
Abstract
Background
The adequacy of association studies for complex diseases depends critically on the existence of linkage disequilibrium (LD) between functional alleles and surrounding SNP markers.
Results
We examined the patterns of LD and haplotype distribution in eight candidate genes for osteoporosis and/or obesity using 31 SNPs in 1,873 subjects. These eight genes are apolipoprotein E (APOE), type I collagen α1 (COL1A1), estrogen receptor-α (ER-α), leptin receptor (LEPR), parathyroid hormone (PTH)/PTH-related peptide receptor type 1 (PTHR1), transforming growth factor-β1 (TGF-β1), uncoupling protein 3 (UCP3), and vitamin D (1,25-dihydroxyvitamin D3) receptor (VDR). Yin yang haplotypes, two high-frequency haplotypes composed of completely mismatching SNP alleles, were examined. To quantify LD patterns, two common measures of LD, D' and r2, were calculated for the SNPs within the genes. The haplotype distribution varied in the different genes. Yin yang haplotypes were observed only in PTHR1 and UCP3. D' ranged from 0.020 to 1.000 with the average of 0.475, whereas the average r2 was 0.158 (ranging from 0.000 to 0.883). A decay of LD was observed as the intermarker distance increased, however, there was a great difference in LD characteristics of different genes or even in different regions within gene.
Conclusion
The differences in haplotype distributions and LD patterns among the genes underscore the importance of characterizing genomic regions of interest prior to association studies.
Similar content being viewed by others

Background
Considerable attention is currently being focused on the genotyping of single nucleotide polymorphisms (SNPs) to search for complex disease susceptibility alleles. The success to detect association between marker alleles and disease critically depends on the extent of linkage disequilibrium (LD) between functional alleles and surrounding markers. Studies on some chromosomes and genome regions have shown that LD is highly variable across human genome and is structured into discrete blocks of sequences separated by hotspots of recombination and/or LD breakdown [1–6]. The LD heterogeneity across the genome is a crucial aspect for genome-wide association studies, which has been emphasized as a powerful approach for complex disease study [7–9]. Due to practical limitations of the expenses, the sample size used on the whole genome scale may not be sufficiently large. For example, the ongoing HapMap represents one of the most comprehensive and largest efforts ever attempted in biomedical research, in which a total of 270 subjects from African, Caucasian and Japanese/Chinese were recruited. However, sample size is an important factor influencing the accuracy of LD evaluation. Small sample size will bias the accuracy of LD estimation [2, 10].
A limited but hypothesis-driven approach for association studies is to focus on the polymorphisms located in or around the candidate genes known to be of potentially functional importance for complex traits (based on their cellular and molecular studies). LD patterns in specific candidate genes need be empirically assessed to facilitate efficient experimental designs and execution of association studies [1, 11]. So far, only limited information is available about the LD patterns and haplotype characteristics in candidate genes for complex diseases [12, 13].
The purpose of this study was to determine the LD and haplotype characteristics in eight candidate genes for the two common complex disorders, osteoporosis and obesity. These eight genes are apolipoprotein E (APOE), type I collagen α1 (COL1A1), estrogen receptor-α (ER-α), leptin receptor (LEPR), parathyroid hormone (PTH)/PTH-related peptide receptor type 1 (PTHR1), transforming growth factor-β1 (TGF-β1), uncoupling protein 3 (UCP3), and vitamin D (1,25-dihydroxyvitamin D3) receptor (VDR).
Results
Haplotype analysis
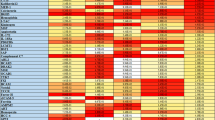
Allele frequencies of the 31 SNPs are shown in Table 1. The information for the main haplotypes whose frequencies were higher than 1% [14], is presented in Table 2. It is clear that the number of observed haplotypes was smaller than that of haplotypes expected. The number of observed haplotypes varied at different loci. In both the PTHR1 gene and the VDR gene, four SNPs were studied. However, seven and twelve haplotypes, respectively, were observed as compared to the expected 16 haplotypes in each gene.
