
Abstract
Background
Inbreeding is among the major concerns in management of local livestock populations. The effective population size of these populations tends to be small, which enhances the risk of fitness reduction and extinction. High-density SNP data make it possible to undertake novel approaches in conservation genetics of endangered breeds and wild populations.
A total of 97 representative samples of domestic and wild pig populations from the Iberian Peninsula, subjected to different levels of threat with extinction, were genotyped with a 60 K SNP panel. Data analyses based on: (i) allele frequency differences; (ii) linkage disequilibrium and (iii) runs of homozygosity were integrated to study population relationships, inbreeding and demographic history.
Results
The domestic pigs analyzed belonged to local Spanish and Portuguese breeds: Iberian ─ including the variants Retinto Iberian, Negro Iberian and Manchado de Jabugo ─, Bisaro and Chato Murciano. The population structure and persistence of phase analysis suggested high genetic relations between Iberian variants, with recent crossbreeding of Manchado de Jabugo with other pig populations. Chato Murciano showed a high frequency of long runs of homozygosity indicating recent inbreeding and reflecting the recent bottleneck reported by historical records. The Chato Murciano and the Manchado de Jabugo breeds presented the lowest effective population sizes in accordance with their status of highly inbred breeds. The Iberian wild boar presented a high frequency of short runs of homozygosity indicating past small population size but no signs of recent inbreeding. The Iberian breed showed higher genetic similarities with Iberian wild boar than the other domestic breeds.
Conclusions
High-density SNP data provided a consistent overview of population structure, demographic history and inbreeding of minority breeds and wild pig populations from the Iberian Peninsula. Despite the very different background of the populations used, we found a good agreement between the different analyses. Our results are also in agreement with historical reports and provide insight in the events that shaped the current genetic variation of pig populations from the Iberian Peninsula. The results exposed will aid to design and implement strategies for the future management of endangered minority pig breeds and wild populations.
Similar content being viewed by others


Background
Progressive population decline has called the attention of the conservation management and scientific communities. Both for wild and domesticated populations alike there is a fear that inbreeding may lead to loss of allelic variation and adverse phenotypic consequences [1]. In addition, loss of variation may lead to reduced response to changing environments, of which genetic susceptibility to novel infectious diseases is a specific concern. Agricultural diversity in particular is of concern for future food safety [2]. Variation conserved in local breeds is often related to important traits that classically are attributed to traditional populations, such as adaptation to the environment and greater resistance to local pathogens.
In addition to these concerns, local populations are often considered to be part of the local culture and history. For instance, local pigs are often linked to local cuisine and the local landscape. The Spanish Iberico or Iberian, and Portuguese Alentejano pigs, for instance, are used to produce highly priced products due to their quality, that in part results from feeding with acorns from sparse Mediterranean oak forests, the so called 'Dehesas’. The wild relative of the pig, the wild boar, on the other hand, plays a significant role in the wildlife of the Iberian Peninsula. It is among the main prey species of an iconic predator from the Iberian Peninsula such as Iberian wolf [3]. Moreover, wild boar is an important reservoir of infectious diseases as relevant as tuberculosis in the Iberian Peninsula [4], and therefore also of concern for public and animal health.
Local pig populations, both wild and domestic, have been highly affected by human-induced changes. Local, usually fat, breeds for instance, were affected by changes in consumer preference in the middle of the 20th century when consumers started to avoid high-fat meat. As a result, a relatively small number of highly productive pig breeds progressively replaced and marginalized the traditional breeds. Many breeds became extinct in the past decades, while many other traditional breeds today face near-extinction either through dwindling population numbers or hybridization with highly productive breeds [5]. At the same time, the increase in woodland across Europe has allowed wild boar populations to increase in many countries, after having been marginalized for centuries [6]. The Iberian Peninsula provides a good representation of local pig populations, both wild and domestic. While sharing the same geography, these populations have undergone different historical events, have different phenotypic attributes, and have a different conservation status. The Iberian pigs have been reared in an extensive traditionally system in the South and West of the Iberian Peninsula for centuries, remaining isolated from the modern breeding practices developed in the late 18th and 19th century in NW Europe [7]. Iberian pigs are related to other Mediterranean pigs of Italy and The Balkans [5], which are thought to have a smaller influence from Asian pigs than the NW European pigs. Conversely, Chato Murciano and Manchado de Jabugo, now both highly endangered populations, and Bisaro resulted from crosses between native pigs from the Iberian Peninsula and foreign pigs at the end of the 19th century [8]. Beside these domestic populations, the Iberian Peninsula is also inhabited by wild boars that may represent the ancestor of these local breeds, and also constitute an important wildlife species of the Iberian Peninsula.
The recent availability of a high-density porcine SNP panel [9] provides an essential tool for genome wide association studies and genomic selection for economically important traits [10, 11]. Besides the use of high-density SNP arrays for economic purposes, these panels have demonstrated their power to assess major questions in conservation genetics [12, 13]. The study of linkage disequilibrium (LD) and genetic distances enable the estimation of effective population size from genetic data [14], which is of major interest in conservation genetics, especially when pedigree information is unavailable as is frequently the case for minority breeds and wild populations. In addition, high-density SNP arrays allow assessing similarities in the patterns of LD across populations (i.e. persistence of phase), providing information about the relatedness of populations [15]. The occurrence of runs of homozygosity (ROH) is indicative of demographic history and recent inbreeding [13, 16]. While the same parameters can be interpreted as signatures of selection on genomic regions [17, 18], when taken as global genomic parameters, they are highly indicative of demographic history [19], if properly corrected for local recombination rate [13]. A genome-wide SNP assay, combined with a detailed recombination map for the species [20] can therefore aid in giving insight into the conservation management of pig populations. Despite the fact that SNP assays are gaining interest for traceability purposes [21, 22], only few studies have used a high-density SNP assays for conservation purposes [1, 12, 23–25].
Here we present a comprehensive study in which high-density SNP data from domestic and wild pigs were used to address questions important to conservation genetics. First, we assessed the relationships between pigs by population structure analysis and by investigating the persistence of LD phase. Secondly, patterns of LD in each population were used together with a high-density recombination map to estimate past and present effective population size. Finally, the number and size of ROH were investigated in each individual. The joint analysis of all those parameters allowed us to obtain reliable and consistent data of population structure, inbreeding and demographic history in each population providing valuable insights for future management strategies in pig from the Iberian Peninsula.
