
Abstract
Background
Infectious Bovine Keratoconjunctivitis (IBK) in beef cattle, commonly known as pinkeye, is a bacterial disease caused by M o r a x e l l a bovis. IBK is characterized by excessive tearing and ulceration of the cornea. Perforation of the cornea may also occur in severe cases. IBK is considered the most important ocular disease in cattle production, due to the decreased growth performance of infected individuals and its subsequent economic effects. IBK is an economically important, lowly heritable categorical disease trait. Mass selection of unaffected animals has not been successful at reducing disease incidence. Genome-wide studies can determine chromosomal regions associated with IBK susceptibility. The objective of the study was to detect single-nucleotide polymorphism (SNP) markers in linkage disequilibrium (LD) with genetic variants associated with IBK in American Angus cattle.
Results
The proportion of phenotypic variance explained by markers was 0.06 in the whole genome analysis of IBK incidence classified as two, three or nine categories. Whole-genome analysis using any categorisation of (two, three or nine) IBK scores showed that locations on chromosomes 2, 12, 13 and 21 were associated with IBK disease. The genomic locations on chromosomes 13 and 21 overlap with QTLs associated with Bovine spongiform encephalopathy, clinical mastitis or somatic cell count.
Conclusions
Results of these genome-wide analyses indicated that if the underlying genetic factors confer not only IBK susceptibility but also IBK severity, treating IBK phenotypes as a two-categorical trait can cause information loss in the genome-wide analysis. These results help our overall understanding of the genetics of IBK and have the potential to provide information for future use in breeding schemes.
Similar content being viewed by others

Background
Infectious Bovine Keratoconjunctivitis (IBK), commonly known as pinkeye, is a highly contagious ocular bacterial cattle disease which occurs in cattle populations throughout the world. IBK is caused by the Gram negative bacterium Moraxella bovis[1] and is characterized by excessive tearing, inflammation of the conjunctiva, and ulceration of the cornea in one or both eyes. As the disease progresses the cornea becomes cloudy or white. In severe cases, perforation of the cornea may occur which can lead to permanent blindness.
IBK is non-fatal; however, it is considered the most important ocular disease in cattle, due to the decreased growth performance of infected individuals. The United State National Animal Health Monitoring System survey [2] and Australian postal survey [3] reported IBK as an economically important disease in animal production. Major economic losses are the result of inappetence and poor weight gain in affected animals suffering from ocular pain and visual impairment [1]. It has been estimated that IBK costs beef producers 150 million US$ in the United States [4] and 22 million AUD$ in Australia [3] per annum.
There has been little research to quantify genetic variation in susceptibility to IBK. Breed differences in susceptibility to IBK have been demonstrated and Hereford cattle were found to be more susceptible compared with all other purebreds (Angus, Braunvieh, Charolais, Gelbvieh, Limousin, Pinzgauer, Red Poll and Simmental) and Bos Indicus breeds [5, 6]. Heritability estimates (0.10 - 0.25) different from zero have been reported, highlighting the presence of within-breed genetic variation in susceptibility to IBK.
Putative QTL on Chromosomes 1 (66 to 110 cM) and 20 (2 to 35 cM) have been reported [7] to be associated with resistance to IBK. Reducing IBK through selection would be advantageous, because genetic gain is cumulative and permanent. Once the loci associated with susceptibility or resistance to IBK are identified, relevant markers can be used for selection in breeding schemes.
In recent years, high-density single nucleotide polymorphism (SNP) genotyping assays for cattle have become available [8]. In addition, statistical methodologies have been developed to make use of these SNP panels to detect association between SNP and economically important traits. The use of genome-wide studies helps identify chromosomal regions associated with disease incidence in immunity-related diseases such as IBK. The objective of this study was to detect SNP markers in linkage disequilibrium (LD) with genetic variants associated with IBK in Angus cattle.
Methods
Ethics statement
All animal procedures were approved by the Iowa State University Animal Care and Use Committee.
Phenotypic data
Records of IBK were collected from 860 animals born and raised in the Iowa State University Angus research herd from spring 2004 through spring 2008. The IBK status of each animal was determined at weaning time and was scored subjectively into five categories for left and right eyes as follows:
where yL/R,i is the left (L) or right (R) eye IBK score of animal i and i=1,…,n (Table 1). Infection status was studied by pooling left and right eye IBK scores in various manners (Table 2).
First, left and right IBK scores were combined to form a classification involving two categories (c=2): Incidence was scored as 1 for both eyes unaffected and as 2 otherwise.
Second, left and right IBK scores were combined into a three category classification (c=3): Incidence was scored as 1 for both eyes unaffected, 2 for a single affected eye and 3 for both affected eyes.
Third, left and right IBK scores were classified in nine categories (c=9): Incidence was scored from 1 to 9 by adding the scores of the left and right eyes (yL,i+yR,i−1).
