes coding for histones H2A, H3, H4 and H2B (1608927_at; 1612573_at; 1613041_at; 1613076_at; 1620332_at; 1622440_at; 1622737_at), a cyclin (1610854_at), a pattelin protein (1610422_at), a GA-induced-like protein (GIP-like) (1610607_at), a putative formin homology (FH) protein (1613373_at) and a DNA polymerase alpha catalytic subunit gene (1607792_at) (Table 1). All these proteins are either related to DNA organization and synthesis or to the cytokinesis process. The down-regulated grapevine GIP gene is homologous to GIP-5 of Petunia hybrida, which is expressed during the cell division phase in stems and corollas [49]. In Arabidopsis patellin1 plays a role in membrane-trafficking when the cell-plate is formed during cytokinesis [50], and formins are plant cytoskeleton-organizing proteins which take part in cytokinesis and in the establishment and maintenance of cell polarity [51]. Very similar effects on cell growth have been reported upon elicitation of parsley cell cultures with an oligopeptide elicitor. Pep 25 provoked the repression of genes regulating the cell cycle, such as cdc2, cyclin and histones [52].
A likely explanation for the repression of cell division would be the need of the cell to use, almost exclusively, the transcription system as well as the available resources to establish a defence-related metabolism.
Conclusion
The transcriptional profiles measured at 2 h and 6 h after DIMEB treatment highlight the fact that this compound is able to induce an early and specific defence response in grapevine liquid cell cultures, supporting the hypothesis of its role as a true elicitor.
The classes of genes modulated by the treatment reveal that DIMEB triggers a signal transduction cascade which activates different families of transcription factors, in turn modulating the effector genes of specific metabolisms. These results thus suggest that in grapevine cells DIMEB induces a stop in cell division, reinforcement of the cell wall and the production of resveratrol and defence proteins (Figure 3). This response largely resembles that occurring upon pathogen attack.

Cellular processes triggered by DIMEB as deduced by transcriptional profiling. Grapevine cell model showing the major genes involved in the cellular processes modulated by DIMEB treatment. Abbreviations: CAD, cinnamyl alchol dehydrogenase; CCoAOMT, caffeoyl-CoA 3-O-methyltransferase; COMT, caffeic acid O-methyltransferase; CCR, cinnamoyl-CoA reductase; F5H, ferulate-5-hydroxylase; PME, pectin methylesterase; PPO, polyphenol oxidase, PR protein, pathogenesis-related protein; STS, stilbene synthase; XET, xyloglucan endotransglycosylase.
Methods
Plant material
Liquid cell cultures of a cross between Vitis riparia and Vitis berlandieri were used to carry out the treatment experiments with DIMEB (50 mM) [21]. Cell cultures were collected 2 h and 6 h after DIMEB treatment from control and treated samples. Cells and medium were separated by centrifugation at 12.000 ×g for 10 min at room temperature.
Total RNA extraction
Total RNA was extracted from control and treated samples using a modified hot-borate method, as described by Moser et al. [53]. DNA traces were removed by DNase I treatment (Sigma-Aldrich, St.Louis, MO, USA) according to the manufacturer's procedure. RNA was isolated from one replicate for the SSH experiment (2 h) and from 3 biological replicates for the microarray experiment (6 h).
cDNA synthesis and SSH library construction
Double-stranded cDNA was synthesized from 0.6 μg of total RNA of the control and treated samples (2 h) using the SMART™ PCR cDNA synthesis kit (Clontech Laboratories, Mountain View, CA) as recommended by the manufacturer.
Suppression subtractive hybridization (SSH) was carried out using the PCR-Select cDNA subtraction Kit (Clontech Laboratories) according to the manufacturer's procedure. The cDNA from the treated sample was used as the "tester" while the cDNA from the control sample was used as the "driver". Following hybridization, the subtracted cDNA molecules were inserted into a pCR® 2.1-TOPO® Vector (Invitrogen, Carlsbad, CA) and then used to transform One Shot® TOP10 Chemically Competent Escherichia coli cells (Invitrogen). Positive transformants, based on blue/white screening, were picked and arrayed in a 384-well plate containing LB medium (Sigma-Aldrich) supplemented with ampicillin (50 μg mL-1) and glycerol (10% v/v). The SSH cDNA library was stored at -80°C.
