
Abstract
Background
DGAT2 is a promising candidate gene for obesity because of its function as a key enzyme in fat metabolism and because of its localization on chromosome 11q13, a linkage region for extreme early onset obesity detected in our sample.
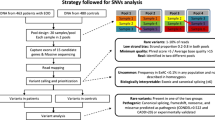
We performed a mutation screen in 93 extremely obese children and adolescents and 94 healthy underweight controls. Association studies were performed in samples of up to 361 extremely obese children and adolescents and 445 healthy underweight and normal weight controls. Additionally, we tested for linkage and performed family based association studies at four common variants in the 165 families of our initial genome scan.
Results
The mutation screen revealed 15 DNA variants, four of which were coding non-synonymous exchanges: p.Val82Ala, p.Arg297Gln, p.Gly318Ser and p.Leu385Val. Ten variants were synonymous: c.-9447A > G, c.-584C > G, c.-140C > T, c.-30C > T, IVS2-3C > G, c.812A > G, c.920T > C, IVS7+23C > T, IVS7+73C > T and *22C > T. Additionally, the small biallelic trinucleotide repeat rs3841596 was identified. None of the case control and family based association studies showed an association of investigated variants or haplotypes in the genomic region of DGAT2.
Conclusion
In conclusion, our results do not support the hypothesis of an important role of common genetic variation in DGAT2 for the development of obesity in our sample. Anyhow, if there is an influence of genetic variation in DGAT2 on body weight regulation, it might either be conferred by the less common variants (MAF < 0.1) or the detected, rare non-synonymous variants.
Similar content being viewed by others


Background
Obesity has become a major public health problem in industrialized countries and its prevalence is still increasing worldwide [1]. Estimates from twin studies attribute up to 80% of human body weight variation to genetic factors [2] and positional candidate gene analyses in linkage peak regions identified in genome wide scans for obesity have been suggested as a means to detect obesity associated genes [i.e. [3–7]]. Examples for positional candidate gene association findings pertain to (a) SLC6A14 on chromosome (chr.) Xq24 [3] which was confirmed by Durand et al. [4] and (b) GAD2 on chr. 10p12 [5] which was confirmed by the same group [6]. In contrast, Swarbrick et al. [7] found no evidence for a relationship between the three GAD2 SNPs and obesity in a sample comprising 2,359 individuals.
A genome wide scan for obesity based on 89 German families, comprising extremely obese children and adolescents and both of their parents and at least one obese sib, identified nine regions with maximum likelihood binomial logarithm of the odd (MLB LOD) scores > 0.7; in an independent confirmation sample of 76 obesity families MLB LOD scores of 0.68 and 0.71 were observed for chromosomes 10p11.23 and 11q13, respectively [8].
The hypothesis of a susceptibility gene for obesity and related phenotypes on chromosome 11q13 was additionally supported by independent linkage studies for BMI and obesity related phenotypes [9–12]. Further support was obtained from chromosomal regions homologous to human chromosome 11q13 in rodents in which quantitative trait loci (QTL) for obesity related phenotypes such as leptin level [13] and BMI [14] were identified. Taken together, there is evidence for a candidate gene for obesity in this chromosomal region.
In earlier studies, we investigated different promising candidate genes on chr.11q, but none of them contributed to the linkage peak [15–17]. Diacylglycerol-O-acyltransferase homolog 2 (DGAT2), another potential candidate gene, is also located on chr. 11q13. DGAT2 is a key enzyme in fat metabolism [18, 19]. It is responsible for the synthesis of triglycerides and catalyzes the reaction that joins diacylglycerol covalently to long chain fatty acyl-CoAs. It was hypothesized that leptin regulates adipocyte size by altering expression patterns of Diacylglycerol O-acyltransferase 1 (DGAT1) and its functional homolog DGAT2 via the CNS to determine the levels of triglyceride synthesis [20]. The deduced 387-amino acid human DGAT2 protein contains at least one transmembrane domain, three potential N-linked glycosylation sites, six potential phosphorylation sites, and a putative glycerol phospholipid domain found in acyltransferases [18]. Although functionally related, DGAT2 shares no sequence homology with the members of the DGAT1 family. The gene was identified via homology search with fungal DGAT subsequent to the finding that Dgat1 knockout mice (Dgat1-/-) were viable and still able to synthesize triglycerides [18, 19, 21].
Dgat2 knockout mice (Dgat2-/-) are lipopenic, their total carcass triglyceride content was reduced by 93% [22]. In contrast to Dgat1-/- mice, where Dgat2 is able to compensate the role of Dgat1 in triglyceride synthesis, Dgat1 was unable to compensate for the absence of Dgat2 in Dgat2-/- mice. Dgat2-/- mice die in the early postnatal period, apparently from abnormalities in energy homeostasis and from impaired permeability barrier function in the skin. The results indicate that Dgat2 is the major enzyme of triglyceride synthesis in mice [22].