50k SNP data
High-density (53,367) SNP genotypes of purebred American Angus cattle were obtained using the Bovine SNP50 Infinium II BeadChip (Illumina, Inc., San Diago CA). The Illumina A/B allele calls were used to compute a covariate for each locus that had values 0, 1, or 2 to represent the number of B alleles. Missing genotypes represented less than 0.2% of total observations and were replaced with average covariate values. All genotypes were retained in the analysis regardless of minor allele frequency.
BayesB threshold model
The threshold model for ordered categorical data assumes the existence of a set of ordered thresholds τ0=−∞<τ1<τ2<…<τc−1<τ c =∞ for the trait and an underlying or latent variable l i for each animal i. When the latent variable for animal i is between thresholds τj−1 and τ j the categorical phenotype y i for animal i takes value j[9, 10].
The BayesB [11] model for genome-wide analysis of continuous traits has previously been extended [12] to analyze categorical phenotypes assuming a threshold model as described below. In this approach, the latent variable l i corresponding to y i , the IBK categorical phenotype, is modeled as follows:
where β is a px1 vector of fixed effects, is a known incidence row vector corresponding to fixed effects in β, K is the number of marker loci, z i k is the value of the covariate for marker k in individual i, α k is the random substitution effect for locus k, with with probability 1- π or α k =0 with probability π. For a locus included in the model, the locus-specific variance, , is assumed to have a scaled inverse chi-square distribution with degrees of freedom ν α =4 and scale parameter , where p k is the B allele frequency of marker k and is the additive genetic variance explained by markers [13, 14]. For a locus not in the model, is set to zero. A flat prior was assumed for the fixed effects and thresholds, and the residual, e i , was assumed to be distributed .
The joint posterior density of β,α,τ, and l[10] is given by:
where and l are the vectors of marker effects, marker variances, thresholds and liabilities.
Given the threshold model, the conditional probability Pr(y i =j|l i ,τ) in equation (6) is either 1 or zero, and following Albert and Chib [15] this probability can be written as:
where I(.) is an indicator function taking the value 1 when expression (.) is true and 0 otherwise.
It is assumed that, given the location parameters β and α, the density function of the latent variable l i in equation (6) is normal:
The density of the joint prior distribution in equation (6) has the form
A Gibbs sampler was used to construct an irreducible, Markov chain with the joint posterior as its stationary distribution [10, 16, 17]. Inferences were made from the Markov chain. In the Gibbs sampler, samples are obtained from the full conditional posteriors rather than the joint posterior [10, 16, 17]. The full conditional posteriors used in this paper are described below.
The full conditional posterior for the fixed effect β m given all other unknowns is a normal distribution:
where
X−m is the matrix X with the column associated with m deleted, and β−m is β with the mth element deleted.
At locus k, following Meuwissen et al[11], the locus-specific marker variance, , and the SNP effect, α k , were sampled from their joint full conditional posterior distribution. The strategy they used was to sample from the marginal of the full conditional posterior and then sample α k from the conditional posterior given the sampled value for [11].
This full-conditional distribution does not have a closed form, and thus the Metropolis-Hasting (MH) algorithm is used to sample . Meuwissen et al[11] used the prior distribution of as the proposal distribution in the MH algorithm. Here we used the proposal distribution described by Habier et al. [14]. Given the sampled value of , the marker effect α k was sampled from its conditional posterior:
where and [14].
Following Cowles [18], the latent variables and thresholds were sampled jointly from their joint full conditional distribution. Cowles [18] samples the thresholds from the marginal of this full conditional posterior, which is obtained by integrating out the latent variables [19]. The marginal full conditional does not have a closed form and an MH algorithm is used to draw samples of the thresholds [18]. Then, given the sampled values of the thresholds, each of the latent variables is sampled from its conditional posterior, which is truncated normal:
as described by Devroye [20].
Genetic variance and heritability
The posterior means of the genetic variance and of the heritability for the latent variable were estimated from the Markov Chain Monte Carlo (MCMC) samples as follows. In each MCMC step, the vector g of breeding values of all animals was sampled as:
where is the sampled value of α k in step t. Now, vector g(t) was used to sample the genetic variance as
where
The heritability h2(t) was then sampled as
The arithmetic means of these samples were used to estimate their posterior means.
Window variance
The genetic variance attributed to a genomic window is defined as follows. First, the breeding value for a genomic window is defined as
where α k is the effect of SNP k and W is the set of indices of SNPs that belong to genomic window w. The genetic variance attributed for genomic window w is now defined as
where
Samples of window variances were obtained using (15) but with g(t) computed with only the sampled SNP effects for window w[21]. These samples were used to compute posterior means and posterior probabilities for percent window variances and were used for inferences on locations of causal variants.