The yin yang haplotypes are defined as two high-frequency haplotypes composed of completely mismatching SNP alleles, i.e., nucleotides differ at every SNP in the haplotype pair [15]. We detected such haplotype phenomenon in two of the eight studied genes (Table 2). In the PTHR1 gene, one pair of yin yang haplotypes, GTAA and ACGG (in the order of SNP20-21-22-23), was observed with frequencies of 34.2% and 52.6%, respectively. A pair of yin yang haplotypes, TCT and CTC (in the order of SNP28-30-31), was also observed in the UCP3 gene with frequencies of 23.5% and 40.5%, respectively.
LD analysis
In this study, we used two most common LD measures, D' and r2, to determine the extent of LD in the genes (Figure 1). The average of D' values within the eight genes was 0.475 ± 0.325 (±SD) with the range of 0.020–1.000, whereas the r2 was 0.158 ± 0.255 (±SD), which ranged from 0.000 to 0.883. Until now, there was no unified standard for meaningful LD between a marker and a functional mutation. Although we do not know whether any of our studied SNPs is functional for osteoporosis and/or obesity, the knowledge of LD patterns revealed through them is useful in inferring effective intermarker distance. When a criterion of D' > 0.33 [4, 16] was used as a meaningful LD, 58.8% (30/51) of the SNP pairs met this criterion with the intermarker physical distance ranging from 138 bp (SNP3-SNP4) to ~122 kb (SNP12-SNP14). If we considered D' = 0.7 as a limit for useful LD [17], only 33.3% (17/51 SNP pairs) were "useful". If a value of r2 > 0.1 was applied as a criterion for meaningful LD [16, 18], 31.4% (16/51 SNP pairs) would be considered in "useful" LD. When we used both D' > 0.33 and r2 > 0.1 as a criterion for meaningful LD [4, 16, 18], these 16 pairs still fell below this threshold. Such "useful" LD was relatively common in a pair-wise physical distance smaller than 8.6 kb, though it was detectable at the distance of 83 kb (SNP13-SNP14).

An exponential decay function was employed to fit the relationship between LD and the physical distance between SNP markers within each gene (SigmaPlot 2000, SPSS Inc., Chicago, IL). X-axis is the physical distance in the unit of base pair.
From Figure 1, the distribution of LD versus physical distance was irregular; however, a decay of LD with increasing distance was still evident. The Spearman correlation coefficient between D' and the distance was -0.70 (P < 0.01). The correlation coefficient was -0.49 (P < 0.01) when LD was quantified by r2. Similar relationship was observed between LD measures and ln(distance), in which the correlation coefficient was -0.68 and -0.47 for D' and r2, respectively. The D'/ln(distance) was calculated for pair-wise SNPs within each gene. The average of D'/ln(distance) varied a lot among these eight genes with the highest value of 0.085 in PTHR1 and the lowest value of 0.001 in TGF-β1. The Spearman correlation coefficient between the two LD measures was 0.81 (P < 0.01).
Discussion
Determining the haplotype and LD characteristics of candidate genes and/or genomic regions has important implications for strategies of complex diseases gene mapping [19]. In the present study, we illustrated the haplotype distribution and LD patterns in eight candidate genes for two complex diseases, obesity and osteoporosis, in 1873 subjects from 405 Caucasian nuclear families.
In this study, the yin yang haplotype pairs, mismatching at every SNP, were observed in two genes, PTHR1 and UCP3, but not in the other six. There are several possible reasons for such differences among genes. Other things being the same, lower LD levels result in smaller yin yang haplotype size, the genomic span per yin yang haplotype region [15]. The different SNP density and LD patterns among genes may be partly responsible for the discrepancies about yin yang haplotypes among genes. Lower minor allele frequencies in some genes may be another reason since minor allele frequency ≥10% is a threshold for yin yang haplotype analysis [15]. Moreover, yin yang haplotypes were affected by the factors that might be different among genes, such as the mutation and recombination rates [15].