Based on both positional as well as on functional arguments, we hypothesized that genetic variations in DGAT2 might alter triglyceride synthesizing activity of the protein in humans. Genetic variations leading to a gain of function of DGAT2 may thus be associated with obesity, whereas variations entailing a reduced function could be relevant in underweight.
Results
Gene structure
To include all potentially relevant exons of DGAT2, its structure was analyzed both in silico and experimentally. Visual inspection of ESTs assembled to the DGAT2 locus in the UCSC genome browser identified two ESTs (BF979495, BF979677) which seemed to harbour alternative/additional exons. The sequences of both ESTs overlap by 200 bp and form a transcript of 1,238 bp. Alignment of this mRNA to genomic DNA revealed the presence of an alternative first noncoding exon of human DGAT2, while exons2–8 are as defined by AB048286 (suppl. table 1). Sequencing of EST BF979677 revealed the presence of an alternative internal exon which is located between exon1 and exon2 as defined in AB048286. Furthermore, by RT-PCR in human adipocyte mRNA, a transcript was identified that comprised 7exons in which exon1 and exons3–8 are as defined by AB048286 while exon 2 is missing. In sum, three alternatively spliced transcripts of the human DGAT2 gene were identified. Including the two previously reported mRNAs (AB048286, ENST00000228027) there are at least five different mRNAs transcribed from this locus [see additional file 1].
Mutation screen
Screening was performed in the coding region, the predicted promoter region and in the identified non-coding 5' exon. The mutation screen in ten fragments comprised 3,079 bps and revealed 15 (14 novel) DNA variants, four of which are coding non-synonymous exchanges: p.Val82Ala, p.Arg297Gln, p.Gly318Ser and p.Leu385Val whereas ten variants are synonymous c.-9447A > G, c.-584C > G, c.-140C > T, c.-30C > T, IVS2–3C > G, c.812A > G, c.920T > C, IVS7+23C > T, IVS7+73C > T and *22C > T (see also table 1). Additionally, a small known biallelic trinucleotide repeat (IVS7+164(TAG)2–3 = rs3841596) located in intron 7 was identified.
Case control association studies
Minor allele frequencies (MAF) of the variants were estimated in sample 1. Most of the variants were rare and it was thus decided to genotype only the more frequent variations rs3841596, rs1017713 and rs3060 in sample 2. Variant -140C > T, located 5' to the translation start, was genotyped in sample 3 which includes sample 2 but is larger and therefore has an improved power (see table 1). Given the sample sizes, the study had a statistical power of more than 80% to detect allelic differences between the respective case and control groups of e.g. 0.17 and 0.1 in MAFs. Genotype distribution in all study samples did not differ from Hardy-Weinberg equilibrium. No significant differences in genotype or allele distributions were found in samples 2 and 3, all nominal p-values were >> 0.05 (see table 1).
Family based association studies
To investigate the contribution of DGAT2 polymorphisms to the linkage peak on chromosome 11q13 [8] SNPs -9447A > G and -140C > T, as well as two additional known variants (rs1017713 (IVS1+212T > C) and rs3060 (*19T > C)) were genotyped in the families contributing to the genome scan peak (sample 4). Neither single marker family based association analyses (PDT) in all 165 families nor in the 48 families contributing to the linkage peak on 11q13, revealed significant evidence for allelic associations (all p-values > > 0.05). Consistent with this finding, subsequent haplotype analyses using FAMHAP did not indicate an associated haplotype (best nominal p-value 0.5 with the zhaomax allcombi option).
Discussion
The linkage scan in 89 families revealed the highest LOD at D11S1313. Subsequent fine-mapping in 76 independent families revealed a combined peak region at position 67.8 – 69.1 Mb (approximately 68.55 - 68.01 cM, UCSC, hg16) between D11S1337 and D11S4095 [8, unpublished data]. DGAT2 is located at 75 Mb and thus close to this peak region. In light of the small sample size, which leads to considerable stochastic variation in the location estimate of linkage peaks [23] and combined with its important role in fat metabolism DGAT2 is a very plausible positional and functional candidate gene for obesity in our sample.