Bioinformatics and identification of candidate genes
SNP effects from genome-wide analysis were obtained using the threshold model in GenSel software [22] and the estimated effects were uploaded into SNPLOTz (http://www.animalgenome.org/tools/snplotz). SNPLOTz visualized the estimated SNP effects and their genomic location and was dynamically linked to Gbrowse (http://www.animalgenome.org/gbrowse), which allowed visualization of SNP with other types of genome features (i.e., annotated genes, curated QTL, transcripts, etc.) [23]. The SNP positions within a chromosome were based on Bos taurus genome assembly (UMD 3.1).
Results and discussion
The distribution of IBK records by left and right eyes are presented in Table 1. The rates for severity of IBK that was scored into five categories from cornea with no apparent lesions to perforation of the cornea were 75, 12, 7, 3 and 3% for left eye and 77, 11, 6, 3 and 3% for right eye. These results indicated similarity between left and right eyes for IBK incidence rates. Rodriguez [24] studied the effects of IBK incidence severity on production traits in Angus breed and found similar incidence rates between left and right eyes.
The distribution of IBK scores classified in two, three or nine categories of infection can be seen in Table 2. The rates of unaffected, affected, only one-eye affected and two-eye affected animals were 63, 37, 26 and 11%, respectively. The severity score of IBK within the nine category classification system varied from 12.8% to 0.58% for infected animals. These results were found to be similar with those from Rodriguez [24]; however, higher than those (3.7%) from Snowder et al.[6], that studied environmental effects and genetic factors influencing the incidence of IBK among nine breeds including Angus. A significant between-breed difference in IBK incidence has been reported [25], with purebred Herefords (22.4%) being more susceptible compared to other pure (Simmental 7.6%, Charolais 6.5%, Angus 3.7%, Limousin 3.4%) and composite breeds. Thus, sufficient genetic variation for resistance to IBK exists in these breeds to consider selection.
Inference on heritability
The posterior distribution of heritability for each of the IBK scores classified into two, three or nine categories is shown in Figure 1, along with the posterior mean (PM), posterior standard deviation and 95% posterior probability interval (PPI), defined to be the range of posterior density falling between the 2.5th and 97.5th percentile.

Posterior inference on heritability of Infectious Bovine Keratoconjunctivitis scores classified within two, three or nine categories in American Angus cattle. PM: Posterior mean, SD: Posterior standard deviation, PPI: 95% posterior probability interval.
The posterior distribution of heritabilities seemed similar across the different classification of IBK scores. Posterior means of heritabilities for two- (0.064) and nine-category (0.064) IBK scores were the same; however, they were slightly lower than 0.066 for three-category IBK scores. The 95% PPI of heritabilities overlapped, which indicates that there is no significant difference among heritability estimates of IBK incidence based on different classifications.
There are very few studies reporting estimates of heritability for IBK incidence in the literature. Our heritability estimates were lower, but in good agreement with the estimates (0.06 - 0.10) obtained from Rodriguez [24]. However, our heritability estimates are lower than the heritability estimates of 0.10-0.25 obtained by Snowder et al.[6] based on the models including only animal effect, and animal and maternal effects for Angus cattle. In the same study, Snowder et al.[6] also found high heritability estimates (0.20-0.28) for Hereford and low heritability (0.0 - 0.13) estimates for Red Poll, Charolais, Simmental, Limousin, Gelbvieh, Pinzgauer, Braunvieh breeds. Ali et al.[26] indicated that estimates of heritability for IBK were small to moderate (0.17 - 0.19) for both pre-weaning and post-weaning calves in a Hereford and Shorthorn composite population. These differences between heritability estimates could be attributed partly by the differences in phenotypes measured, failure to accurately distinguish between phenotypes of healthy and sick animals; false assumption that disease observed is the primary infection, lack of knowledge of the influence of passive immunity on disease incidence, susceptibility of disease biased by time, age, or season dependency and/or differences in the models of analyses used in these studies [6],[24].
Effect of different categorisation of infectious bovine keratoconjunctivitis on the whole-genome analysis
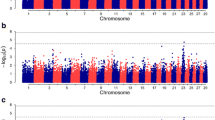
Genome-wide Manhattan plots that display the proportion of genetic variance explained by all 2,648 1-Mb SNP windows with respect to their genomic positions are shown for IBK scores classified as two, three or nine categories in Figure 2. Additional Table S1, S2 and S3 in the Additional file 1 also present the information about the top thirty 1-Mb SNP windows explaining 0.20% or more of the genetic variance observed in IBK classifications. The top thirty 1-Mb SNP windows explained 13.3, 14.5 and 14.0% of genetic variance observed in IBK scores classified as two, three or nine categories, respectively. As seen from Figure 2 and Tables in the Additional file 1, whole-genome analysis for each categorisation (two, three or nine) of IBK scores indicated that similar genomic regions were associated with IBK incidence although the variance explained by each region differed slightly according to the categorisation used.