Amplification of cDNA inserts and spotting on filters
The SSH library clones were cultured overnight at 37°C in a 384-well plate with LB medium and ampicillin (50 μg mL-1). A small aliquot (1 μl) of each liquid culture was then transferred into four 96-well plates containing PCR mix and used as template to amplify the corresponding cDNA inserts. PCR reactions (95°C for 15 min, 94°C for 45 sec, 68°C for 45 sec, 72°C for 2 min for 35 cycles, 72°C for 7 min) contained 300 nM Nested Primer PCR 1 and 300 nM Nested Primer PCR 2R (Clontech Laboratories), 0.5 U HotStartTaq DNA polymerase (Qiagen, Shanghai, China), 200 μM dNTPs, 1.5 M betain (Sigma-Aldrich) and 80 μM Cresol Red (Sigma-Aldrich). The 40 μl PCR reactions were then concentrated by overnight incubation at 37°C. The human nebulin cDNA (NM_004543) was PCR amplified in the same way to serve as a positive control. One microliter of each concentrated cDNA insert together with one microliter of a 2 ng/μl solution of amplified nebulin were transferred onto 8 × 12 cm Hybond+ nylon membranes (Amersham, GE Healthcare Bio-Sciences AB, Little Chalfont, UK) using a manual 96-pin tool. The samples were arrayed in duplicate according to a 4 × 4 grid pattern. Before and after spotting, membranes were denatured on Whatmann 3 MM paper saturated with denaturation buffer (0.5 M NaOH, 1.5 M NaCl) for 15 min. Membranes were then neutralised on Whatmann 3 MM paper saturated with neutralization buffer (1.5 M NaCl, 0.5 M Tris-HCl, pH 7.2) for 15 min, rinsed in 2× SSC, air dried and crosslinked at 80°C for 2 h.
Target labelling
To assess whether the isolated clones were truly positive, they were hybridized with the same total RNAs used for SSH library construction. The RNAs were DIG-labelled by reverse transcription according to Vernon et al. [54] with the following modifications: 7.5 μl of PCR DNA Labelling MIX 10× (Roche, Basel, Switzerland) and 1.5 μl of 50 μM of Oligo(dT)20 were added to 5 μg of total RNA of each sample (tester and driver). After incubation of the two samples at 65°C for 10 min and then on ice for 2 min, a mix of 6 μl of RT Buffer 5× (Invitrogen), 3 μl of 0.1 M DTT (Invitrogen), 1.5 μl of RNase OUT (40 U/μl) (Invitrogen) and 1.5 μl of Superscript II (200 U/μl) (Invitrogen) was added to each sample. Reverse transcription was performed at 42°C for 1 h and then continued for a further hour after addition of another 1.5 μl of Superscript II (200 U/μl) (Invitrogen). The reaction was stopped by incubation at 70°C for 15 min and was followed by treatment with 1.5 μl of RNase H (2 U/μl) (Invitrogen) at 37°C for 20 min. The digoxigenin-labelled probe of the control target was synthesized by PCR amplification of a portion of human nebulin cDNA cloned in pBluescript II SK/KS (-) (Stratagene) in the presence of PCR DNA Labelling MIX 10×. PCR reaction was carried out in 50 μl using 7 ng/μl of pBluescript II SK (-) containing human nebulin cDNA as template and the primers nebulin-for 5'-CAGGAGACTATTACAGGTTT-3' and nebulin-rev 5'-ACCCATAGGCAGCTTGAGAA-3', according to the manufacturer's procedure. PCR conditions were 95°C for 15 min, 35 cycles of 94°C for 45 sec, 52°C for 45 sec, 72°C for 1 min, followed by 72°C for 7 min.
Hybridization, washing and detection
Two filters were incubated with 20 ml of pre-hybridization solution (5× SSC, 0.1% (w/v) N-lauroylsarcosine, 0.02% (w/v) SDS, 1% (v/v) blocking solution in 1× acid maleic buffer) at 72°C for 30 min. Two different probes were prepared: the first was obtained by mixing the DIG-labelled "tester" DNA (30 μl) with the DIG-labelled human nebulin (2 μl), the second by mixing the DIG-labelled "driver" DNA (30 μl) with the DIG-labelled human nebulin (2 μl). After a short denaturation step (95°C for 3 min) the two probes were incubated separately with one filter each overnight at 68°C in hybridization solution (20 ml, 5× SSC, 0.1% (w/v) N-lauroylsarcosine, 0.02% (w/v) SDS, 1% (v/v) blocking solution in 1× acid maleic buffer). After hybridization, four high-stringency washings at 68°C for 20 min (2× SSC, 0.5% (w/v) SDS) followed by two low-stringency washings (0.2× SSC, 0.5% (w/v) SDS) at 68°C for 20 min, were carried out. Chemiluminescence was detected by 30-min exposure to Kodak® BioMax Light Film (Kodak, Rochester, NY) after incubation with anti-DIG antibodies and CDP-Star, according to the manufacturer's procedure (Roche).