So far, LD was inferred either by using unrelated random sample or family data. The LD values evaluated from these two different data sets were correlated, but in some instances they were quite different. Family subjects contain more information than unrelated random individuals, and the inference of LD using family data is more accurate [4]. Sample size is another important factor influencing the accuracy of LD evaluation. Generally, larger sample size can minimize sampling error and produce more accurate evaluation of LD [20]. In this study, haplotypes were reconstructed based on the nuclear families in a larger sample rather than unrelated individuals as the other studies [12, 18, 21, 22]. This increased the precision of LD evaluation.
In the present study, two most common measurements of LD, D' and r2, were used, both measurements varying between 0 and 1. According to their calculating formula [23], they are positively correlated, as observed in this study, but sometimes they are quite different. For example, D' value for SNP6-SNP8 was 1.000, while the value for r2 was only 0.004. Such a discrepancy between the two measures was also observed by Tiret et al [17]. One possible explanation for the discrepancy is that the rare allele frequency of SNP8 was very low, only 1.9%, and r2 is more sensitive to allele frequencies than D' [23].
Although a trend of decay of LD with increasing physical distance was observed in the present study, there was a great variation in LD with distance. For example, the physical distances for SNP33-SNP34 in VDR and SNP20-SNP21 in PTHR1 were both about 5 kb, however, the D' values were 0.062 and 0.964, and the r2 were 0.004 and 0.826, respectively. In the ER-α gene, the physical distance of SNP14-SNP15 was about 2.6 times that of SNP13-SNP14, while the D' values were 0.675 and 0.028, and r2 were 0.599 and 0.019, respectively. Such irregularity has been observed in the other studies [6, 17, 18, 24] and is expected, since LD is also affected by other factors, such as natural selection, the rates of recombination and mutation, and gene conversion [25].
Several limitations of this study should be acknowledged. Compared with some recent analyses [4, 14, 26], we only selected a limited number of SNPs in each gene, which may bring some difficulty to define the LD characteristics. Though 15-kb chromosome-wide resolution for LD pattern and haplotype structure study is acceptable but not ideal [27]. In addition, the yin yang haplotype analysis might be affected by the limited SNPs and genes examined. The SNPs were chosen based on the information from available literature and public databases rather than evenly distributed within gene. Another limitation is that this study was restricted to Caucasians of European descent, which limited the ability to perform cross-population comparisons for yin yang haplotype coverage and haplotype diversities etc. Since genetic diversity and LD extent differ between populations, studying contrasted populations is informative. However, if the purpose is to provide guidelines for the design of association studies in a given population, information drawn from that population should be preferable [5], especially because Caucasians are more susceptible to osteoporosis and obesity compared with other populations [28, 29].
In conclusion, the haplotype distribution was different among the eight studied candidate genes for osteoporosis and/or obesity. The LD characteristics were different between genes or even at different regions within gene, though LD decays with the increase of physical distance. The differences in haplotype distributions and LD patterns among genes underscore the importance of characterizing each locus of interest prior to association studies.
Methods
Subjects
The study subjects came from the ongoing genetic studies of osteoporosis and obesity that have been approved by the Creighton University Institutional Review Board. All subjects were Caucasians of European origin. We have recruited 405 nuclear families composed of 1,873 healthy individuals, including 840 parents, 744 daughters and 389 sons. All individuals signed informed-consent documents before entering the project.