A mutation screen in the coding region of the gene, the predicted promoter sequence and a 5' non-coding exon (altogether 3,079 bp) revealed 15 genetic variants, 14 of which were novel. Twelve of the variants were rare (MAF = 1%) and would thus have a too low statistical power to allow for a comparison in a case control association analysis. Nonetheless, these rare variants might have an impact on the phenotype. Four coding non-synonymous variants were detected: p.Val82Ala occurred once in an extremely obese male, whereas p.Arg297Gln, p.Gly318Ser and p.Leu385Val were detected in underweight controls. [1] The conservative amino acid (aa) exchange p.Val82Ala is located in a predicted transmembrane domain of the DGAT2 protein [18]. This position is situated within an area highly conserved among the selected species with Val82 being unchanged for more than 1 billion years of evolution. While this non-synonymous variant seemingly does not affect the predicted transmembrane domain (aa 73 to aa 95), altered function may be the consequence as already postulated for other genes [24]. Moreover, for the very same aa substitution positioned within a transmembrane domain (TM) an inactivating variant in TM2 of the monocarboxylate transporter 8 [25] as well as an activating variant in TM1 of the lutropin receptor [26] had been described. Therefore although Val82Ala is a conservative exchange it has been shown that a Valin to Alanin substitution is able to materially affect membrane protein functions in both an activating as well as in an inactivating manner. Hence, assuming that a gain of function might well lead to obesity, it is reasonable to consider the Valin to Alanin substitution in DGAT2 as a potential cause for the patient's remarkably increased BMI (see table 2). [2] Arg297Gln is a non conservative amino acid exchange. In contrast to arginine, glutamine has an amide-side group that is able to form hydrogen bonds, which might influence protein structure. However, positioned in a region of little evolutionary conservation characterised by a difference in amino acid sequence length between mammals and plants and a non-conservative amino acid exchange between these kingdoms (basic polar arginine in mammals vs. neutral unpolar methionine in plants) an exchange of the wt arginine vs. also polar but neutral glutamine does not suggest a functional consequence of this substitution. [3] The substitution of glycine to serine at position 318 is also non-conservative. During evolution persisted at this position a neutral unpolar amino acid; therefore an exchange by polar serine may be functionally relevant. However, several amino acids flanking position 318 show little conservation; therefore the patient's remarkably low BMI as consequence of this amino acid substitution seems rather speculative. [4] The exchange of leucine to valine at position 385 is conservative. The non reactive aliphatic side chains of leucine and valine that are important for hydrophobic bonds within the protein are not affected. Functional studies of these variants in DGAT2 have to be performed to clarify the effect of the detected variants on body weight regulation.
There is no indication that the rare synonymous variants might have an effect on body weight regulation. Variant c.-584C > G in the putative promoter region is located in a potential binding site for the transcription factor ARP-1 (COUP-TF II), which might participate in regulation of lipid metabolism and cholesterol synthesis [27] and is assumed to negatively influence PPARα gene transcription [28]. Two variants were detected in untranslated regions (-30C > T in the 5'UTR and *22C > T in the 3'UTR). These variants may influence mRNA stability, but as they are rare, we assumed that they have no major effect on common obesity under a "common disease common variant"-perspective given that the estimated MAF of each variant was 1/186 = 0.54% (95% confidence interval 0.014%...2.96%). The intronic variants IVS2–3C > G, IVS7+23C > T and IVS7+73C > T are also rare and neither affect any consensus splice site nor do they introduce cryptic splice sites. None of the case control and family based association studies showed an association of investigated variants or haplotypes in the genomic region of DGAT2.
Starting off with a mutation screen of the coding sequence and the 5'flanking region we were investigating both case control samples and independent samples with families contributing to a linkage peak. However, due to insufficient statistical power to explore the less common variants (MAF < 0.1), our study design only allows evaluation of common variants.
In conclusion, our results do not support the hypothesis of an important role of common genetic variation in DGAT2 for the development of obesity in our sample. One may thus speculate that if there is an influence of genetic variation in DGAT2 on body weight regulation, it might either be the less common synonymous or non-coding variants that play an important role.
Methods
Study subjects
The ascertainment strategy for the extremely obese and underweight study groups was previously described in detail [29]. Briefly, extremely obese German index patients were ascertained at German hospitals specialized in inpatient treatment of extreme obesity in children and adolescents. All index patients had an age- and gender-specific BMI ≥90th percentile as previously determined in a representative German population sample [30]. The BMIs of the underweight students were below the 15th percentile whereas normal weight controls had BMIs between the 40th and the 60th age- and gender-specific percentile. Mean BMI and age and the respective standard deviations are provided below. Written informed consent was given by all participants and, in the case of minors, their parents. This study was approved by the Ethics Committee of the University of Marburg.
The coding exons of DGAT2, the predicted promoter region and an additionall non-coding 5' exon were screened in a 'screening sample' (sample 1) comprising 93 extremely obese children and adolescent cases (48.4 % females, mean BMI 34.4 ± 5.0 kg/m2; mean age 14.1 ± 2.0 yrs) and 94 healthy underweight controls (36.2 % females, mean BMI 18.5 ± 1.2 kg/m2; mean age 25.5 ± 4.0 yrs). Identified sequence variants were genotyped in sample 2, comprising both the initial groups (sample 1) and additional 87 cases (51.7 % females, mean BMI 36.9 ± 7.0 kg/m2; mean age 14.6 ± 2.8 yrs) as well as 93 healthy underweight controls (52.7 % females, mean BMI 18.3 ± 1.0 kg/m2; mean age 25.7 ± 3.8 yrs). Finally, in order to increase the power to detect association for one variant (-140C > T), sample 2 was further extended (sample 3). Sample 3 comprised a total of 361 extremely obese cases (53.2 % females, mean BMI 34.7 ± 6.3 kg/m2; mean age 14.4 ± 2.6 yrs) and a total of 445 control subjects comprising 278 underweight students (50.7 % females, mean BMI 18.2 ± 1.1 kg/m2; mean age 25.0 ± 3.7 yrs) and 167 normal weight controls (60.5 % females, mean BMI 21.8 ± 1.1 kg/m2; mean age 24.6 ± 2.4 yrs).