Sequencing of transcripts identified by SSH
Following the screening procedure, the 289 positive clones were amplified, as described above for filter production, but without betain and Cresol Red in the PCR reaction mix. Five microliters of each PCR reaction were purified from primers and nucleotides using 1.5 μl of ExoSAP-IT™ (Amersham) at 37°C for 1 h. The reaction was stopped at 75°C for 15 min. Three nanograms for every 100 bp of amplified fragment were used for the sequencing reaction with Nested PCR Primer 1. Sequencing of 243 positively amplified clones was outsourced to the BMR Sequencing Service of C.R.I.B.I. (University of Padua, Padua, Italy) [55]. Electropherograms were analyzed with Phred [56, 57] to assign a quality score and with a perl script using the UniVec Database [58] to identify any vector and adaptors sequences. Interspersed repeats and low complexity DNA sequences were identified through analysis with RepeatMasker [59]. The sequences were then organized in transcript consensus sequences (clusters) using the CAP3 DNA sequence program [60].
Affymetrix GeneChip experiments
Total RNA of the control and treated cells after 6 h of DIMEB treatment (3 biological replicates for each type of sample) were used to hybridize 6 different GeneChip®Vitis vinifera Genome Arrays (Affymetrix, Santa Clara, CA). Ten micrograms of total RNA for each replicate were purified as described above (Total RNA extraction), subjected to further purification using "RNeasy" columns (Qiagen) and sent to an external service (IFOM-IEO Campus for ONCOGENOMICS, Milan, Italy) for labelling and hybridization. RNA samples passed the quality check as determined by electrophoresis run on a Agilent BioAnalyzer (Agilent, Palo Alto, CA, USA). Biotin-labelling, hybridization, washing, staining and scanning procedures were performed according to the Affymetrix technical manual. Analysis of raw data was performed using the open source software of the Bioconductor project [61, 62] with the statistical R programming language [63, 64]. The quality of the hybridization reactions was checked using the affyPLM package. Intensity distribution of PM for each chip and the quality of the 3 biological replicates of both control and treated conditions were analyzed with the functions and plots (histogram and MA plots) of the affy package [65–67]. Background adjustment, normalization and summarization were performed using gcrma and the affy package. Data, before and after application of the gcrma algorithm [68], were compared through the graphical representation of box-plots and MA plots. Probe sets which were not expressed or were non-differentially expressed between the two conditions considered were eliminated in a filtering step based on the inter-quantile range method (IQR = 0.25) using the genefilter package. A two-class paired SAM analysis (Δ = 0.9; FDR = 13.3%) [69] was performed using the probe sets resulting from the filtering procedure in order to identify differentially expressed probe sets between the control and treated conditions. A fold-change of two was then applied.
Functional annotation of the SSH transcripts and Affymetrix probesets
Protein sequences encoded by the SSH transcripts or by the representative sequence of each probeset as provided by the NetAffx Analysis Center [70] were predicted using a consensus generated by three different CDS predictors [71]. Blastp analyses [72] of the polypeptides obtained from the predicted CDSs were performed by searching against the UniProt database [73]. GO terms (molecular function, biological process and cellular component) [23] were linked at every consensus sequence on the basis of the results of the Blastp analysis (Additional files 1 and 2). The sequences were organized in main functional categories according to the GO term biological process (Additional files 3 and 4). In cases of non significant Blastp results (Evalue <1e-8; sequence alignment length <75% of the query polypeptide length), these were classified as "No hits found".
The SSH transcripts were deposited at the NCBI database [74] under the sequence IDs reported in the Additional file 1. Both SSH transcripts and probesets were also referred to corresponding Tentative Consensus sequences obtained by a search (BlastN) against the Grape Gene Index database [75] and to the corresponding genomic locus on Pinot Noir clone ENTAV 115 [76] (Additional files 1 and 2).