Results
A total of 97 pigs from domestic and wild autochthonous populations from the Iberian Peninsula were genotyped with the Porcine SNP60 Beadchip [9]. The SNPs located on the sex chromosomes and those with more than 5% missing genotypes were excluded from the analysis, resulting in a total of 47,594 SNPs used for the analysis.
Population structure
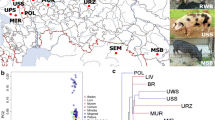
The Principal Component Analysis (PCA) revealed four main clusters represented by wild boar, Iberian, Bisaro and Chato Murciano (Figure 1). Among populations, Chato Murciano was the most divergent breed, showing a pairwise Fst ≥ 0.22 with all populations except for Bisaro where it was 0.18. The variants of Iberian ─Retinto, Negro Iberian, Manchado de Jabugo and five unclassified Iberian pigs─ showed low Nei’s genetic distances between them (≤ 0.06) and low Fst ( ≤ 0.05), and likewise the two populations of wild boar from Spain and Portugal (0.04 and 0.06 respectively). Among domestic pigs, Iberian variants showed the lowest Nei’s genetic distance to wild boar (0.10 - 0.12). The results from Nei’s genetic distances and the pairwise Fst between populations are detailed in Additional file 1.

Different population groups defined with PCA analysis. WB, wild boar; IB, Iberian pig; IB_Unk, Iberian unidentified variant; NI, Negro Iberian; RE, Retinto; MJ, Manchado de Jabugo; BI, Bisaro; CM, Chato Murciano.
The Bayesian clustering algorithms implemented in the Structure software assigned all individuals to clusters that coincide to their population of origin, with the exception of one Manchado de Jabugo pig (MJ_02) that was placed in the cluster of the other Iberian variants. Pairwise genetic distances between individuals (Additional file 2) and PCA analysis done using the Iberian pig data only (Additional file 3) confirmed this finding. K values from 2 to 7 were tested (Figure 2). The optimal K-value was estimated using the method described by Evanno et al. [26], indicating that K = 4 was the most parsimonious number of clusters (Additional file 4) in full agreement with the PCA analysis. Chato Murciano and Bisaro appeared as differentiated clusters when K = 2-3, while Iberian and wild pigs shared the same cluster for those K values. The Iberian cluster (yellow) contributed to all the other populations, in particular to Bisaro (25%) and wild pigs from Spain (12%) (Additional file 4). No differentiation between wild boar from Portugal and Spain was apparent, nor between the Iberian, Retinto and Negro Iberian variants, for any of the K values tested. However, signs of admixture from an unspecified origin were observed in Manchado de Jabugo for K ≥ 5. Thus, for subsequent analysis, samples were grouped as follows: Bisaro, Chato Murciano, Manchado de Jabugo, Iberian pig and wild boar. In addition, the Iberian variants Retinto and Negro Iberico were also analyzed separately. Finally, for population-based analyses the pig MJ_02 was removed since it fell outside of any of the groups considered in the analysis.

Graphic representation of estimated membership coefficients for each individual for K =2-7. Each color represents the proportion of the genome assigned to each assumed cluster.
Linkage disequilibrium among populations
A total of 24,703 SNPs in wild boar, 29,856 SNPs in Bisaro, 33,454 in Chato Murciano, 27,858 in Iberian and 26,246 in Manchado de Jabugo were used to estimate LD for all SNP pairs less than 3 Mbp apart (Table 1). Pairwise r2 values were averaged over all 18 autosomes and plotted as a function of increasing genetic distance in all populations studied (Figure 3; Additional file 1). The persistence of LD as the distance between loci increased and the strength of LD, varied widely between populations and between chromosomes. The decay of LD as a function of marker distance was greater in wild boar (r2 < 0.2 within 0.1 Mbp) than in the domestic breeds and showed the lowest average r2 across all genetic distances. Among the domestic breeds, LD was the lowest in Iberian (r2 < 0.2 within 0.2 Mbp). By contrast, Manchado de Jabugo and Chato Murciano had the most pronounced extent of LD at short genetic distances, although LD decreased faster in Chato Murciano than in Manchado de Jabugo for genetic distances higher than 1 Mbp.

Decline of LD measure by r2 against distance in bases across the 18 autosomes . WB, wild boar; IB, Iberian; MJ, Manchado de Jabugo; BI, Bisaro; CM, Chato Murciano.
Persistence of phase
The persistence of LD phase was calculated as the Pearson correlation (r) between SNP pairs in all possible population pairs. Similar to LD, r decreased as the distance between markers increased. This was observed for all pairs of populations, although at different degrees (Figure 4; Additional file 5). Bisaro and Chato Murciano showed the greatest correlation of phase at short genetic distance. However, for SNP pairs spaced more than 1.5 Mb apart, Iberian and Manchado de Jabugo showed the highest correlation of phase. Correlations between the other pairs of domestic pig populations (CM-MJ, CM-IB, BI-IB, BI-MJ) tended to be similar. The persistence of phase found between wild boar and all domestic pigs was lower than between domestic populations.

Correlation of Phase between populations for SNP pairs grouped by distance across the whole genome. The pairs between wild boar and domestic pigs (WB – IB/MJ/CM/BI) were uniformly plotted in gray for ease of reading. WB, wild boar; IB, Iberian; MJ, Manchado de Jabugo; BI, Bisaro; CM, Chato Murciano.
Current effective population size
The mean values of LD for all 1 Mb bins across the entire genome were used to estimate the current effective population size (Ne) implementing the equation r2 = 1/(4Nec +1) [27]. This estimation was performed taking into account the recombination for each of these bins [20]. Large Ne was observed for the wild boar population (Table 2). Among the domestic breeds, Iberian also had a high Ne (Ne = 151 ± 84) while Manchado de Jabugo and Chato Murciano had smaller effective population sizes (Ne = 46 ± 50 and 59 ± 31 respectively). Nevertheless, notice the large SD of these estimates.
Past effective population size
The past Ne at generation T, where T = 1/2c [14], was similarly estimated for each bin of 1 Mb and sorted based on decreasing recombination rate values. This approach allowed studying Ne from as few as 5 to 20,000 generations ago (Figure 5). Similar to the estimation of the current Ne, wild boar tended to have the highest past Ne, followed by Iberian pigs. A noteworthy drop in Ne was observed in wild boar 10,000 – 20,000 generations ago, with a decrease of Ne from over 70,000 to below 30,000. The Ne increased rapidly in Iberian pigs at ~3,500 - 5,000 showing a maximum Ne at ~3,500 generations ago (Ne ~ 12,000). This increase in Ne was not observed in any other population (Additional file 6).