Plot of the proportion of window variance accounted for by genome locations for two, three or nine IBK scores in American Angus cattle. Each spot on the plot indicates the proportion of genetic variance contributed by a SNP window defined based on Bos taurus genome assembly (UMD 3.1). The colors represent SNP windows from chromosome 1 to X.
Two-category classification of IBK was performed by considering all animals whose IBK status was at least as severe as the threshold level to be ‘affected’, while considering all animals whose IBK status was less severe than the threshold level to be ‘unaffected’. It was also assumed that animals with IBK had the same levels of exposure to the genetic risk factors. In the two-category classification, however, some animals considered ‘affected’ could be in more severe states than other ‘affected’ animals [27]. This additional information could be used in the multiple-category classification of IBK, whereas it was lost upon two-category classification. In the linkage analyses of two-, three- and multiple-categorical traits, Corbet el al.[27] indicated that two-category classification of multiple-categorical traits could lead to crippling power loss, especially in the case of many loci of small effects. In particular, if the underlying genetic factors for any trait confer not only IBK susceptibility but also IBK severity, treating IBK phenotypes as multi-categorical could provide additional information in genome-wide analysis.
Association analysis for infectious bovine keratoconjunctivitis
Figure 2 and Tables in the Additional file 1 indicated many genomic regions with different degree of association with IBK incidence in American Angus breed. Casas and Stone [7] had previously reported presence of QTL for IBK tolerance on chromosome 1 and 20. Casas and Snowder [25] further indicated that the region on chromosome 20 might be associated with general resistance to bacterial diseases (including IBK). In this present study, several SNP windows associated with IBK disease status were identified on chromosome 1 (Additional file 1). However, three SNP windows ([109.01 Mb] - [109.87 Mb], [110.04 Mb] - [110.98 Mb] and [112.04 Mb] - [113.00 Mb]) on chromosome 1 overlapped previously described QTL region for IBK susceptibility [7]. There are two annotated genes within these 1-Mb SNP windows on chromosome 1; PTX3 (pentraxin 3) and IL12A (interleukin 12A, natural killer cell stimulatory factor 1, cytotoxic lymphocyte maturation factor 1, p35).
PTX3 has been reported to be rapidly produced and released by several cell types, in particular by mononuclear phagocytes, dendritic cells, fibroblasts and endothelial cells [28],[29] and recognizes microbial moieties, opsonizes fungi and selected Gram-positive and Gram-negative bacteria and activates complement. Opsonization resulted in facilitated pathogen recognition and innate immune cell activation; moreover, opsonization by PTX3 is likely to be involved in the activation of an appropriate adaptive immune response [29]. IL12A, an immunomodulatory cytokine secreted by antigen presenting cells, is critical for differentiation of T helper (Th)1 and Th2 lymphocytes [30]. IL12 has been shown to augment the growth of activated T- and natural killer (NK)-cells [31], stimulate interferon gamma (IFN- γ) production by T-cells and NK cells, and suppress the expansion of Th2 cell clones [32]. This information about PTX3 and IL12A genes indicates that they could be strong biological as well as positional candidates involved in response to IBK infections.
It is important to note that in the present study there were no common 1-Mb SNP windows on chromosome 20 for IBK incidence. While this result was not consistent with the results from the previous studies [7],[25] about IBK incidence, the difference may be in part due to design differences between this study (a single breed of purebred Bos taurus animals) and the previous studies (a single Bos taurus x Bos indicus sire bred to multiple breeds of cows). These differences in results may indicate different metabolic pathways or different segregating QTL (within breed vs. across breed) for IBK incidence.
In the present work, five 1-Mb SNP windows (Table 3), which were identified as being associated with IBK incidence, were determined as common windows within the thirty SNP windows for IBK classifications (Tables in Additional file 1): rs109448194 - rs42270183 and rs41642303 - rs110857971 on chromosome 2, rs108956311 - rs43705367 on chromosome 12, rs29021773 - rs109429649 on chromosome 13 and rs41966737 - rs41640647 on chromosome 21. The percentage of genetic variance explained by these five 1-Mb SNP windows based on two-, three- and nine-category of IBK incidence were 0.30, 0.45 and 0.27%, and 0.89, 0.28 and 0.32% for two SNP windows on chromosome 2; 0.90, 1.01 and 1.11% for SNP window on chromosome 12; 1.42, 0.50 and 0.37% for SNP window on chromosome 13 and 0.40, 0.26 and 0.29% for SNP window on chromosome 21, respectively. These results showed that same genomic regions could have different degree of association with IBK incidence based on the two-, three-, or nine-category classification of IBK incidence, which means IBK could have separate sets of genes controlling affection and severity of the incidence.