Real-time reverse transcription (RT)-PCR
To validate the SSH and microarray data, 12 genes and 5 genes identified by SSH and GeneChip array respectively, were also analyzed by quantitative RT-PCR experiments (Additional file 5). Specific primers were designed to generate 100–200 bp PCR products (Additional file 5). The actin gene (TC45156) was used to normalize the data (actin forward: 5'-TCCTTGCCTTGCGTCATCTAT-3'; actin reverse: 5'-CACCAATCACTCTCCTGCTACAA-3') since in preliminary trials it appeared to be constantly expressed in the RNA samples subjected to gene expression analyses. For RT-PCR, total RNA from control and treated samples of the SSH experiment and from 3 biological replicates of control and treated samples of the GeneChip experiments were used. DNA traces were removed with DNase I treatment (Sigma-Aldrich) according to the manufacturer's procedure. Reverse transcription reactions and real-time RT-PCR reactions were performed using the SuperScript™ III Platinum® Two-Step qRT-PCR Kit with SYBR® Green (Invitrogen) according to the manufacturer's protocols with minor modification (300 nM of each primer in a final volume of 12.5 μl). PCR reactions contained 20 ng of cDNA and were replicated 3 times (technical replicates). Amplification reactions were performed with an ABI PRISM® 7000 Sequence Detection System (Applied Biosystems). The following thermal profile was used: 50°C for 2 min; 95°C for 10 min; 40 cycle of 95°C for 15 sec and 55°C for 1 min. Data were analysed with the ABI PRISM® 7000 SDS Software (Applied Biosystems). PCR reaction efficiencies were calculated with the LinRegPCR program [77]. For all the consensus sequences, the differential expression between treated and control samples was expressed as a ratio calculated with the Pfaffl equation [78]. The overall standard error of the mean normalized expression was obtained by applying the error calculation based on Taylor's series as developed for REST© software [79].
Data Availability
All microarray expression data are available at EBI ArrayExpress under the series entry E-MEXP-2114.
Abbreviations
- DIMEB:
-
(heptakis(2,6-di-O-methyl)-β-cyclodextrin)
- SSH:
-
Suppression subtractive hybridization
- cDNA:
-
Complementary DNA
- CDS:
-
Coding Sequence
- EST:
-
Expressed Sequence Tag
- GO:
-
Gene Ontology
- NCBI:
-
National Center for Biotechnology Information
- SAM:
-
Significance Analysis of Microarrays
- RT-PCR:
-
Real time polymerase chain reaction.
References
Dixon RA, Harrison MJ: Activation, structure and organization of genes involved in microbial defence in plants. Adv Genet. 1990, 28: 165-234.
Jeandet P, Douillt-Breuil AC, Bessis R, Debord S, Sbaghi M, Adrian M: Phytoalexins from the Vitaceae: biosynthesis, phytoalexin gene expression in transgenic plants, antifungal activity, and metabolism. J Agr Food Chem. 2002, 50 (10): 2731-2741. 10.1021/jf011429s.
Derckel JP, Baillieul F, Manteau S, Audran JC, Haye B, Lambert B, Legendre L: Differential induction of grapevine defenses by two strains of Botrytis cinerea. Phytopathology. 1999, 89 (3): 197-203. 10.1094/PHYTO.1999.89.3.197.
Harborne JB: The comparative biochemistry of phytoalexin induction in plants. Biochem Syst Ecol. 1999, 27 (4): 335-367. 10.1016/S0305-1978(98)00095-7.
Langcake P, Pryce RJ: Production of resveratrol by Vitis Vinifera and other members of Vitaceae as a response to infection or injury. Physiol Plant Pathol. 1976, 9 (1): 77-86. 10.1016/0048-4059(76)90077-1.
Stein U, Hoos G: Induktions- und Nachweismethoden für Stilbene bei Vitaceen. Vitis. 1984, 23: 179-184.
Dercks W, Creasy LL: The significance of stilbene phytoalexins in the Plasmopara viticola grapevine interaction. Physiol Mol Plant Pathol. 1989, 34 (3): 189-202. 10.1016/0885-5765(89)90043-X.
Langcake P, Cornford CA, Pryce RJ: Identification of pterostilbene as a phytoalexin from Vitis Vinifera leaves. Phytochemistry. 1979, 18: 1025-1027. 10.1016/S0031-9422(00)91470-5.
Gatto P, Vrhovsek U, Muth J, Segala C, Romualdi C, Fontana P, Pruefer D, Stefanini M, Moser C, Mattivi F, Velasco R: Ripening and genotype control stilbene accumulation in healthy grapes. J Agr Food Chem. 2008, 56 (24): 11773-11785. 10.1021/jf8017707.
Adrian M, Jeandet P, Veneau J, Weston LA, Bessis R: Biological activity of resveratrol, a stilbenic compound from grapevines, against Botrytis cinerea, the causal agent for gray mold. J Chem Ecol. 1997, 23 (7): 1689-1702. 10.1023/B:JOEC.0000006444.79951.75.
Langcake P, Pryce RJ: Production of resveratrol and viniferins by grapevines in response to UV irradiation. Phytochemistry. 1977, 6: 1193-1196. 10.1016/S0031-9422(00)94358-9.
Schubert R, Fischer R, Hain R, Schreier PH, Bahnweg G, Ernst D, Sandermann H: An ozone-responsive region of the grapevine resveratrol synthase promoter differs from the basal pathogen-responsive sequence. Plant Mol Biol. 1997, 34 (3): 417-426. 10.1023/A:1005830714852.