To investigate the potential genetic effects of variants in DGAT2 on body weight regulation; SNPs rs1017713, rs3060, -9447A > G and -140C > T were genotyped in a family based association analysis, the respective markers were also genotyped in the 165 genome scan families (sample 4) described previously [8] to test for linkage. Sample 4 is independent of samples 1–3. The aim of our study was the investigation of associations of common DGAT2 variants with extreme early-onset obesity.
Promoter prediction and evaluation of gene structure
Promoter sequence was predicted by PromoterInspector, Mammalian Promoter Prediction Software from Genomatix, [31]. Analyses were based on human genome assemblies hg15 and hg16 [32] and the corresponding ENSEMBL genome browser [33]. cDNA clone sequences of Unigene cluster Hs.334305 representative for the human DGAT2 gene were downloaded from NCBI [34] and assembled using GAP4 [35]. DGAT2 transcripts were aligned to human genomic sequence using Sim4 [36]. Two known human mRNAs mapped to the DGAT2 locus in genome assemblies hg15 and hg16. One of these, AB048286 (2,439 bp) formed the basis for RefSeq entry NM_032564, the annotation status of which was provisional. The second mRNA AL834287 (2,347 bp) was 92 bp shorter at its 5'end than AB048286. Nonetheless, both transcripts harbour 8 exons; and as defined by AB048286, the human DGAT2 at chr. 11q13.5 covers 32,766 bp with a coding region (CDS) of 1167 bp extending from exon1 to exon8. In the corresponding Ensembl genome browser [33] there were also two transcripts assigned to the DGAT2 locus (ENST00000289503, 1,545 bp; ENST00000228027, 2,238 bp). The former entry harboured 8 exons as found in AB048286 while the latter contained only 7 exons, i.e. exon5 was missing which indicated the presence of at least one alternatively spliced DGAT2 transcript.
Sequencing
Human cDNA clone BF979958 was obtained from RZPD [37] and cultured by standard methods [38]. Sequencing was performed using vector primers and BigDye Terminator Cycle Sequencing v2.0 kit (Applied Biosystems, Weiterstadt, Germany). Sequencing reactions were electrophoresed on ABI 3700 automated sequencers. Base calling was performed using phred [39, 40]. Sequence assembly was done using phrap [41]. Trace files were inspected visually in GAP4. RT-PCR: Primers located in exons 1 and 8 of DGAT2 as defined by reference sequence NM_032564 were used in a nested PCR approach (PCR I: 1F [ACCCTCATAGCCGCCTACTC], 1R [AGGTTAGCTGAGCCACCCAG]; PCR II: 2F [CTCATAGCCGCCTACTCC], 2R [CTAGAACAGGGCAAGCTGGA]) on human multiple tissue cDNA (Clontech, Heidelberg, Germany) or adipocyte mRNA [42]. Omniscript RT Kit (QIAGEN, Hilden, Germany) was used for reverse transcription. PCR products were cloned into pCR2.1-TOPO (Invitrogen, Karlsruhe, Germany). Sequencing of recombinant clones, sequence assembly, trace file inspection and alignment to genomic sequence was done as described above.
Mutation screen
A mutation screen was performed in the 8 coding exons of human DGAT2 and also in the predicted promoter region and a non-coding 5' exon. For PCR amplification, primers corresponding to intron sequences were used in order to detect potential splice site variants [for PCR primers see additional file 2]. Mutation screens of exon 6 and 8 were performed using denaturing high performance liquid chromatography (dHPLC) analysis on a Transgenomic WAVE® system [Transgenomic, Cheshire, UK; 43]. The optimal melting temperatures for separation of homo- and heteroduplices were deduced from the melting temperature of the PCR-amplicon using WAVEmaker software, version 4.0 (Transgenomic, Cheshire, UK). All chromatograms were compared with chromatograms of sequenced wild-type samples. PCR amplicons showing a peak appearance different to the wild-type pattern were sequenced (SeqLab, Göttingen, Germany). To detect mutations in exons 1–5, 7, the promoter region and the non-coding 5' exon standard nonisotopic single-strand conformation polymorphism analyses (SSCP) was performed [44]. 15% acrylamide gels (Q-BIOgene, Heidelberg, Germany; 37.5:1) were run at 600 V for 16 h at 4°C and for 5.5 h at ambient temperature; all gels were silver stained. The sensitivity of dHPLC has been described to be approximately 95% [45] and that of SSCP about 97% when using two temperatures [46]. All SSCP patterns were compared with patterns of sequenced wild-type samples. Samples that showed a pattern different from that of the wild-types were re-sequenced (Seq Lab, Göttingen, Germany). The nomenclature of the described variants follows den Dunnen and Antonarakis [47] and NM_032564.