Several candidate genes in the five 1-Mb SNP windows were identified and listed with their functions in Table 4. Examining the first 1-Mb SNP window [19.10 - 19.97 Mb] on chromosome 2 indicated two genes; AGPS (alkylglycerone phosphate synthase), NFE2L2 (nuclear factor (erythroid-derived 2)-like 2). AGPS is a peroxisomal enzyme which is required for the synthesis of plasmalogens. Liegel et al.[33] indicated that a mutation based on a G to A substitution at the +5 position of intron 14 in the AGPS gene resulted in severe plasmalogen deficiency which is the cause of cataracts and male sterility in the blind sterile 2 mice having spontaneous autosomal recessive mutation. NFE2L2 (also called NRF2) is a basic leucine zipper transcription factor that mediates the cytoprotective cellular antioxidant response [34]. Wangy et al. indicated that NFE2L2 was an essential transcription factor in protecting living organisms from oxidative stress-related disease. Ungvari et al.[35] showed that polymorphisms in the regulatory regions of NFE2L2 are associated with susceptibility to infection-induced asthma in the study of relationship between air pollution, NFE2L2 gene polymorphisms and childhood asthma in a Hungarian population. They also found remarkable differences in the genotype distributions of these polymorphisms between different polluted regions, which indicate an environment-dependent regulation of the antioxidant defense mechanisms. This NFE2L2 gene could be associated with susceptibility to IBK incidence because several factors including the environment, season, fly concentration, the presence of the pathogen, strain of the pathogen and background of the animal play a role in the incidence, penetrance, and severity of IBK [36]. In addition, Schwalfenber [37] reported the role of vitamin D in the prevention of viral, fungal and bacterial (such as ocular) infections. He indicated that 1,25(OH)2D, active form of vitamin D, has been shown to increase the induction of genes encoding for human β defensin which promotes resistance to eye infection by P. aeruginosa. Epidemiological studies have linked vitamin D deficiency to increased rates of cancer, as well as autoimmune and infectious diseases [37]. Vitamin D-mediated protection from pro-oxidant stress was determined to be indirect due to the induction of NFE2L2[38] and it was found that NFE2L2 expression was down-regulated in prostate cancer and suppression of NFE2L2 promotes prostate tumor development in TRAMP mice [39].
The second SNP window [22.09 - 22.77 Mb] harbors 3 genes; WIPF1 (WAS/WASL interacting protein family, member 1), OLA1 (Obg-like ATPase 1), SP3 (Sp3 transcription factor). WIPF1 encodes a protein that plays an important role in the organization of the actin cytoskeleton. Wickramarachchi et al.[40] indicated that deficiency of actin regulators can result in defects limited to the immune system or sometimes to a single immune cell type and lack of an actin cytoskeletal regulator can cause immunodeficiency, autoimmunity, autoinflammatory disease, or a combination of these manifestations. In addition, Dustin and Cooper [41] pointed out that the actin cytoskeleton seems to play two critical roles in the activation of T cells: T cell shape development and movement, including formation of the immunological synapse and the formation of a scaffold for signaling components. OLA1 is a negative regulator of the antioxidative process. Knockdown of OLA1 in human cells elicited an increased resistance to oxidizing agents. Zhanga et al.[42] also reported that knockdown of OLA1, a newly discovered regulatory protein of oxidative stress response, inhibits cell migration and invasion ability in breast cancer cells. Sp factors are able to stimulate transcription from proximal promoters or from distal enhancers. They can also physically interact with other transcription factors. van Loo et al.[43] studied the impact of the absence of the widely expressed transcription factor SP3 on the developing hematopoietic system in the mouse. They showed that the absence of SP3 results in cell-autonomous differentiation defects in the erythroid and myeloid cell lineage. WIPF1, OLA1and SP3 genes are found to be associated with defects in immune system or cell lineage making them biological candidate for IBK disease.
Two genes were identified within the SNP window [53.01 - 53.99 Mb] on chromosome 12; SCEL (sciellin), EDNRB (endothelin receptor type B). Sciellin is a protein that is encoded by the SCEL gene. it is a precursor of the cornified enveloped. The cornified envelope is an insoluble protein complex formed under the plasma membrane in the uppermost layers of stratified squamous epithelium and plays a major role in the barrier properties of the stratum corneum [44]. Champliaud et al.[45] reported proteins interacting with sciellin and identified vitamin D-upregulated protein 1 (VDUP1), which plays multiple roles in a wide range of cellular processes such as proliferation or apoptosis. Recently, it has been reported that VDUP1 is also involved in the immune system via positive regulation of natural killer development [46]. The endothelin receptor B (EDNRB) gene encodes a g-protein-coupled receptor-mediated endothelin, inducing development and transformation of the neural crest cell-specific lineage. Hypermethylation of the EDNRB promotor has been shown in multiple tumor types, and EDNRB has been proposed as a putative tumor suppressor gene [47].