Adrian M, Jeandet P, Bessis R, Joubert JM: Induction of phytoalexin (resveratrol) synthesis in grapevine leaves treated with aluminum chloride (AlCl3). J Agr Food Chem. 1996, 44 (8): 1979-1981. 10.1021/jf950807o.
Radman R, Saez T, Bucke C, Keshavarz T: Elicitation of plants and microbial cell systems. Biotechnol Appl Bioc. 2003, 37 (Pt 1): 91-102. 10.1042/BA20020118.
Liswidowati , Melchior F, Hohmann F, Schwer B, Kindl H: Induction of stilbene synthase by Botrytis cinerea in cultured grapevine cells. Planta. 1991, 183 (2): 307-314. 10.1007/BF00197803.
Tassoni A, Fornale S, Franceschetti M, Musiani F, Michael AJ, Perry B, Bagni N: Jasmonates and Na-orthovanadate promote resveratrol production in Vitis vinifera cv. Barbera cell cultures. New Phytol. 2005, 166 (3): 895-905. 10.1111/j.1469-8137.2005.01383.x.
Lijavetzky D, Almagro L, Belchi-Navarro S, Martínez-Zapater J, Bru R, Pedreno MA: Synergistic effect of methyljasmonate and cyclodextrin on stilbene biosynthesis pathway gene expression and resveratrol production in Monastrell grapevine cell cultures. BMC Res Notes. 2008, 1: 132-10.1186/1756-0500-1-132.
Aziz A, Poinssot B, Daire X, Adrian M, Bezier A, Lambert B, Joubert JM, Pugin A: Laminarin elicits defense responses in grapevine and induces protection against Botrytis cinerea and Plasmopara viticola. Mol Plant Microbe Interact. 2003, 16 (12): 1118-1128. 10.1094/MPMI.2003.16.12.1118.
Morales M, Bru R, Garcia-Carmona F, Barcelo AR, Pedreno MA: Effect of dimethyl-beta-cyclodextrins on resveratrol metabolism in Gamay grapevine cell cultures before and after inoculation with Xylophilus ampelinus. Plant Cell Tiss Org. 1998, 53 (3): 179-187. 10.1023/A:1006027410575.
Bru R, Selles S, Casado-Vela J, Belchi-Navarro S, Pedreno MA: Modified cyclodextrins are chemically defined glucan inducers of defense responses in grapevine cell cultures. J Agr Food Chem. 2006, 54 (1): 65-71. 10.1021/jf051485j.
Zamboni A, Vrhovsek U, Kassemeyer HH, Mattivi F, Velasco R: Elicitor-induced resveratrol production in cell cultures of different grape genotypes (Vitis spp.). Vitis. 2006, 45 (2): 63-68.
Cao WX, Epstein C, Liu H, DeLoughery C, Ge NX, Lin JY, Diao R, Cao H, Long F, Zhang X, Chen YD, Wright PS, Busch S, Wenck M, Wong K, Saltzman AG, Tang ZH, Liu L, Zilberstein A: Comparing gene discovery from Affymetrix GeneChip microarrays and Clontech PCR-select cDNA subtraction: a case study. BMC Genomics. 2004, 5 (1): 26-10.1186/1471-2164-5-26.
The Gene Ontology. [http://www.geneontology.org/]
Stone JM, Collinge MA, Smith RD, Horn MA, Walker JC: Interaction of a protein phosphatase with an Arabidopsis serine-threonine receptor kinase. Science. 1994, 266 (5186): 793-795. 10.1126/science.7973632.
Song WY, Wang GL, Chen LL, Kim HS, Pi LY, Holsten T, Gardner J, Wang B, Zhai WX, Zhu LH, Fauquet C, Ronald P: A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science. 1995, 270 (5243): 1804-1806. 10.1126/science.270.5243.1804.
Pastuglia M, Swarup R, Rocher A, Saindrenan P, Roby D, Dumas C, Cock JM: Comparison of the expression patterns of two small gene families of S gene family receptor kinase genes during the defence response in Brassica oleracea and Arabidopsis thaliana. Gene. 2002, 282 (1–2): 215-225. 10.1016/S0378-1119(01)00821-6.
Halfter U, Ishitani M, Zhu JK: The Arabidopsis SOS2 protein kinase physically interacts with and is activated by the calcium-binding protein SOS3. Proc Natl Acad Sci USA. 2000, 97 (7): 3735-3740. 10.1073/pnas.040577697.
Laxalt AM, Munnik T: Phospholipid signalling in plant defence. Curr Opin Plant Biol. 2002, 5 (4): 332-338. 10.1016/S1369-5266(02)00268-6.