Genotyping
High-throughput genotyping for two additional intronic SNPs (rs1017713, rs3060,) as well as for variants -9447A > G and -140C > T entering the family based association studies was performed as described earlier [48] using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). For case control association studies, genotyping of SNPs -9447A > G and c.920T > C was perfomed via tetra-ARMS-PCR [49] [see additional file 3]. For all other SNPs [see additional file 3], PCR with subsequent diagnostic restriction fragment length polymorphism analyses (RFLP) was used. PCR products were run on ethidium bromide-stained 2.5% agarose gels. Positive controls for the variant alleles and a negative control (water) were run on each gel. To validate the genotypes, allele determinations were rated independently by at least two experienced individuals. Discrepancies were resolved unambiguously either by reaching consensus or by retyping. Missings were retyped twice. Genotyping success rate was above 99%. Genotyping of rs3841596, a biallelic trinucleotide repeat was carried out using fluorescence-based semi-automated technique on an automated DNA sequencing machine (LiCor 4200-2; MWG-Biotech, Ebersberg, FRG). Analyses and assignment of the marker alleles were done with ONE-Dscan Version 1.3 software (MWG-Biotech).
In silico evaluation of non-synonymous variants
To gain information about putative functional relevance of an amino acid substitution, public sequence database [34] was mined for full length mammalian and more distant related DGAT2 orthologs where particular attention was given to species surpassing oil production. These data were utilized to determine the evolutionary conservation of the DGAT2 amino acid sequence. Protein sequence alignment was carried out via Omiga (Oxford Molecular Ltd.). Transmembrane domains were predicted in silico [50].
Statistics
Associations in the case control sample were analyzed by Cochran-Armitage trend test for genotype frequencies and Fisher's exact test for alleles. Family based association analyses were performed using the pedigree transmission disequilibrium test [PDT; 51]. Analyses of linkage disequilibrium (LD) between the investigated polymorphisms as well as haplotype associations in the families were investigated by FAMHAP v16 [e.g. 52]. All reported p-values are nominal. Due to lack of p-values < 0.05 (see below), adjustment for multiple testing was considered unnecessary.
Abbreviations
- BMI:
-
body mass index
- CNS:
-
central nervous system
- CNTF:
-
ciliary neurotrophic factor
- DGAT:
-
diacylglycerol O-acyltransferase homolog
- EST:
-
expressed sequence tag
- GAD2:
-
glutamate decarboxylase 2
- GAL:
-
galanin
- MAF:
-
minor allele frequency
- MLB LOD:
-
maximum likelihood binomial logarithm of the odd
- QTL:
-
quantitative trait locus
- SLC6A14:
-
solute carrier family 6 member 14
- SNP:
-
single nucleotide polymorphism
- TM:
-
transmembrane domain
- UCP:
-
uncoupling protein
- UTR:
-
untranslated region
References
WHO: Obesity. Preventing and managing the global epidemic. Report of a WHO consultation on obesity. Geneva. 1998
Maes HH, Neale MC, Eaves LJ: Genetic and environmental factors in relative body weight and human adiposity. Behav Genet. 1997, 27: 325-351. 10.1023/A:1025635913927.
Suviolahti E, Oksanen LJ, Ohman M, Cantor RM, Ridderstrale M, Tuomi T, Kaprio J, Rissanen A, Mustajoki P, Jousilahti P, Vartiainen E, Silander K, Kilpikari R, Salomaa V, Groop L, Kontula K, Peltonen L, Pajukanta P: The SLC6A14 gene shows evidence of association with obesity. J Clin Invest. 2003, 112: 1762-72. 10.1172/JCI200317491.
Durand E, Boutin P, Meyre D, Charles MA, Clement K, Dina C, Froguel P: Polymorphisms in the amino acid transporter solute carrier family 6 (neurotransmitter transporter) member 14 gene contribute to polygenic obesity in French Caucasians. Diabetes. 2004, 53 (9): 2483-6. 10.2337/diabetes.53.9.2483. Erratum in: Diabetes 2005; 54: 587
Boutin P, Dina C, Vasseur F, Dubois S, Corset L, Seron K, Bekris L, Cabellon J, Neve B, Vasseur-Delannoy V, Chikri M, Charles MA, Clement K, Lernmark A, Froguel P: GAD2 on chromosome 10p12 is a candidate gene for human obesity. PLoS Biol. 2003, 1: E68-10.1371/journal.pbio.0000068.
Meyre D, Boutin P, Tounian A, Deweirder M, Aout M, Jouret B, Heude B, Weill J, Tauber M, Tounian P, Froguel P: Is glutamate decarboxylase 2 (GAD2) a genetic link between low birth weight and subsequent development of obesity in children?. J Clin Endocrinol Metab. 2005, 90: 2384-90. 10.1210/jc.2004-1468.