Genotyping
After searching public SNP databases, such as dbSNP http://www.ncbi.nlm.nih.gov/SNP/, and available literature, 37 SNPs in the eight candidate genes for obesity and/or osteoporosis were chosen. The selection criteria were as follows: 1) functional relevance and importance; 2) position in or around the gene; and 3) their use in previous genetic studies. Information about the 37 SNPs is presented in Table 1. The genotyping procedure for all SNPs was similar, involving polymerase chain reaction (PCR) and invader assay reaction (Third Wave Technology, Madison, WI). PCR was performed in a 10 μl reaction mixture. The sequences of the PCR primers for all 37 SNPs are presented in Table 1. After amplification, an invader reaction was performed in a 7.5 μl reaction volume. The genotype for every sample was called according to the ratio of the fluorescence intensity of the two dyes, which was read using a Cytofluor 4000 multi-well plate reader (Applied Biosystems, Foster City, CA). Program PedCheck http://watson.hgen.pitt.edu/register/soft_doc.html was employed to verify Mendelian inheritance of all marker alleles within each family [30].
We initially genotyped all 37 SNPs in a random sample of 190 to 380 subjects. The minor allele frequency of six SNPs was found less than 1.0%. Because the precision and variance of LD estimates suffer from low allele frequencies [6], these six SNPs were excluded from further genotyping and data analysis. Finally, 31 SNPs were analyzed in this study. Among them, 14 were located in exons, 12 in introns, and 5 in untranslated regions. SNP23 in the TGF-β1 gene is an insertion/deletion polymorphism of a cytosine (+/- C) and the other 30 SNPs are nucleotide substitutions, including 25 transitions and 5 transversions. These 31 SNPs totally spanned ~451 kb. The average intermarker distance was 63 kb (ranging from 138 bp to 291 kb).
Statistical analysis
Allele frequencies of each SNP were estimated in 1,873 subjects using a maximum likelihood method implemented in the program SOLAR which is available at http://www.sfbr.org/sfbr/public/software/solar. The haplotypes of each gene for all subjects were reconstructed using the program Genehunter version 2.1 http://www.hgmp.mrc.ac.uk/Registered/Option/genehunter.html. Genehunter extracts complete multipoint inheritance information to infer maximum likelihood haplotypes for all individuals in nuclear families [31]. Haplotype frequencies were estimated from the unrelated subjects (parents of the nuclear families).
Two normalized measures, D' and r2, were used to characterize the LD patterns within the studied candidate gene. Pair-wise D' values are computed as: D' = D/Dmax, Dmax = min(f1+f+2, f+1f2+) when D > 0, and Dmax = min(f1+f+1, f+2f2+) when D < 0, where f's are sample estimates of SNP frequencies [32]. r2 is quantified as r2 = (f11f22 - f12f21)2/[(f11 + f12)(f21 + f22)(f11 + f21)(f12 + f22)], where f's are sample estimates of haplotype frequencies [23]. An exponential decay function was employed to fit the relationship between LD (both D' and r2) and the physical distance between SNP markers within each gene (SigmaPlot 2000, SPSS Inc., Chicago, IL). To quantify the relationship between D' and r2 both as index of LD, correlation coefficient between them was calculated using SPSS software.
Author's contribution
JRL performed SNP genotyping, combined the results of analysis and drafted the manuscript. LJZ performed SNP genotyping and data analysis. PYL and YL performed statistical analysis. VD contributed to the manuscript preparation. HS, YJL, YYZ, DHX and PX performed SNP genotyping. HWD conceived of the study, and participated in its design and coordination. All authors read and approved the final manuscript.
References
Ardlie KG, Kruglyak L, Seielstad M: Patterns of linkage disequilibrium in the human genome. Nat Rev Genet. 2002, 3: 299-309. 10.1038/nrg777.
Reich DE, Cargill M, Bolk S, Ireland J, Sabeti PC, Richter DJ, Lavery T, Kouyoumjian R, Farhadian SF, Ward R, Lander ES: Linkage disequilibrium in the human genome. Nature. 2001, 411: 199-204. 10.1038/35075590.
Wall JD, Pritchard JK: Haplotype blocks and linkage disequilibrium in the human genome. Nat Rev Genet. 2003, 4: 587-597. 10.1038/nrg1123.
Moffatt MF, Traherne JA, Abecasis GR, Cookson WO: Single nucleotide polymorphism and linkage disequilibrium within the TCR alpha/delta locus. Hum Mol Genet. 2000, 9: 1011-1019. 10.1093/hmg/9.7.1011.