Ulker B, Somssich IE: WRKY transcription factors: from DNA binding towards biological function. Curr Opin Plant Biol. 2004, 7 (5): 491-498. 10.1016/j.pbi.2004.07.012.
Gu YQ, Wildermuth MC, Chakravarthy S, Loh YT, Yang CM, He XH, Han Y, Martin GB: Tomato transcription factors Pti4, Pti5, and Pti6 activate defense responses when expressed in Arabidopsis. Plant Cell. 2002, 14 (4): 817-831. 10.1105/tpc.000794.
Schiermeyer A, Thurow C, Gatz C: Tobacco bZIP factor TGA10 is a novel member of the TGA family of transcription factors. Plant Mol Biol. 2003, 51 (6): 817-829. 10.1023/A:1023093101976.
Zhang YL, Fan WH, Kinkema M, Li X, Dong XN: Interaction of NPR1 with basic leucine zipper protein transcription factors that bind sequences required for salicylic acid induction of the PR-1 gene. Proc Natl Acad Sci USA. 1999, 96 (11): 6523-6528. 10.1073/pnas.96.11.6523.
Richter H, Pezet R, Viret O, Gindro K: Characterization of 3 new partial stilbene synthase genes out of over 20 expressed in Vitis vinifera during the interaction with Plasmopara viticola. Physiol Mol Plant Pathol. 2005, 67 (3–5): 248-260.
Campbell EJ, Schenk PM, Kazan K, Penninckx IAMA, Anderson JP, Maclean DJ, Cammue BPA, Ebert PR, Manners JM: Pathogen-responsive expression of a putative ATP-binding cassette transporter gene conferring resistance to the diterpenoid sclareol is regulated by multiple defense signaling pathways in Arabidopsis. Plant Physiol. 2003, 133 (3): 1272-1284. 10.1104/pp.103.024182.
Yazaki K: Transporters of secondary metabolites. Curr Opin Plant Biol. 2005, 8 (3): 301-307. 10.1016/j.pbi.2005.03.011.
Whetten R, Sederoff R: Lignin biosynthesis. Plant Cell. 1995, 7 (7): 1001-1013. 10.1105/tpc.7.7.1001.
Ferrari S, Galletti R, Denoux C, De Lorenzo G, Ausubel FM, Dewdney J: Resistance to Botrytis cinerea induced in Arabidopsis by elicitors is independent of salicylic acid, ethylene, or jasmonate signaling but requires PHYTOALEXIN DEFICIENT3. Plant Physiol. 2007, 144 (1): 367-379. 10.1104/pp.107.095596.
Park SW, Kaimoyo E, Kumar D, Mosher S, Klessig DF: Methyl salicylate is a critical mobile signal for plant systemic acquired resistance. Science. 2007, 318 (5847): 113-116. 10.1126/science.1147113.
Shulaev V, Silverman P, Raskin I: Airborne signalling by methyl salicylate in plant pathogen resistance. Nature. 1997, 386 (6626): 738-738.
Broekaert WF, Delauré SL, De Bolle MF, Cammue BP: The role of ethylene in host-pathogen interactions. Annu Rev Phytopathol. 2006, 44: 393-416. 10.1146/annurev.phyto.44.070505.143440.
Ecker JR, Davis RW: Plant defense genes are regulated by ethylene. Proc Natl Acad Sci USA. 1987, 84 (15): 5202-5206. 10.1073/pnas.84.15.5202.
Marcos JF, Gonzalez-Candelas L, Zacarias L: Involvement of ethylene biosynthesis and perception in the susceptibility of citrus fruits to Penicillium digitatum infection and the accumulation of defence-related mRNAs. J Exp Bot. 2005, 56 (418): 2183-2193. 10.1093/jxb/eri218.
Buschges R, Hollricher K, Panstruga R, Simons G, Wolter M, Frijters A, vanDaelen R, vanderLee T, Diergaarde P, Groenendijk J, Topsch S, Vos P, Salamini F, Schulze-Lefert P: The barley mlo gene: A novel control element of plant pathogen resistance. Cell. 1997, 88 (5): 695-705. 10.1016/S0092-8674(00)81912-1.
Consonni C, Humphry ME, Hartmann HA, Livaja M, Durner J, Westphal L, Vogel J, Lipka V, Kemmerling B, Schulze-Lefert P, Somerville SC, Panstruga R: Conserved requirement for a plant host cell protein in powdery mildew pathogenesis. Nat Genet. 2006, 38 (6): 716-720. 10.1038/ng1806.