Estimated effective population size (Ne) over time. WB, wild boar; NI, Negro Iberian; RE, Retinto; MJ, Manchado de Jabugo; BI, Bisaro; CM, Chato Murciano.
Runs of homozygosity
ROH of a minimum of 10 kbp containing at least 20 homozygous SNPs were studied in each individual separately. All individuals included in this study showed ROH. However, there were marked differences between populations in terms of number and length of ROH (Figure 6). The sums of all ROH per animal allowed the estimation of the percentage of the genome covered by ROH in each population (Additional file 7). The Chato Murciano had the largest mean proportion of its genome covered by ROH (29%). Other populations had mean values lower than 20%, with Bisaro displaying the lowest mean proportion (10%). The mean of the total number of ROH per population was higher in Chato Murciano and wild boar (34 and 30 respectively) than in Iberian and Manchado de Jabugo (26 and 24 respectively). The Bisaro breed showed the lowest mean number of ROH (13). Regarding the length of ROH, approximately 36% of the Chato Murciano pigs analyzed had long ROH (> 100 Mbp) and 92% of the pigs of this breed had ROH in the range of 50–100 Mbp, making Chato Murciano the population with the highest proportion of long ROH. By contrast, none of the wild boars analyzed had long ROH, and only 20% of wild pigs analyzed contained ROH in the range of 50–100 Mbp, indicating that wild boar had shorter ROH than the other populations. Manchado de Jabugo and Bisaro contained fewer ROH than the other populations, with Manchado de Jabugo displaying a higher proportion of long ROH than Bisaro. Twenty-five percent of Manchado de Jabugo pigs showed long ROH and 75% contained ROH in the range of 50–100 Mbp. These percentages are 6% and 33% respectively in Bisaro. Finally, Iberian had values intermediate to these breeds, since 16% of Iberian pigs displayed long ROH and 58% in the range of 50 – 100 Mb. All the pigs analyzed showed ROH shorter than 50 Mbp.

Average of number of ROH Vs. average of length of ROH. Each dot represents an individual. WB, wild boar; IB, Iberian; MJ, Manchado de Jabugo; BI, Bisaro; CM, Chato Murciano.
A Pearson’s correlations matrix was made including LD, length of ROH and recombination rate. We found a positive correlation between mean values of LD and length of ROH per chromosome (ρ = 0.70, p < 0.002) while the correlation was negative between lengths of ROH and recombination rates (ρ = - 0.67, p < 0.003).
Discussion
High density SNP analysis can provide information on past and current population demography. LD is largely affected by population history and demography [28–30], constituting a potential tool to be applied to genetic population management. Specifically, LD can be used to estimate past and present effective population size [31] and to study persistence of LD phase [24]. The availability of a large number of SNPs, allows the study of parameters that can be directly relevant to assess effects of inbreeding such as the occurrence of Runs of Homozygosity (ROH). In addition, high-density SNP arrays are expected to improve the accuracy to assess population structure and relationship among populations [32–34]. Nevertheless, the applications of high-density SNP assays for investigations into genetic population management of minority breeds and wild populations in pig are scarce.
Relationships between populations
Understanding the relationships among and within populations of livestock is a necessary first step to establish conservation priorities and strategies [35, 36]. Population structure analysis, based on differences in allele frequencies, has been a proven method to assess relationships between populations [23, 37]. We combined this widely used approach with the estimates of persistence of phase as a measure of relationship between populations [24]. The different methods implemented to assess the relationships between populations showed a high degree of congruence.
The results obtained from the population structure and persistence of phase analyses indicate closer relationships between Chato Murciano and Bisaro, and between Iberian pigs and wild boar. This observed division seems to reflect the classical separation of pig populations from the Iberian Peninsula into two origins: the Celtic type and the Iberian type pigs [5]. Most of Celtic type breeds from the Iberian Peninsula are now extinct or are highly endangered. The Bisaro pig is a representative of this group [38]. All the variants of Iberian, which include Retinto, Negro Iberico and Manchado de Jabugo, belong to the Iberian type. Although the similarity between Chato Murciano and Bisaro could be due to a Celtic origin ─ or at least a mixed origin ─ of Chato Murciano, it is possible that the differentiation between the two groups of pigs actually differentiate admixed and non-admixed populations.
The high Pearson correlations for persistence of phase at long genetic distances detected between Manchado de Jabugo and other Iberian variants is typical for subpopulations of the same breed [15]. Furthermore, structure analysis confirmed the close genetic relationship between variants of Iberian pig but also signs of genetic admixture in Manchado de Jabugo. This is agreement with historical records documenting that Manchado de Jabugo is a variant of the Iberian crossed with foreign pigs [8].
Inbreeding and effective population size
The analysis of Ne and ROH can be used to address major issues in conservation genetics such as effects of genetic drift and inbreeding [39]. The small population size inferred for the majority of local populations enhances the effect of consanguinity and genetic drift, which could compromise the long-term viability of the populations [40]. With the absence of pedigree data in many local breeds, genetic marker data can be used as a surrogate to estimate current and past Ne, for instance through exploring the extent of LD [31]. Despite the interest in Ne for conservation of populations, the estimation of this parameter is remarkably complex [41]. The estimation of Ne assumes an ideal population that is isolated, without migration, with random mating and with constant linear population growth [14]. Although it is recognized that these assumptions are generally violated in natural populations, estimation of Ne is widely used. The estimation of recent Ne has been computed using linked [31, 42] and unlinked [43] genetic markers. We estimated Ne separately for 1 Mb bins containing information of recombination rates in order to obtain more information of demographic history [42]. While this method may provide a greater temporal dimension of Ne[44] it may make it difficult to interpret the results of current Ne[41]. Additionally, our estimate of current Ne must be treated with care due to the low sample sizes, especially in those populations with a sample size lower than 15 animals (i.e. Manchado de Jabugo and Retinto).
ROH have been used to infer the history of consanguinity in human populations [39, 45] and cattle [16]. These studies have demonstrated that long ROH are related with high consanguinity levels and also have shown the existence of a good correlation with pedigree inbreeding coefficients [16, 46].