Stephens JC, Schneider JA, Tanguay DA, Choi J, Acharya T, Stanley SE, Jiang R, Messer CJ, Chew A, Han JH, Duan J, Carr JL, Lee MS, Koshy B, Kumar AM, Zhang G, Newell WR, Windemuth A, Xu C, Kalbfleisch TS, Shaner SL, Arnold K, Schulz V, Drysdale CM, Nandabalan K, Judson RS, Ruano G, Vovis GF: Haplotype variation and linkage disequilibrium in 313 human genes. Science. 2001, 293: 489-493. 10.1126/science.1059431.
Abecasis GR, Noguchi E, Heinzmann A, Traherne JA, Bhattacharyya S, Leaves NI, Anderson GG, Zhang Y, Lench NJ, Carey A, Cardon LR, Moffatt MF, Cookson WO: Extent and distribution of linkage disequilibrium in three genomic regions. Am J Hum Genet. 2001, 68: 191-197. 10.1086/316944.
Risch N, Merikangas K: The future of genetic studies of complex human diseases. Science. 1996, 273: 1516-1517.
Weiss KM, Clark AG: Linkage disequilibrium and the mapping of complex human traits. Trends Genet. 2002, 18: 19-24. 10.1016/S0168-9525(01)02550-1.
Jorde LB: Linkage disequilibrium and the search for complex disease genes. Genome Res. 2000, 10: 1435-1444. 10.1101/gr.144500.
Teare MD, Dunning AM, Durocher F, Rennart G, Easton DF: Sampling distribution of summary linkage disequilibrium measures. Ann Hum Genet. 2002, 66: 223-233. 10.1046/j.1469-1809.2002.00108.x.
Stephens M, Smith NJ, Donnelly P: A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001, 68: 978-989. 10.1086/319501.
Bonnen PE, Wang PJ, Kimmel M, Chakraborty R, Nelson DL: Haplotype and linkage disequilibrium architecture for human cancer-associated genes. Genome Res. 2002, 12: 1846-1853. 10.1101/gr.483802.
Zhu X, Yan D, Cooper RS, Luke A, Ikeda MA, Chang YP, Weder A, Chakravarti A: Linkage disequilibrium and haplotype diversity in the genes of the renin-angiotensin system: findings from the family blood pressure program. Genome Res. 2003, 13: 173-181. 10.1101/gr.302003.
Croucher PJ, Mascheretti S, Hampe J, Huse K, Frenzel H, Stoll M, Lu T, Nikolaus S, Yang SK, Krawczak M, Kim WH, Schreiber S: Haplotype structure and association to Crohn's disease of CARD15 mutations in two ethnically divergent populations. Eur J Hum Genet. 2003, 11: 6-16. 10.1038/sj.ejhg.5200897.
Zhang J, Rowe WL, Clark AG, Buetow KH: Genomewide Distribution of High-Frequency, Completely Mismatching SNP Haplotype Pairs Observed To Be Common across Human Populations. Am J Hum Genet. 2003, 73: 1073-1081. 10.1086/379154.
Kruglyak L: Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet. 1999, 22: 139-144. 10.1038/9642.
Tiret L, Poirier O, Nicaud V, Barbaux S, Herrmann SM, Perret C, Raoux S, Francomme C, Lebard G, Tregouet D, Cambien F: Heterogeneity of linkage disequilibrium in human genes has implications for association studies of common diseases. Hum Mol Genet. 2002, 11: 419-429. 10.1093/hmg/11.4.419.
Nakajima T, Jorde LB, Ishigami T, Umemura S, Emi M, Lalouel JM, Inoue I: Nucleotide diversity and haplotype structure of the human angiotensinogen gene in two populations. Am J Hum Genet. 2002, 70: 108-123. 10.1086/338454.