One gene is located within SNP window [14.36 - 14.92 Mb] on chromosome 13; VAMP7 (vesicle-associated membrane protein 7). VAMP7 inhibits the release of lytic granules and severely impairs natural killer (NK) cell cytotoxic activity which is used to eliminate cancer and virus-infected cells. Furthermore, VAMP7 is involved in IFN secretion in NK cells, which indicates that VAMP7 is involved in many fusion processes and thus plays a general function in NK cell activity [48]. In addition, this SNP window lies in the region where putative QTLs that affect Bovine spongiform encephalopathy at 22.997-61.84 cM resides [49]. These findings indicate that these genes could be candidates for IBK susceptibility based on their functions.
The SNP window [19.00 - 19.98 Mb] on chromosome 21 harbors three genes that encode proteins; NTRK3 (neurotrophic tyrosine kinase, receptor, type 3), MIR1179 (microRNA mir-1179), MIR7-1 (microRNA mir-7-1). NTRK3 gene encodes a member of the NTRK family. These neurotrophins (NTs) receptors are best known for their role in the differentiation and survival of various types of neurons [50]. In the study of latitude-driven adaptation for both schizophrenia and vitamin D related genes, Amato et al. found 9 genes including NTRK3 in common among those ones related to latitude, vitamin D and schizophrenia. MicroRNAs (miRNAs) are small 21-23 nucleotide-long noncoding RNAs involved in several biological process including development, differentiation, apoptosis, survival, motility, invasion and proliferation. Many miRNAs are implicated as proto-oncogenes or as tumor suppressors and aberrantly expressed in various cancer types. MIR7 has been characterized as a tumor suppressor in several human cancers. This SNP window also lies in the region where putative QTLs that affect clinical mastitis at 12.60-35.89 cM and somatic cell scores at 12.601-29.77 cM, have been identified by Schulman et al.[51] and Schnabel et al.[52], respectively. These results indicate that genetic variant(s) that reside in this chromosomal region could be associated with bacterial disease such as IBK in cattle.
Conclusions
The present analyses, using Bovine SNP50 Infinium II BeadChips, identified several 1-Mb SNP regions and genes within these regions that were associated with IBK. They may be of interest for breeding schemes as they could be used to identify animals susceptible to IBK.
IBK disease is a lowly heritable complex trait that is polygenic in nature, where many loci with small effects are expected and disease incidence is affected by the environment. Such traits require a large number of individuals to enhance the power of the genome-wide analysis and obtain good statistical support for the detection of the causal loci.
References
McConnel CS, Shum L, House JK: Infectious bovine keratoconjunctivitis antimicrobial therapy. Aust Vet J. 2007, 85: 65-69. 10.1111/j.1751-0813.2006.00080.x.
NAHMS: Part III. Reference of 1997, beef cow-calf production management and disease control. USDA,APHIS, Natl Anim Health Monit Syst. 1998, [http://www.aphis.usda.gov/animal_health/nahms/beefcowcalf/downloads/beef97/Beef97_dr_PartIII.pdf]
Slatter D, Edwards M, Hawkins C, et al: A national survey of the accurrence of infectious bovine keratoconjunctivitis. Aust Vet J. 1982, 59: 65-68. 10.1111/j.1751-0813.1982.tb02728.x.
Hansen R: New tools in the battle against pinkeye. Nevada Livest. Prod. Annu. Update. 2001, Univ. of Nevada–Reno, UNR Coop. Ext. SP 01–01, 5-8.
Frisch JE: The relative incidence and effect of bovine infectious keratoconjunctivitis in bos indicus and bos taurus cattle. Anim Prod. 1975, 21: 265-274. 10.1017/S0003356100030737.
Snowder GD, Van Vleck LD, Cundiff LV, Bennett GL: Genetic and environmental factors associated with incidence of infectious bovine keratoconjunctivitis in preweaned beef calves. J Anim Sci. 2005, 83: 507-518.
Casas E, Stone RT: Putative quantitative trait loci associated with the probability of contracting infectious bovine keratoconjunctivitis. J Anim Sci. 2006, 84: 3180-3184. 10.2527/jas.2006-200.
Matukumalli LK, Lawley CT, Schnabel RD, Taylor JF, Allan MF, Heaton MP, O’Connell J, Moore SS, Smith TP, Sonstegard TS, Van Tassell CP: Development and characterization of a high density SNP genotyping assay for cattle. PLoS One. 2009, 4 (4): e5350-10.1371/journal.pone.0005350.
Wright S: An analysis of variability in number of digits in an inbred strain of guinea pigs. Genetics. 1934, 19: 506-536.
Sorensen DA, Gianola D: Likelihood, Bayesian and MCMC methods in Quantitative Genetics. 2002, New York: Springer-Verlag, New York, Inc;
Meuwissen THE, Hayes BJ, E GM: Prediction of total genetic value using genome wide dense marker maps. Genetics. 2001, 157: 1819-1829.
Villanueva B, Fernandez J, Garcia-Cortes LA, Varona L, Daetwyler HD, Toro MA: Accuracy of genome-wide evaluation for diease resistance in aquaculture breeding programs. J Anim Sci. 2011, 89: 3433-3442. 10.2527/jas.2010-3814.