Cosgrove DJ: Enzymes and other agents that enhance cell wall extensibility. Annu Rev Plant Phys. 1999, 50: 391-417. 10.1146/annurev.arplant.50.1.391.
Shani Z, Dekel M, Roiz L, Horowitz M, Kolosovski N, Lapidot S, Alkan S, Koltai H, Tsabary G, Goren R, Shoseyov O: Expression of endo-1,4-beta-glucanase (cel1) in Arabidopsis thaliana is associated with plant growth, xylem development and cell wall thickening. Plant Cell Rep. 2006, 25 (10): 1067-1074. 10.1007/s00299-006-0167-9.
Dixon DC, Seagull RW, Triplett BA: Changes in the accumulation of alpha-tubulin and beta-tubulin isotypes during cotton fiber development. Plant Physiol. 1994, 105 (4): 1347-1353.
Ebel C, Gomez LG, Schmit AC, Neuhaus-Url G, Boller T: Differential mRNA degradation of two beta-tubulin isoforms correlates with cytosolic Ca2+ changes in glucan-elicited soybean cells. Plant Physiol. 2001, 126 (1): 87-96. 10.1104/pp.126.1.87.
Ben-Nissan G, Lee JY, Borohov A, Weiss D: GIP, a Petunia hybrida GA-induced cysteine-rich protein: a possible role in shoot elongation and transition to flowering. Plant J. 2004, 37 (2): 229-238.
Peterman TK, Ohol YM, McReynolds LJ, Luna EJ: Patellin1, a novel Sec14-like protein, localizes to the cell plate and binds phosphoinositides. Plant Physiol. 2004, 136 (2): 3080-3094. 10.1104/pp.104.045369.
Favery B, Chelysheva LA, Lebris M, Jammes F, Marmagne A, de Almeida-Engler J, Lecomte P, Vaury C, Arkowitz RA, Abad P: Arabidopsis formin AtFH6 is a plasma membrane-associated protein upregulated in giant cells induced by parasitic nematodes. Plant Cell. 2004, 16 (9): 2529-2540. 10.1105/tpc.104.024372.
Logemann E, Wu SC, Schroder J, Schmelzer E, Somssich IE, Hahlbrock K: Gene activation by UV light, fungal elicitor or fungal infection in Petroselinum crispum is correlated with repression of cell cycle-related genes. Plant J. 1995, 8 (6): 865-876.
Moser C, Gatto P, Moser M, Pindo M, Velasco R: Isolation of functional RNA from small amounts of different grape and apple tissues. Mol Biotechnol. 2004, 26 (2): 95-99. 10.1385/MB:26:2:95.
Vernon SD, Unger ER, Rajeevan M, Dimulescu IM, Nisenbaum R, Campbell CE: Reproducibility of alternative probe synthesis approaches for gene expression profiling with arrays. J Mol Diagn. 2000, 2 (3): 124-127.
BMR Genomics. [http://bmr.cribi.unipd.it/]
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8 (3): 175-185.
Ewing B, Green P: Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8 (3): 186-194.
The UniVec Database. [http://www.ncbi.nlm.nih.gov/VecScreen/UniVec]
RepeatMasker. [http://www.repeatmasker.org/]
Xuang X, Madan A: CAP3: a DNA sequence assembly program. Genome Res. 1999, 9: 868-877. 10.1101/gr.9.9.868.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J: Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004, 5 (10): R80-10.1186/gb-2004-5-10-r80.
The Bioconductor project. [http://www.bioconductor.org/]
Ihaka R, Gentleman RC: R: a language for data analysis and graphics. J Comput Graph Stat. 1996, 5 (3): 299-314. 10.2307/1390807.
The R Project for Statistical Computing. [http://www.r-project.org/]
Bolstad BM, Irizarry RA, Astrand M, Speed TP: A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003, 19 (2): 185-193. 10.1093/bioinformatics/19.2.185.
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP: Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003, 4 (2): 249-264. 10.1093/biostatistics/4.2.249.
Gautier L, Cope L, Bolstad BM, Irizarry RA: affy – analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004, 20 (3): 307-315. 10.1093/bioinformatics/btg405.
Wu ZJ, Irizarry RA: Preprocessing of oligonucleotide array data. Nat Biotechnol. 2004, 22 (6): 656-658. 10.1038/nbt0604-656b.
Tusher VG, Tibshirani R, Chu G: Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001, 98 (9): 5116-5121. 10.1073/pnas.091062498.
Affymetrix. [http://www.affymetrix.com/index.affx]
Wasmuth JD, Blaxter ML: prot4EST: translating expressed sequence tags from neglected genomes. BMC Bioinformatics. 2004, 5: 187-10.1186/1471-2105-5-187.
Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25 (17): 3389-3402. 10.1093/nar/25.17.3389.
The UniProt Database. [http://www.uniprot.org/]
The NCBI database. [http://www.ncbi.nlm.nih.gov/]
The DFCI Grape Gene Index. [http://compbio.dfci.harvard.edu/tgi/cgi-bin/tgi/gimain.pl?gudb=grape]
Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, Pruss D, Pindo M, Fitzgerald LM, Vezzulli S, Reid J, Malacarne G, Iliev D, Coppola G, Wardell B, Micheletti D, Macalma T, Facci M, Mitchell JT, Perazzolli M, Eldredge G, Gatto P, Oyzerski R, Moretto M, Gutin N, Stefanini M, Chen Y, Segala C, Davenport C, Dematte L, Mraz A, Battilana J, Stormo K, Costa F, Tao Q, Si-Ammour A, Harkins T, Lackey A, Perbost C, Taillon B, Stella A, Solovyev V, Fawcett JA, Sterck L, Vandepoele K, Grando SM, Toppo S, Moser C, Lanchbury J, Bogden R, Skolnick M, Sgaramella V, Bhatnagar SK, Fontana P, Gutin A, Peer Van de Y, Salamini F, Viola R: A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS ONE. 2007, 2 (12): e1326-10.1371/journal.pone.0001326.
Ramakers C, Ruijter JM, Deprez RHL, Moorman AFM: Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett. 2003, 339 (1): 62-66. 10.1016/S0304-3940(02)01423-4.
Pfaffl MW: A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29 (9): e45-10.1093/nar/29.9.e45.
Pfaffl MW, Horgan GW, Dempfle L: Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30 (9): e36-10.1093/nar/30.9.e36.
Acknowledgements
We wish to thank Dr. H H Kassermeyer for cell supply and helpful discussion. This work was supported by the Fondo Unico of the Provincia Autonoma di Trento (Resveratrol Project).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
AZ made a substantial contribution to conception, data collection and interpretation and manuscript drafting. PG participated in data analysis and manuscript writing. AC contributed to sequence analysis and annotation. SP participated in data analysis and manuscript revision. RV critically revised the manuscript. CM contributed to data interpretation and manuscript writing. RV (Velasco) and FM participated in the project's design and coordination. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2009_2247_MOESM1_ESM.xls
Additional file 1:Functional annotation of the transcripts identified by SSH. Cluster ID, Cluster length, GenBank Accession Number at NCBI [74], NCBI Sequence ID of the corresponding genomic locus on Pinot Noir clone ENTAV 115 [76], reference Tentative Consensus sequence in Grape Gene Index [75], GO terms, Ontology type [23], UniProtID [73], description and E-value are reported for each sequence. (XLS 58 KB)
12864_2009_2247_MOESM2_ESM.xls
Additional file 2:Functional annotation of differentially expressed probe sets. AffyID, Fold change, reference sequence accession numbers, NCBI Sequence ID of the corresponding genomic locus on Pinot Noir clone ENTAV 115 [76], reference Tentative Consensus sequence in Grape Gene Index [75], GO terms, Ontology type [23] and UniProtID [73] description are reported for each probe set. (XLS 114 KB)
12864_2009_2247_MOESM3_ESM.doc
Additional file 3:Functional category distribution of 127 transcripts modulated at 2 h. Each transcript is grouped in a single functional category defined by Gene Ontology "Biological process" terms [23]. Number and percentage of transcripts are reported for each main category. "No hits found" refers to transcripts with no significant homology to UniProt proteins. (DOC 43 KB)
12864_2009_2247_MOESM4_ESM.doc
Additional file 4:Functional category distribution of 223 upregulated and 148 downregulated probe sets. Each probe set is grouped in a single functional category defined by Gene Ontology "Biological process" terms [23]. Number and percentage of probe sets is reported for each main category. "No hits found" refers to probe sets with no significant homology to Uniprot proteins. (DOC 57 KB)
12864_2009_2247_MOESM5_ESM.xls
Additional file 5:Real-time RT-PCR validation of a set of genes identified in the SSH experiment or in the microarray experiment. ClusterID or AffyID, description, RT-PCR relative expression value (treated vs. control) and sequences of forward and reverse primers are reported for each experiment. RT-PCR data for SSH validation are expressed as means ± SE of three technical replicates, while RT-PCR data for microarray validation are expressed as means ± SE of three biological replicates. (XLS 28 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zamboni, A., Gatto, P., Cestaro, A. et al. Grapevine cell early activation of specific responses to DIMEB, a resveratrol elicitor. BMC Genomics 10, 363 (2009). https://doi.org/10.1186/1471-2164-10-363
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-10-363