Weale ME, Depondt C, Macdonald SJ, Smith A, Lai PS, Shorvon SD, Wood NW, Goldstein DB: Selection and evaluation of tagging SNPs in the neuronal-sodium-channel gene SCN1A: implications for linkage-disequilibrium gene mapping. Am J Hum Genet. 2003, 73: 551-565. 10.1086/378098.
Guo SW: Linkage disequilibrium measures for fine-scale mapping: a comparison. Hum Hered. 1997, 47: 301-314.
Peterson RJ, Goldman D, Long JC: Effects of worldwide population subdivision on ALDH2 linkage disequilibrium. Genome Res. 1999, 9: 844-852. 10.1101/gr.9.9.844.
Albagha OM, Tasker PN, McGuigan FE, Reid DM, Ralston SH: Linkage disequilibrium between polymorphisms in the human TNFRSF1B gene and their association with bone mass in perimenopausal women. Hum Mol Genet. 2002, 11: 2289-2295. 10.1093/hmg/11.19.2289.
Devlin B, Risch N: A comparison of linkage disequilibrium measures for fine-scale mapping. Genomics. 1995, 29: 311-322. 10.1006/geno.1995.9003.
Taillon-Miller P, Bauer-Sardina I, Saccone NL, Putzel J, Laitinen T, Cao A, Kere J, Pilia G, Rice JP, Kwok PY: Juxtaposed regions of extensive and minimal linkage disequilibrium in human Xq25 and Xq28. Nat Genet. 2000, 25: 324-328. 10.1038/77100.
Ardlie K, Liu-Cordero SN, Eberle MA, Daly M, Barrett J, Winchester E, Lander ES, Kruglyak L: Lower-than-expected linkage disequilibrium between tightly linked markers in humans suggests a role for gene conversion. Am J Hum Genet. 2001, 69: 582-589. 10.1086/323251.
Zabetian CP, Buxbaum SG, Elston RC, Kohnke MD, Anderson GM, Gelernter J, Cubells JF: The structure of linkage disequilibrium at the DBH locus strongly influences the magnitude of association between diallelic markers and plasma dopamine beta-hydroxylase activity. Am J Hum Genet. 2003, 72: 1389-1400. 10.1086/375499.
Dawson E, Abecasis GR, Bumpstead S, Chen Y, Hunt S, Beare DM, Pabial J, Dibling T, Tinsley E, Kirby S, Carter D, Papaspyridonos M, Livingstone S, Ganske R, Lohmussaar E, Zernant J, Tonisson N, Remm M, Magi R, Puurand T, Vilo J, Kurg A, Rice K, Deloukas P, Mott R, Metspalu A, Bentley DR, Cardon LR, Dunham I: A first-generation linkage disequilibrium map of human chromosome 22. Nature. 2002, 418: 544-548.
Liu YZ, Liu YJ, Recker RR, Deng HW: Molecular studies of identification of genes for osteoporosis: the 2002 update. J Endocrinol. 2003, 177: 147-196.
Chagnon YC, Rankinen T, Snyder EE, Weisnagel SJ, Perusse L, Bouchard C: The human obesity gene map: the 2002 update. Obes Res. 2003, 11: 313-367.
O'Connell JR, Weeks DE: PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998, 63: 259-266. 10.1086/301904.
Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES: Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet. 1996, 58: 1347-1363.
Lewontin RC: The interaction of selection and linkage. I. General considerations; heterotic models. Genetics. 1964, 49: 49-67.
Acknowledgements
The investigators were partially supported by grants from Health Future Foundation of the USA, the National Institute of Health, the State of Nebraska Cancer and Smoking Related Disease Research Program, the State of Nebraska Tobacco Settlement Fund, US Department of Energy, Chinese National Science Foundation, and the Ministry of Education of P. R. China.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Long, JR., Zhao, LJ., Liu, PY. et al. Patterns of linkage disequilibrium and haplotype distribution in disease candidate genes. BMC Genet 5, 11 (2004). https://doi.org/10.1186/1471-2156-5-11
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2156-5-11