The existence of Chato Murciano pigs with a high number of long ROH shows the importance of recent inbreeding and thus low individual genetic diversity. Indeed, we observed three Chato Murciano pigs that had more than 45% of the genome covered by ROH, but also pigs with much lower percentage. This observation is consistent with known management strategies of this breed [23] and in agreement with a strong bottleneck described for this breed about 20 years ago when the entire breed consisted of only 30–40 breeding animals. The high number of long ROH also indicates that this breed has not recently been extensively crossed with other breeds otherwise the long ROH would have broken down. The frequency of long ROH in Manchado de Jabugo was similar to the other Iberian variants. Recent admixture between Manchado de Jabugo and other pig breeds, as observed in the structure analysis, may have resulted in the break-down of long (>100 Mbp) homozygous haplotypes. Despite the fact that Manchado de Jabugo is highly endangered with extinction as suggested from the small census population size (http://dad.fao.org/), this population did not show signs of high levels of consanguinity, likely because of its admixed origin. Thus, the conservation program currently implemented in Manchado de Jabugo is effective and necessary to assure the future viability of this population. What is also evident, however, is that this management strategy has gone at the expense of the historical genetic distinctiveness of the breed. By contrast, Bisaro showed signs of low consanguinity in agreement with its mixed origin and the strict conservation program implemented in this breed [47]. Although the Iberian pigs generally showed relatively low percentage of the genome covered by ROH, a few individuals showed a high coverage by ROH [48]. This can be expected in this heterogeneous breed which consists of local populations and different color forms.
The agreement between our observations and expectations based on historic reports highlights that analyzing the structure of ROH can aid in assessing levels of current consanguinity, and historic events such as bottlenecks in local pig populations. Furthermore, the assessment of ROH at the individual level has practical implications in conservation programs. Animals displaying high levels of ROH, for instance, could be excluded or given lower priority for breeding purposes in endangered populations. However, it must be taken into account that the 60 K SNP panel applied may underestimate the number of small ROH due to ascertainment bias [13] and may inflate the length of the longest ROH [16]. Yet Bosse et al. [13] and Purfield et al. [16] concluded that high-density SNP panels allow an appropriate estimation of ROH, especially for the analysis of large ROH.
Demographic history
The study of demographic history provides a better understanding of the current risk of inbreeding, and might facilitate predicting the effects of future changes in effective population size. Despite the fact that estimation of demography implies the simplification of a complex biological reality, estimation of effective population size based on LD and recombination rate provides useful predictions and consistent comparisons between populations [31, 49]. It must be considered that the estimation of recent Ne is more inaccurate than the estimation ancient Ne owing to the increase in the variability of Ne values as the length of the segment used for the estimation increase [14]. The estimation of past Ne for wild boar tended to be higher than for domestic populations, with important drops in Ne. Moreover, wild boar had a very high number of short ROH and no long ROH. A high number of short ROH has been related with a reduced population size in the past and low inbreeding in recent times [39]. This pattern could be explained by the bottlenecks that occurred in Europe in the last century [50], that would have reduced the Ne of wild boar drastically. Moreover, continuous events of formation of subpopulations and migration between them, favored by the lack of geographic barriers across the Iberian Peninsula, and even occasional admixture with domestic pigs, could have avoided high inbreeding in wild boar populations. Genetic signs of migration between subpopulations of Iberian wild boars have been described in Portugal [51].
The low recombination rate observed in large parts of the porcine genome essentially allows a much wider window in the past effective population size. Assuming a generation interval of approximately 2 years [24]. We observed a distinct drop of the Ne between 20,000 and 10,000 generations ago exclusively in wild boar that seems to reflect the sharp population decrease during the Last Glacial Maximum [52]. The increase in population size observed exclusively in Iberian pigs around 4000–4500 generations ago (see Additional file 6) is consistent with the time frame of the domestication in Europe [53, 54]. Recently the role of Europe as domestication centre has been dismissed [53, 54]. In this scenario of domestication, European domestic pigs appeared as a result of repeated events of admixture between domestic pigs imported from Near Eastern regions around 8,000 years ago [55] and wild pigs from Europe [53]. On one hand, the fact that Iberian pig allowed the study domestication events implies that this breed represents a suitable model to study domestication in Europe, reinforcing the need to preserve the breed and avoid admixture with other pig populations. On the other hand, it confirms historic reports and previous studies with mtDNA showing that Iberian pigs did not originate from crosses with other breeds [56]. Admixture events may mask genetic signs of past demographic events [54] explaining why other domestic breeds such as Bisaro and Chato Murciano showed a different pattern of past Ne. Studies using Next Generation sequence data are needed to support and increase the accuracy of past Ne estimations in Iberian pig.
Structure analysis showed that Iberian pigs contributed to the wild boar genetic stock (Figure 2). This fact together with the genetic distances and Fst values between wild boar and Iberian pigs provide support that Iberian may have been crossbreeding with wild boar until medieval times [57]. This is in agreement with results for the European breeds studied by Groenen et al.[52], who describe a complex history in European breeds and incomplete lineage sorting, supporting admixture between wild and domestic pigs in Europe. It must be kept in mind that Iberian pigs were traditionally bred outdoors, which has enabled crossbreeding between wild and domestic pigs. Intriguingly, this is unlikely to have affected only the domesticated pigs, but rather also the wild boar of the Iberian Peninsula, as suggested by recent investigations on introgression from domesticated into wild populations [12].
Conclusions
This study provides a comprehensive picture of demographic history, population structure and inbreeding of wild and domestic pig populations from the Iberian Peninsula as well as their relevance in conservation genetics. The occurrence of ROH in Chato Murciano was very high in some individuals, which may be due to a recent bottleneck and also highlights the lack of a well-designed genetic management program. Manchado de Jabugo showed a relatively high heterozygosity. This is unexpected given the extremely low census population size, and most likely reflects recent admixture with commercial pig breeds observed in this population. Conservation programs need to be maintained and carefully designed in order to avoid further loss of genetic distinctiveness. The study of Ne and ROH in Bisaro indicated high genetic diversity of this breed as a result of its mixed origin and the efforts carried out to preserve this breed. We observed that the Iberian breed may represent a good model to assess genetic signs of past demographic events as domestication, being an additional argument for the need to preserve Iberian pig breed and to avoid crossbreeding with other breeds. Previous evidence supporting the Iberian breed as being closely related to wild boar were confirmed and further evidence was provided for recurrent crossbreeding between these populations in the past. The analysis of wild populations from different regions of the Iberian Peninsula indicates natural migrations of wild pigs across the Iberian Peninsula as well as low levels of inbreeding in Iberian wild boar.