Swarbrick MM, Waldenmaier B, Pennacchio LA, Lind DL, Cavazos MM, Geller F, Merriman R, Ustaszewska A, Malloy M, Scherag A, Hsueh WC, Rief W, Mauvais-Jarvis F, Pullinger CR, Kane JP, Dent R, McPherson R, Kwok PY, Hinney A, Hebebrand J, Vaisse C: Lack of support for the association between GAD2 polymorphisms and severe human obesity. PLoS Biol. 2005, 3: e315-10.1371/journal.pbio.0030315.
Saar K, Geller F, Ruschendorf F, Reis A, Friedel S, Schauble N, Nurnberg P, Siegfried W, Goldschmidt HP, Schafer H, Ziegler A, Remschmidt H, Hinney A, Hebebrand J: Genome scan for childhood and adolescent obesity in German families. Pediatrics. 2003, 111: 321-7. 10.1542/peds.111.2.321.
Norman RA, Thompson DB, Foroud T, Garvey WT, Bennett PH, Bogardus C, Ravussin E: Genomewide search for genes influencing percent body fat in Pima Indians: suggestive linkage at chromosome 11q21-q22. Pima Diabetes Gene Group. Am J Hum Genet. 1997, 60: 166-73.
Pratley RE, Thompson DB, Prochazka M, Baier L, Mott D, Ravussin E, Sakul H, Ehm MG, Burns DK, Foroud T, Garvey WT, Hanson RL, Knowler WC, Bennett PH, Bogardus C: An autosomal genomic scan for loci linked to prediabetic phenotypes in Pima Indians. J Clin Invest. 1998, 101: 1757-64.
Watanabe RM, Ghosh S, Langefeld CD, Valle TT, Hauser ER, Magnuson VL, Mohlke KL, Silander K, Ally DS, Chines P, Blaschak-Harvan J, Douglas JA, Duren WL, Epstein MP, Fingerlin TE, Kaleta HS, Lange EM, Li C, McEachin RC, Stringham HM, Trager E, White PP, Balow J, Birznieks G, Chang J, Eldridge W: The Finland-United States investigation of non-insulin-dependent diabetes mellitus genetics (FUSION) study. II. An autosomal genome scan for diabetes-related quantitative-trait loci. Am J Hum Genet. 2000, 67: 1186-200.
Hirschhorn JN, Lindgren CM, Daly MJ, Kirby A, Schaffner SF, Burtt NP, Altshuler D, Parker A, Rioux JD, Platko J, Gaudet D, Hudson TJ, Groop LC, Lander ES: Genomewide linkage analysis of stature in multiple populations reveals several regions with evidence of linkage to adult height. Am J Hum Genet. 2001, 69: 106-16. 10.1086/321287.
Almind K, Kulkarni RN, Lannon SM, Kahn CR: Identification of interactive loci linked to insulin and leptin in mice with genetic insulin resistance. Diabetes. 2003, 52: 1535-43. 10.2337/diabetes.52.6.1535.
Chung WK, Zheng M, Chua M, Kershaw E, Power-Kehoe L, Tsuji M, Wu-Peng XS, Williams J, Chua SC, Leibel RL: Genetic modifiers of Leprfa associated with variability in insulin production and susceptibility to NIDDM. Genomics. 1997, 41: 332-44. 10.1006/geno.1997.4672.
Munzberg H, Tafel J, Busing B, Hinney A, Ziegler A, Mayer H, Siegfried W, Matthaei S, Greten H, Hebebrand J, Hamann A: Screening for variability in the ciliary neurotrophic factor (CNTF) gene: no evidence for association with human obesity. Exp Clin Endocrinol Diabetes. 1998, 106: 108-12.
Schauble N, Geller F, Siegfried W, Goldschmidt H, Remschmidt H, Hinney A, Hebebrand J: No evidence for involvement of the promoter polymorphism -866 G/A of the UCP2 gene in childhood-onset obesity in humans. Exp Clin Endocrinol Diabetes. 2003, 111: 73-6. 10.1055/s-2003-39232.
Schauble N, Reichwald K, Grassl W, Bechstein H, Muller HC, Scherag A, Geller F, Utting M, Siegfried W, Goldschmidt H, Blundell J, Lawton C, Alam R, Whybrow S, Stubbs J, Platzer M, Hebebrand J, Hinney A: Human galanin (GAL) and galanin 1 receptor (GALR1) variations are not involved in fat intake and early onset obesity. J Nutr. 2005, 135: 1387-92.
Cases S, Stone SJ, Zhou P, Yen E, Tow B, Lardizabal KD, Voelker T, Farese RV: Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J Biol Chem. 2001, 276: 38870-6. 10.1074/jbc.M106219200.
Lardizabal KD, Mai JT, Wagner NW, Wyrick A, Voelker T, Hawkins DJ: DGAT2 is a new diacylglycerol acyltransferase gene family: purification, cloning, and expression in insect cells of two polypeptides from Mortierella ramanniana with diacylglycerol acyltransferase activity. J Biol Chem. 2001, 276: 38862-9. 10.1074/jbc.M106168200.