Fernando RL, Habier D, Sticker C, Dekkers JCM, Totir LR: Genomic selection. Acta Agriculturae Scand. 2007, 57: 192-195.
Habier D, Fernando RL, Kizilkaya K, J GD: Extension of the Bayesian alphabet for genomic selection. BMC Bioinformatics. 2011, 12 (186): 1-12.
Albert J, Chib S: Bayesian analysis of binary and polychotomous response data. J Am Stat Assoc. 1993, 88: 669-679. 10.1080/01621459.1993.10476321.
Geman D, Geman S: Stochastic relaxation, Gibbs distributions, and the Bayesian restoration of images. IEEE Trans Pattern Anal Intel. 1984, 6: 721-741.
Gelfand AE, Smith AFM: Sampling-based approaches to calculating marginal densities. J Amer Stat Ass. 1990, 85: 398-409. 10.1080/01621459.1990.10476213.
Cowles MK: Accelerating Monte Carlo Markov Chain convergence for cumu-lative link generalized linear models. Stat Comp. 1996, 6: 101-111. 10.1007/BF00162520.
Chen MH, Shao QM, Ibrahim JG: Monte Carlo Methods in Bayesian Computation. 2000, New York: Springer-Verlag;
Devroye L: Non-Uniform Random Variate Generation. 1986, New York: Springer-Verlag;
Fernando RL, Garrick D: Bayesian methods applied to GWAS. Genome-Wide Association Studies and Genomic Prediction. 2013
Fernando R, Garrick D: GenSel-User manual for a portfolio of genomic selection related analyses. [http://taurus.ansci.iastate.edu]
Hu Z, Fernando R, Garrick DJ, Reecy JM: SNPLOTz: a generic genome plot tool to aid the SNP association studies. BMC Bioinformatics. 2010, Suppl4: P4-
Rodríguez JE: Infectious Bovine Keratoconjunctivitis in Angus cattle. Master’s thesis. 2006
Casas E, Snowder GD: Putative quantitative trait loci associated with bovine pathogenic disease incidence. J Anim Sci. 2008, 86: 2455-2460. 10.2527/jas.2008-0933.
Ali AA, Thomson PC, J OC: Genetic parameters of infectious bovine keratoconjunctivitis and its relationship with weight and parasite infestations in Australian tropical Bos taurus cattle. Genet Sel Evol. 2012, 44 (22):
Corbet J, Gu C, Rice J, Reich T, Province M, Rao D: Power loss for linkage analysis due to the dichotomization of trichotomous phenotypes. Hum Hered. 2004, 57: 21-27. 10.1159/000077386.
Garlanda C, Bottazzi B, Bastone A, Mantovani A: Pentraxins at the crossroads between innate immunity, imflammation, matrix deposition, and female fertility. Annu Rev Immunol. 2005, 23: 337-366. 10.1146/annurev.immunol.23.021704.115756.
Battazzi B, Garlanda C, Salvatori G, Jeannin P, Manfredi A, Mantovani A: Pentraxins as a key component of innate immunity. Current Opinion in Immunology. 2006, 18: 10-15. 10.1016/j.coi.2005.11.009.
Szabo SJ, Jacobson NG, Dighe AS, Gubler U, Murphy KM: Developmental commitment to the Th2 lineage by extinction of IL-12 signaling. Immunity. 1995, 2: 665-675. 10.1016/1074-7613(95)90011-X.
Robertson MJ, Soiffer RJ, Wolf SF, Manley TJ, Donahue C, Young D, Herrmann SH, Ritz J: Response of human natural killer (NK) cells to NK cell stimulatory factor (NKSF): cytolytic activity and proliferation of NK cells are differentially regulated by NKSF. J Exp Med. 1992, 175: 779-788. 10.1084/jem.175.3.779.
Chung F: Anti-inflammatory cytokines in asthma and allergy interleukin-10, interleukin-12, interferon-gamma. Mediators Inflamm. 2001, 10: 51-59. 10.1080/09629350120054518.
Liegel R, Chang B, Dubielzig R, Sidjanin D: Blind sterile 2 (bs2), a hypomorphic mutation in Agps, results in cataracts and male sterility in mice. Mol Genet Metab. 2011, 103: 51-59. 10.1016/j.ymgme.2011.02.002.
Giudice A, Montella M: Activation of the Nrf2–ARE signaling pathway a promising strategy in cancer prevention. Bioessays. 2006, 28: 169-181. 10.1002/bies.20359.
Ungvari I, Hadadi E, Virag V, Nagy A, Kiss A, Kalmar A, Zsigmond G, Semsei AF, Falus A, Szalai C: Relationship between air pollution, NFE2L2 gene polymorphisms and childhood asthma in a Hungarian population. J Community Genet. 2012, 3: 25-22. 10.1007/s12687-011-0075-8.