Methods
Animals and sampling
A total of 97 unrelated pig samples were collected from populations of the Iberian Peninsula, and genomic DNA was extracted by standard protocols. The study included 18 wild boars (WB) from different regions of Portugal (n = 11) and Spain (n = 7), and 79 domestic pigs. The domestic pigs utilized for the analysis belonged to three local breeds: Iberian ─including a Retinto Iberian variant (RE, n = 11), a Negro Iberian variant (NI, n = 15), an unidentified Iberian variant (IB, n = 5), and Manchado de Jabugo (MJ, n = 8) ─, Bisaro (BI, n = 15) and Chato Murciano (CM, n = 25).
SNP genotyping
High-density SNP genotyping was performed using the Porcine SNP60 Beadchip (IlluminaInc, USA) designed to genotype 62,163 SNPs [9], according to manufacturer’s protocol. For this study, only SNPs mapped to one of the 18 autosomes on Sus scrofa build 10.2 and with less than 5% missing genotypes were included in the analysis.
Data analysis
Allele Sharing Distances were calculated using PLINK v1.07 [45]. Nei’s genetic distances [58] and Fst values between populations were calculated using the Power marker software [59]. The pairwise distances between individuals were used to construct a Neighbor-Joining tree in Mega 5.03 [60]. The admixture model implemented by the program Structure v2.0 [61] was used to examine relatedness among pig populations and population stratification. K values (number of assumed clusters) from two to seven were tested. Consistent results were obtained by using a burning period of 100,000 followed by 100,000 Markov chain Monte Carlo (MCMC) repetitions. The analysis was replicated and the most likely number of clusters was determined by the Evanno method [26] using the web server Structure Harvester [62]. Moreover, to obtain further detail of the population structure, the PCA was performed using the program Eigenstrat [[63]].
Linkage Disequilibrium analysis
Markers significantly deviating from Hardy-Weinberg equilibrium (P < 0.001) and with a MAF lower than 0.05 were excluded from LD analysis using PLINK v1.07 [64]. LD (r2) was estimated for all marker pairs less than 3Mbp apart across all populations and in each autosomal chromosome independently using Haploview 4.2 [65]. Graphic display of r2vs. distance per chromosome and means plot of r2 in each breed vs. each chromosome were made in R environment (http://www.r-project.org/).
Persistence of phase
To calculate the persistence of phase and the time since two breeds diverged we followed the procedure implemented by Badke et al.[24]. Briefly, the SNP data was split into groups of SNPs with pairwise marker distance of 100 kbp, and the pairwise Pearson correlation between SNP was estimated across the 10 possible pairs of population.
Effective Population size
Effective population sizes were calculated in all populations implementing the equation r2 = 1/(4Nec +1) → Ne = (1/4c) * (1/r2 -1), where r2 is the LD, c is the marker distance in Morgans between SNP and Ne is the effective population size [27]. Additionally, past effective population size at generation T was calculated by the approximation T = 1/2c[14]. This formula implies that regions of low recombination rate allow the study of ancient Ne.
Previous authors [25] tended to apply the generalization 1 Mb ~ 1 cM to calculate Ne, but this assumption may lead to incorrect estimates of Ne. Recombination rate varies considerably across and within porcine chromosomes [20], to an even larger extent than observed in other mammals [13]. Instead, we used the averaged high-density recombination map described by Tortereau et al.[20]. The effective population size estimates were derived by averaging multiple genomic regions in order to have a better approximation of the effective population size [49]. Towards this end, the chromosomes were divided in 1 Mb bins containing information of recombination rates and average r2 for all possible pairs of SNPs included in each bin. Mean values and standard deviations among bins were subsequently used to estimate past and present effective population size. The approximation of past Ne assumes that c is much larger than the mutation rate (~10-8 per locus and generation) [15] so bins with c < 10-6 where not considered for past Ne estimation.
Runs of homozygosity
The software PLINK v1.07 [64] was used to detect ROH for individuals separately. The ROH were defined by a minimum of 10 kbp in size and 20 homozygous SNPs. One heterozygous SNP was permitted in ROH, so that the length of the ROH was not disrupted by an occasional heterozygote. In addition, minimum SNP density of 1 SNP/Mb and a largest possible gap between SNPs of 1 Mb were predefined in order to assure that the ROH were not affected by the SNP density.
Number of ROH, total length of ROH and the average of ROH length in each animal were calculated for each chromosome and the mean across animals was estimated for each breed. Those ROH longer than 100 Mbp were categorized as long ROH. The percentage of the total genome length affected by ROH in each animal was also inferred.
Availability of supporting data
The data sets supporting the results of this article are included within the article (and its additional files).
References
Muir WM, Wong GK-S, Zhang Y, Wang J, Groenen MAM, Crooijmans RPMA, Megens H-J, Zhang H, Okimoto R, Vereijken A, Jungerius A, Albers GAA, Lawley CT, Delany ME, MacEachern S, Cheng HH: Genome-wide assessment of worldwide chicken SNP genetic diversity indicates significant absence of rare alleles in commercial breeds. Proc Natl Acad Sci USA. 2008, 105: 17312-17317. 10.1073/pnas.0806569105.
FAO: Building on gender agrobiodiversity and local knowledge. Food Agr Organ Unit Nation. 2006, 1: 1-5.
Barja I: Prey and prey-age preference by the iberian wolf canis lupus signatus in a multiple-prey ecosystem. Wildl Biol. 2009, 15: 147-154. 10.2981/07-096.
Beltrán-Beck B, Ballesteros C, Vicente J, De la Fuente J, Gortázar C: Progress in oral vaccination against tuberculosis in its main wildlife reservoir in Iberia, the Eurasian wild boar. Vet Med Int. 2012, 2012: 978501-
Megens HH-J, Crooijmans RPMA, San Cristobal M, Hui X, Li N, Groenen MAM, Magali SC, Xiao H, Ning L, Martien AMG: Biodiversity of pig breeds from China and Europe estimated from pooled DNA samples: differences in microsatellite variation between two areas of domestication. Genet Sel Evol. 2008, 40: 103-128.
Sáez-Royuela , Telleria JL: The increased population of the wild boar (Sus scrofa L.) in Europe. Mamm Rev. 1986, 16: 97-101. 10.1111/j.1365-2907.1986.tb00027.x.