Suzuki R, Tobe K, Aoyama M, Sakamoto K, Ohsugi M, Kamei N, Nemoto S, Inoue A, Ito Y, Uchida S, Hara K, Yamauchi T, Kubota N, Terauchi Y, Kadowaki T: Expression of DGAT2 in white adipose tissue is regulated by central leptin action. J Biol Chem. 2005, 280: 3331-7. 10.1074/jbc.M410955200.
Smith SJ, Cases S, Jensen DR, Chen HC, Sande E, Tow B, Sanan DA, Raber J, Eckel RH, Farese RV: Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking Dgat. Nat Genet. 2000, 25: 87-90. 10.1038/75651.
Stone SJ, Myers HM, Watkins SM, Brown BE, Feingold KR, Elias PM, Farese RV: Lipopenia and skin barrier abnormalities in DGAT2-deficient mice. J Biol Chem. 2004, 279: 11767-76. 10.1074/jbc.M311000200.
Cordell HJ: Sample size requirements to control for stochastic variation in magnitude and location of allele-sharing linkage statistics in affected sibling pairs. Ann Hum Genet. 2001, 65: 491-502. 10.1046/j.1469-1809.2001.6550491.x.
Partridge , Liu , Kim , Bowie : Transmembrane domain helix packing stabilizes integrin alphaIIbbeta3 in the low affinity state. J Biol Chem. 2005, 280: 7294-300. 10.1074/jbc.M412701200.
Biebermann H, Ambrugger P, Tarnow P, von Moers A, Schweizer U, Grueters A: Extended clinical phenotype, endocrine investigations and functional studies of a loss-of-function mutation A150V in the thyroid hormone specific transporter MCT8. Eur J Endocrinol. 2005, 153: 359-66. 10.1530/eje.1.01980.
Gromoll J, Partsch CJ, Simoni M, Nordhoff V, Sippell WG, Nieschlag E, Saxena BB: A mutation in the first transmembrane domain of the lutropin receptor causes male precocious puberty. J Clin Endocrinol Metab. 1998, 83: 476-80. 10.1210/jc.83.2.476.
Ladias JA, Karathanasis SK: Regulation of the apolipoprotein AI gene by ARP-1, a novel member of the steroid receptor superfamily. Science. 251: 561-5. 10.1126/science.1899293.
Torra IP, Chinetti G, Duval C, Fruchart JC, Staels B: Peroxisome proliferator-activated receptors: from transcriptional control to clinical practice. Curr Opin Lipidol. 2001, 12: 245-54. 10.1097/00041433-200106000-00002. Review
Hinney A, Lentes KU, Rosenkranz K, Barth N, Roth H, Ziegler A, Hennighausen K, Coners H, Wurmser H, Jacob K, Romer G, Winnikes U, Mayer H, Herzog W, Lehmkuhl G, Poustka F, Schmidt MH, Blum WF, Pirke KM, Schafer H, Grzeschik KH, Remschmidt H, Hebebrand J: Beta 3-adrenergic-receptor allele distributions in children, adolescents and young adults with obesity, underweight or anorexia nervosa. Int J Obes Relat Metab Disord. 1997, 21: 224-30. 10.1038/sj.ijo.0800391.
Hebebrand J, Himmelmann GW, Heseker H, Schafer H, Remschmidt H: Use of percentiles for the body mass index in anorexia nervosa: diagnostic, epidemiological, and therapeutic considerations. Int J Eat Disord. 1996, 19: 359-69. 10.1002/(SICI)1098-108X(199605)19:4<359::AID-EAT4>3.0.CO;2-K.
Genomatix. [http://www.genomatix.de]
UCSC Genome Bioinformatics. [http://genome.ucsc.edu/]
ENSEMBL genome browser. [http://www.ensembl.org]
The National Centre for Biotechnology Information (NCBI). [http://www.ncbi.nlm.nih.gov]
Bonfield JK, Smith K, Staden R: A new DNA sequence assembly program. Nucleic Acids Res. 1995, 23: 4992-4999. 10.1093/nar/23.24.4992.
Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W: A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998, 8: 967-74.
RZPD German Resource Center for Genome Research (RZPD). [http://www.rzpd.de/]
Sambrook J, Fritsch EF, Maniatis T: Molecular Cloning, A Laboratory Manual. 1989, Cold Spring Harbor (N.Y.) Laboratory Press, 2
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8: 175-185.
Ewing B, Green P: Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8: 186-194.
Phragment assembly program (phrap). [http://www.phrap.org/phrap.docs/phrap.html]
Wabitsch M, Brenner RE, Melzner I, Braun M, Moller P, Heinze E, Debatin KM, Hauner H: Characterization of a human preadipocyte cell strain with high capacity for adipose differentiation. Int J Obes Relat Metab Disord. 2001, 25: 8-15. 10.1038/sj.ijo.0801520.