Brown M, Brightman A, Fenwick B, Rider M: Infectious bovine keratoconjunctivitis: A review. J Vet Med. 1998, 12: 259-266.
Schwalfenberg G: Not enough vitamin D for Canadians. Can Fam Physician. 2007, 53: 841-854.
Wild A, Moinova H, RT M: Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem. 1999, 274: 33627-33636. 10.1074/jbc.274.47.33627.
Frohlich D, Mccabe M, Arnold R, Day M: The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene. 2008, 27: 4353-4362. 10.1038/onc.2008.79.
Wickramarachchi D, Theofilopoulos A, Kono D: Immune pathology associated with altered actin cytoskeleton regulation. Autoimmunity. 2010, 43: 64-75. 10.3109/08916930903374634.
Dustin M, Cooper J: The immunological synapse and the actin cytoskeleton: molecular hardware for T cell signaling. Nat Immunol. 2101, 1: 23-29.
Zhanga J, Rubioa V, Zheng S, Shi Z: Knockdown of OLA1, a regulator of oxidative stress response, inhibits motility and invasion of breast cancer cells. J Zhejiang Univ Sci B. 2009, 10 (11): 796-804. 10.1631/jzus.B0910009.
van Loo PF, Bouwman P, Ling K, Middendorp G, Suske G, Grosveld F, Dzierzak E, Philipsen S, Hendriks RW: Impaired hematopoiesis in mice lacking the transcription factor Sp3. Blood. 2003, 102 (3): 858-866. 10.1182/blood-2002-06-1848.
Nemes Z, Steinert P: Bricks and mortar of the epidermal barrier. Exp Mol Med. 1999, 31: 5-19. 10.1038/emm.1999.2.
Champliaud M, Viel A, Baden H: The Expression of Vitamin D-Upregulated Protein 1 in Skin and its Interaction with Sciellin in Cultured Keratinocytes. J Invest Dermatol. 2003, 121 (4): 781-785. 10.1046/j.1523-1747.2003.12539.x.
Jeon J, Yoon S, Choi I, JW C: Vitamin D3 upregulated protein 1 (VDUP1) is a regulator for redox signaling and stress-mediated diseases. J Dermatol. 2006, 33: 662-669. 10.1111/j.1346-8138.2006.00156.x.
Tao K, Wu C, Wu K, Li W, Han G, Shuai X, Wang G: Quantitative analysis of promoter methylation of the EDNRB gene in gastric cancer. Med Oncol. 2012, 29: 107-112. 10.1007/s12032-010-9805-8.
Krzewski K, Gil-Krzewska A, Watts J, Stern JN, Strominger JL: VAMP4-and VAMP7-expressing vesicles are both required for cytotoxic granule exocytosis in NK cells. Eur J Immunol. 2011, 41 (11): 3323-3329. 10.1002/eji.201141582.
Zhang C, De Koning DJ, Hernandez-Sanchez J, Haley CS, Williams JL, Wiener P: Mapping of multiple quantitative trait loci affecting bovine spongiformencephalopathy. Genetics. 2004, 167 (4): 1863-1872. 10.1534/genetics.104.026401.
Levi-Montalcini R, Hamburger V: Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J Exp Zool. 1951, 116: 321-361. 10.1002/jez.1401160206.
Schulman NF, Viitala SM, de Koning DJ, Virta J, Maki-Tanila A, Vilkki JH: Quantitative trait loci for health traits in Finnish Ayrshire cattle. J Dairy Sci. 2004, 87 (2): 443-449. 10.3168/jds.S0022-0302(04)73183-5.
Schnabel RD, Sonstegard TS, Taylor JF, Ashwell MS: Whole-genome scan to detect QTL for milk production, conformation, fertility andfunctional traits in two US Holstein families. Anim Genet. 2005, 36 (5): 408-416. 10.1111/j.1365-2052.2005.01337.x.
Acknowledgements
Authors would like to thank Iowa State University Center for Integrated Animal Genomics for financial support of phenotype collections. The authors acknowledge Scientific and Technological Research Council of Turkey (TUBITAK) for financial support. Authors would like to thank Mary Sue Mayes for assistance with phenotype collections and processing DNA samples for genotyping.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KK developed the model and part of the program, and carried out the statistical analysis. RGT and JMR did the data preparation and helped with the statistical analysis. DJG and RLF developed the GenSel program used for analysis. KK, RGT, DJG, RLF and JMR helped to draft the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12863_2012_1115_MOESM1_ESM.pdf
Additional file 1: Top thirty 1-Mb SNP windows from genome-wide association study of IBK classified in two, three or nine categories.(PDF 34 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kizilkaya, K., Tait, R.G., Garrick, D.J. et al. Genome-wide association study of infectious bovine keratoconjunctivitis in Angus cattle. BMC Genet 14, 23 (2013). https://doi.org/10.1186/1471-2156-14-23
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2156-14-23