Alves E, Fernández AI, Barragán C, Ovilo C, Rodríguez C, Silió L: Inference of hidden population substructure of the Iberian pig breed using multilocus microsatellite data. Spanish J Agr Res. 2006, 4: 37-46.
García Dory M, Martínez S, Orozco F: Guía de campo de las razas autóctonas españolas. 1990, Madrid: Alianza Editorial
Ramos AM, Crooijmans RPMA, Affara NA, Amaral AJ, Archibald AL, Beever JE, Bendixen C, Churcher C, Clark R, Dehais P, Hansen MS, Hedegaard J, Hu ZL, Kerstens HH, Law AS, Megens H-J, Milan D, Nonneman DJ, Rohrer GA, Rothschild MF, Smith TPL, Schnabel RD, Van Tassell CP, Taylor JF, Wiedmann RT, Schook LB, Groenen MAM: Design of a high density SNP genotyping assay in the pig using SNPs identified and characterized by next generation sequencing technology. PloS One. 2009, 4: e6524-10.1371/journal.pone.0006524.
Duijvesteijn N, Knol EF, Merks JWM, Crooijmans RPMA, Groenen MAM, Bovenhuis H, Harlizius B: A genome-wide association study on androstenone levels in pigs reveals a cluster of candidate genes on chromosome 6. BMC Genet. 2010, 11: 42-
Ponsuksili S, Murani E, Brand B, Schwerin M, Wimmers K: Integrating expression profiling and whole-genome association for dissection of fat traits in a porcine model. J Lipid Res. 2011, 52: 668-678. 10.1194/jlr.M013342.
Goedbloed DJ, Megens HJ, Van Hooft P, Herrero-Medrano JM, Lutz W, Alexandri P, Crooijmans RPMA, Groenen MAM, Van Wieren SE, Ydenberg RC, Prins HHT: Genome-wide single nucleotide polymorphism analysis reveals recent genetic introgression from domestic pigs into Northwest European wild boar populations. Mol Ecol. 2012, 22: 856-866.
Bosse M, Megens H-J, Madsen O, Paudel Y, Frantz LAF, Schook LB, Crooijmans RPMA, Groenen MAM: Regions of homozygosity in the porcine genome: consequence of demography and the recombination landscape. PLoS Genet. 2012, 8: e1003100-10.1371/journal.pgen.1003100.
Hayes BJ, Visscher PM, McPartlan HC, Goddard ME: Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res. 2003, 13: 635-643. 10.1101/gr.387103.
PW DRa, Hayes BJ, Spelman RJ, Goddard ME: Linkage disequilibrium and persistence of phase in Holstein-Friesian, Jersey and Angus cattle. Genetics. 2008, 179: 1503-1512. 10.1534/genetics.107.084301.
Purfield DC, Berry DP, McParland S, Bradley DG: Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13: 70-
Amaral AJ, Ferretti L, Megens H-J, Crooijmans RPMA, Nie H, Ramos-Onsins SE, Perez-Enciso M, Schook LB, Groenen MAM: Genome-wide footprints of pig domestication and selection revealed through massive parallel sequencing of pooled DNA. PloS One. 2011, 6: e14782-10.1371/journal.pone.0014782.
Rubin C-J, Megens H-J, Barrio AM, Maqbool K, Sayyab S, Schwochow D, Wang C, Carlborg O, Jern P, Jorgensen CB, Archibald AL, Fredholm M, Groenen MAM, Andersson L: Strong signatures of selection in the domestic pig genome. Proc Natl Acad Sci USA. 2012, 109: 19529-19536. 10.1073/pnas.1217149109.
Amaral AJ, Megens H-J, Crooijmans RPMA, Heuven HCM, Groenen MAM: Linkage disequilibrium decay and haplotype block structure in the pig. Genetics. 2008, 179: 569-579. 10.1534/genetics.107.084277.
Tortereau F, Servin B, Frantz L, Megens H-J, Milan D, Rohrer G, Wiedmann R, Beever J, Archibald AL, Schook LB, Groenen MAM: A high density recombination map of the pig reveals a correlation between sex-specific recombination and GC content. BMC Genom. 2012, 13: 586-10.1186/1471-2164-13-586.
Wilkinson S, Archibald AL, Haley CS, Megens H-J, Crooijmans RPMA, Groenen MAM, Wiener P, Ogden R: Development of a genetic tool for product regulation in the diverse British pig breed market. BMC Genom. 2012, 13: 580-10.1186/1471-2164-13-580.
Ramos AM, Megens HJ, Crooijmans RPMA, Schook LB, Groenen MAM: Identification of high utility SNPs for population assignment and traceability purposes in the pig using high-throughput sequencing. Anim Genet. 2011, 42: 613-620. 10.1111/j.1365-2052.2011.02198.x.
Herrero-Medrano JM, Megens HJ, Crooijmans RP, Abellaneda JM, Ramis G: Farm-by-farm analysis of microsatellite, mtDNA and SNP genotype data reveals inbreeding and crossbreeding as threats to the survival of a native Spanish pig breed. Anim Genet. 2013, 44: 259-266. 10.1111/age.12001.
Badke YM, Bates RO, Ernst CW, Schwab C, Steibel JP: Estimation of linkage disequilibrium in four US pig breeds. BMC Genom. 2012, 13: 24-10.1186/1471-2164-13-24.
Uimari P, Tapio M: Extent of linkage disequilibrium and effective population size in Finnish Landrace and Finnish Yorkshire pig breeds. J Anim Sci. 2011, 89: 609-614. 10.2527/jas.2010-3249.
Evanno G, Regnaut S, Goudet J: Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol. 2005, 14: 2611-2620. 10.1111/j.1365-294X.2005.02553.x.
Sved JA: Linkage disequilibrium of chromosome segments. Theor Popul Biol. 1971, 141: 125-141.
Reich DE, Cargill M, Bolk S, Ireland J, Sabeti PC, Richter DJ, Lavery T, Kouyoumjian R, Farhadian SF, Ward R, Lander ES: Linkage disequilibrium in the human genome. Nature. 2001, 411: 199-204. 10.1038/35075590.
Megens H-J, Crooijmans RPMA, Bastiaansen JWM, Kerstens HHD, Coster A, Jalving R, Vereijken A, Silva P, Muir WM, Cheng HH, Hanotte O, Groenen MAM: Comparison of linkage disequilibrium and haplotype diversity on macro- and microchromosomes in chicken. BMC Genet. 2009, 10: 86-
Ardlie KG, Kruglyak L, Seielstad M: Patterns of linkage disequilibrium in the human genome. Nat Rev Genet. 2002, 3: 299-309. 10.1038/nrg777.