Oefner PJ, Underhill PA: DNA mutation detection using denaturing high-performance liquid chromatography (DHPLC). Curr Prot Hum Genet. 1998, 7: 1-10.
Hinney A, Schmidt A, Nottebom K, Heibult O, Becker I, Ziegler A, Gerber G, Sina M, Gorg T, Mayer H, Siegfried W, Fichter M, Remschmidt H, Hebebrand J: Several mutations in the melanocortin-4 receptor gene including a nonsense and a frameshift mutation associated with dominantly inherited obesity in humans. J Clin Endocrinol Metab. 1999, 84: 1483-6. 10.1210/jc.84.4.1483.
Ellis LA, Taylor CF, Taylor GR: A comparison of fluorescent SSCP and denaturing HPLC for high throughput mutation scanning. Hum Mutat. 2000, 15: 556-64. 10.1002/1098-1004(200006)15:6<556::AID-HUMU7>3.0.CO;2-C.
Salazar LA, Hirata MH, Hirata RD: Increasing the sensitivity of single-strand conformation polymorphism analysis of the LDLR gene mutations in brazilian patients with familial hypercholesterolemia. Clin Chem Lab Med. 2002, 40: 441-5. 10.1515/CCLM.2002.075.
den Dunnen JT, Antonarakis SE: Nomenclature for the description of human sequence variations. Hum Genet. 2001, 109: 121-4. 10.1007/s004390100505.
Wang HJ, Geller F, Dempfle A, Schauble N, Friedel S, Lichtner P, Fontenla-Horro F, Wudy S, Hagemann S, Gortner L, Huse K, Remschmidt H, Bettecken T, Meitinger T, Schafer H, Hebebrand J, Hinney A: Ghrelin receptor gene: identification of several sequence variants in extremely obese children and adolescents, healthy normal-weight and underweight students, and children with short normal stature. J Clin Endocrinol Metab. 2004, 89: 157-62. 10.1210/jc.2003-031395.
Ye S, Dhillon S, Ke X, Collins AR, Day IN: An efficient procedure for genotyping single ucleotide polymorphisms. Nucleic Acids Res. 2001, 29: E88-8. 10.1093/nar/29.17.e88.
Martin ER, Monks SA, Warren LL, Kaplan NL: A test for linkage and association in general pedigrees: the pedigree disequilibrium test. Am J Hum Genet. 2000, 67: 146-54. 10.1086/302957.
Becker T, Knapp M: A powerful strategy to account for multiple testing in the context of haplotype analysis. Am J Hum Genet. 2004, 75: 561-70. 10.1086/424390.
Hebebrand J, Himmelmann GW, Heseker H, Schafer H, Remschmidt H: Use of percentiles for the body mass index in anorexia nervosa: diagnostic, epidemiological, and therapeutic considerations. Int J Eat Disord. 1996, 19: 359-69. 10.1002/(SICI)1098-108X(199605)19:4<359::AID-EAT4>3.0.CO;2-K.
Acknowledgements
The participation of the probands and their families is gratefully acknowledged. This project was supported by the Deutsche Forschungsgemeinschaft, Germany (DFG) grant HE 1446/4-1, the European Union (Framework VI; LSHM-CT2003-503041), and the National Genome Research Net (NGFN2) "Obesity and Related Disorders".
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
SF designed and carried out PCR and mutation screening, performed genotyping and drafted the manuscript. KR and MP performedpromoter prediction, evaluation of gene structure and resequencing. AS performed statistical analyses. HBr and HBi performed in silico evaluation of non-synonymous variants. AKW and RF participated in the study design and helped drafting the manuscript. KK and TM performed the high-throughput genotyping. MW contributed mRNA for sequencing analyses and participated in the study design. AH and JH conceived the study, and participated in its design and coordination and drafted the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12863_2006_496_MOESM1_ESM.doc
Additional File 1: mRNAs transcribed from human DGAT2 locus. list of mRNAs transcribed from human DGAT2 locus, including status, evidence and gene structure defined by mRNA (DOC 28 KB)
12863_2006_496_MOESM2_ESM.doc
Additional File 2: PCR primer for mutation screen in DGAT2. list of PCR primer for mutation screen in DGAT2 exons (DOC 28 KB)
12863_2006_496_MOESM3_ESM.doc
Additional File 3: Genotyping information: Genotypes were generated via RFLP, tetra-arms PCR [3] and MALDI-TOF. Genotyping information for investigated SNPs including primer, genotyping methods and restriction enzymes (DOC 40 KB)
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Friedel, S., Reichwald, K., Scherag, A. et al. Mutation screen and association studies in the Diacylglycerol O-acyltransferase homolog 2 gene (DGAT2), a positional candidate gene for early onset obesity on chromosome 11q13. BMC Genet 8, 17 (2007). https://doi.org/10.1186/1471-2156-8-17
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2156-8-17