Tenesa A, Navarro P, Hayes BJ, Duffy DL, Clarke GM, Goddard ME, Visscher PM: Recent human effective population size estimated from linkage disequilibrium. Genome Res. 2007, 17: 520-526. 10.1101/gr.6023607.
Downing T, Imamura H, Decuypere S, Clark TG, Coombs GH, Cotton JA, Hilley JD, De DS, Maes I, Mottram JC, Quail MA, Rijal S, Sanders M, Stark O, Sundar S, Vanaerschot M, Hertz-fowler C, Dujardin J, Berriman M: Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res. 2011, 21: 2143-2156. 10.1101/gr.123430.111.
Gautier M, Laloë D, Moazami-Goudarzi K: Insights into the genetic history of French cattle from dense SNP data on 47 worldwide breeds. PloS One. 2010, 5: e13038-10.1371/journal.pone.0013038.
Decker JE, Pires JC, Conant GC, McKay SD, Heaton MP, Chen K, Cooper A, Vilkki J, Seabury CM, Caetano AR, Johnson GS, Brenneman RA, Hanotte O, Eggert LS, Wiener P, Kim J-J, Kim KS, Sonstegard TS, Van Tassell CP, Neibergs HL, McEwan JC, Brauning R, Coutinho LL, Babar ME, Wilson GA, McClure MC, Rolf MM, Kim J, Schnabel RD, Taylor JF: Resolving the evolution of extant and extinct ruminants with high-throughput phylogenomics. Proc Natl Acad Sci USA. 2009, 106: 18644-18649. 10.1073/pnas.0904691106.
Berthouly C, Maillard JC, Pham Doan L, Nhu Van T, Bed’Hom B, Leroy G, Hoang Thanh H, Laloë D, Bruneau N, Vu Chi C, Nguyen Dang V, Verrier E, Rognon X: Revealing fine scale subpopulation structure in the Vietnamese H’Mong cattle breed for conservation purposes. BMC Genet. 2010, 11: 45-
Druml T, Salajpal K, Dikic M, Urosevic M, Grilz-Seger G, Baumung R: Genetic diversity, population structure and subdivision of local Balkan pig breeds in Austria, Croatia, Serbia and Bosnia-Herzegovina and its practical value in conservation programs. Genet Sel Evol. 2012, 44: 5-10.1186/1297-9686-44-5.
McKay SD, Schnabel RD, Murdoch BM, Matukumalli LK, Aerts J, Coppieters W, Crews D, Dias Neto E, Gill CA, Gao C, Mannen H, Wang Z, Van Tassell CP, Williams JL, Taylor JF, Moore SS: An assessment of population structure in eight breeds of cattle using a whole genome SNP panel. BMC Genet. 2008, 9: 37-
Royo LJ, Alvarez I, Fernández I, Pérez-Pardal L, Alvarez-Sevilla A, Godinho R, Ferrand N, Goyache F: Genetic characterisation of Celtic-Iberian pig breeds using microsatellites. Proceedings of the 6 th International Symposium on the Mediterranean Pig: 11-13 October 2007. Edited by: Università di Bologna, Italy. 2007, Messina, Italy, 32-35.
Kirin M, McQuillan R, Franklin CS, Campbell H, McKeigue PM, Wilson JF: Genomic runs of homozygosity record population history and consanguinity. PloS One. 2010, 5: e13996-10.1371/journal.pone.0013996.
Frankham R, Ralls K: Conservation biology: inbreeding leads to extinction. Nature. 1998, 392: 441-442. 10.1038/33022.
Wang J: Estimation of effective population sizes from data on genetic markers. Phil Trans Roy Soc Lond B Biol Sci. 2005, 360: 1395-1409. 10.1098/rstb.2005.1682.
Hill WG: Estimation of effective population size from data on linkage disequilibrium. Genet Res. 1981, 38: 209-216. 10.1017/S0016672300020553.
Park L: Effective population size of current human population. Genet Res. 2011, 31: 1-10.
Waples RS, England PR: Estimating contemporary effective population size on the basis of linkage disequilibrium in the face of migration. Genetics. 2011, 189: 633-644. 10.1534/genetics.111.132233.
Pemberton TJ, Absher D, Feldman MW, Myers RM, Rosenberg NA, Li JZ: Genomic patterns of homozygosity in worldwide human populations. Am J Hum Genet. 2012, 91: 275-292. 10.1016/j.ajhg.2012.06.014.
Mcquillan R, Leutenegger A, Abdel-rahman R, Franklin CS, Pericic M, Barac-lauc L, Smolej-narancic N, Janicijevic B, Polasek O, Tenesa A, Macleod AK, Farrington SM, Rudan P, Hayward C, Vitart V, Rudan I, Wild SH, Dunlop MG, Wright AF, Campbell H, Wilson JF: Runs of homozygosity in European populations. Am J Hum Genet. 2008, 83: 359-372. 10.1016/j.ajhg.2008.08.007.
Alves PC, Pinheiro I, Godinho R, Vicente J, Gortázar C, Scandura M: Genetic diversity of wild boar populations and domestic pig breeds (Sus scrofa) in South-western Europe. Biol J Linn Soc. 2010, 101: 797-822. 10.1111/j.1095-8312.2010.01530.x.
Esteve-Codina A, Kofler R, Himmelbauer H, Ferretti L, Vivancos AP, Groenen MAM, Folch JM, Rodríguez MC, Pérez-Enciso M: Partial short-read sequencing of a highly inbred Iberian pig and genomics inference thereof. Heredity. 2011, 107: 256-264. 10.1038/hdy.2011.13.
Stumpf MPH, McVean GAT: Estimating recombination rates from population-genetic data. Nat Rev Genet. 2003, 4: 959-968. 10.1038/nrg1227.
Apollonio M, Randi E, Toso S: The systematics of the wildboar (Sus scrofa L.) in Italy. Bollettino di Zoologia. 1988, 3: 213-221.
Ferreira E, Souto L, Soares AMVM, Fonseca C: Genetic structure of the wild boar population in Portugal: evidence of a recent bottleneck. Mammalian Biology - Zeitschrift für Säugetierkunde. 2009, 74: 274-285.
Groenen MAM, Archibald AL, Uenishi H, Tuggle CK, Takeuchi Y, Rothschild MF, Rogel-Gaillard C, Park C, Milan D, Megens H-J, Li S, Larkin DM, Kim H, Frantz LAF, Caccamo M, Ahn H, Aken BL, Anselmo A, Anthon C, Auvil L, Badaoui B, Beattie CW, Bendixen C, Berman D, Blecha F, Blomberg J, Bolund L, Bosse M, Botti S, Bujie Z, Bystrom M, Capitanu B, et al: Analyses of pig genomes provide insight into porcine demography and evolution. Nature. 2012, 491: 393-398. 10.1038/nature11622.
Ottoni C, Flink LG, Evin A, Geörg C, De Cupere B, Van Neer W, Bartosiewicz L, Linderholm A, Barnett R, Peters J, Decorte R, Waelkens M, Vanderheyden N, Ricaut F-X, Cakirlar C, Cevik O, Hoelzel AR, Mashkour M, Karimlu AFM, Seno SS, Daujat J, Brock F, Pinhasi R, Hongo H, Perez-Enciso M, Rasmussen M, Frantz L, Megens H-J, Crooijmans R, Groenen MAM, et al: Pig domestication and human-mediated dispersal in western Eurasia revealed through ancient DNA and geometric morphometrics. Mol Biol Evol. 2013, 30: 824-832. 10.1093/molbev/mss261.
Larson G, Burger J: A population genetics view of animal domestication. Trends Genet. 2013, 29: 197-205. 10.1016/j.tig.2013.01.003.
Larson G, Albarella U, Dobney K, Rowley-Conwy P, Schibler J, Tresset A, Vigne J-D, Edwards CJ, Schlumbaum A, Dinu A, Balaçsescu A, Dolman G, Tagliacozzo A, Manaseryan N, Miracle P, Van Wijngaarden-Bakker L, Masseti M, Bradley DG, Cooper A: Ancient DNA, pig domestication, and the spread of the Neolithic into Europe. Proc Natl Acad Sci USA. 2007, 104: 15276-15281. 10.1073/pnas.0703411104.
Alves E, Ovilo C, Rodríguez MC, Silió L: Mitochondrial DNA sequence variation and phylogenetic relationships among Iberian pigs and other domestic and wild pig populations. Anim Genet. 2003, 34: 319-324. 10.1046/j.1365-2052.2003.01010.x.
Van Asch B, Pereira F, Santos LS, Carneiro J, Santos N, Amorim A: Mitochondrial lineages reveal intense gene flow between Iberian wild boars and South Iberian pig breeds. Anim Genet. 2012, 43: 35-41. 10.1111/j.1365-2052.2011.02222.x.
Nei M: Genetic distance between populations. Am Nat. 1972, 106: 283-292. 10.1086/282771.
Liu K, Muse SV: PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics. 2005, 21: 2128-2129. 10.1093/bioinformatics/bti282.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007, 24: 1596-1599. 10.1093/molbev/msm092.
Pritchard JK, Stephens M, Donnelly P: Inference of population structure using multilocus genotype data. Genetics. 2000, 155: 945-959.
Earl DA, vonHoldt BM: Structure harvester: a website and program for visualizing structure output and implementing the Evanno method. Conservat Genet Resour. 2011, 4: 359-361.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D: Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006, 38: 904-909. 10.1038/ng1847.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, De Bakker PIW, Daly MJ, Sham PC: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007, 81: 559-575. 10.1086/519795.
Barrett JC, Fry B, Maller J, Daly MJ: Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005, 21: 263-265. 10.1093/bioinformatics/bth457.
Acknowledgements
This project was financially supported by European Research Council under the European Community's Seventh Framework Programme (FP7/2007-2013)/ERC grant #ERC-2009-AdG: 249894 (Sel Sweep project).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests
Authors’ contributions
JMHM analyzed the data and wrote the manuscript. HJM designed and conceived this study. MAMG critically review the manuscript. MB designed the ROH analysis and supervised the corresponding writing parting of the manuscript. MPE collected samples and helped draft the manuscript. RPMAC and GRV collected the genetic data and participated in the supervision of the study. All authors read and approved the final manuscript.
Electronic supplementary material
12863_2013_1368_MOESM1_ESM.pdf
Additional file 1: Table S1: Fst pairwise (below the diagonal) and Nei’s genetic distances (above the diagonal) between populations. Table S2: Average r2 value for SNP spaced 0.5, 1.0, 1.5, 2.0, 2.5 and 3.0. (PDF 39 KB)
12863_2013_1368_MOESM2_ESM.png
{kind=link}
Additional file 2: Neighbor-Joining tree of pig populations constructed from individual pairwise genetic distance. WBPOR, wild boar from Portugal; WBSPN, wild boar from Spain; RE, Retinto Iberian; NI, Negro Iberian, IB, Iberian (unidentified variant); MJ, Manchado de Jabugo; BI, Bisaro; CM, Chato Murciano. (PNG 192 KB)
12863_2013_1368_MOESM3_ESM.zip
Additional file 3: PCA analysis using exclusively Iberian pigs. IB_Unk, Iberian unknown variant; NI, Negro Iberian; RE, Retinto; MJ, Manchado de Jabugo. (ZIP 4 KB)
12863_2013_1368_MOESM4_ESM.pdf
Additional file 4: STRUCTURE analysis and Evanno method to determine the optimal number of clusters; Membership coefficient of the breeds tested in the four clusters inferred by STRUCTURE software.(PDF 105 KB)
Additional file 5: Persistence of phase for intervals of 100 kb ranging from 0 to 10 Mb.(XLSX 28 KB)
12863_2013_1368_MOESM6_ESM.zip
Additional file 6: Estimated effective population size (Ne) over time: detail of 10,000 generations ago. WB, wild boar; NI, Negro Iberian; RE, Retinto; MJ, Manchado de Jabugo; BI, Bisaro; CM, Chato Murciano. (ZIP 3 KB)
12863_2013_1368_MOESM7_ESM.pdf
Additional file 7: Estimation of the percentage covered by ROH in each population. WB, wild boar; IB, Iberian; MJ, Manchado de Jabugo; BI, Bisaro; CM, Chato Murciano. (PDF 9 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Herrero-Medrano, J.M., Megens, HJ., Groenen, M.A. et al. Conservation genomic analysis of domestic and wild pig populations from the Iberian Peninsula. BMC Genet 14, 106 (2013). https://doi.org/10.1186/1471-2156-14-106
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2156-14-106