
Abstract
Background
Polymorphisms of the mannose-binding lectin gene (MBL2) affect the concentration and functional efficiency of the protein. We recently used haplotype-specific sequencing to identify 23 MBL2 haplotypes, associated with enhanced susceptibility to several diseases.
Results
In this work, we applied the same method in 288 and 470 chromosomes from Gabonese and European adults, respectively, and found three new haplotypes in the last group. We propose a phylogenetic nomenclature to standardize MBL2 studies and found two major phylogenetic branches due to six strongly linked polymorphisms associated with high MBL production. They presented high Fst values and were imbedded in regions with high nucleotide diversity and significant Tajima's D values. Compared to others using small sample sizes and unphased genotypic data, we found differences in haplotyping, frequency estimation, Fu and Li's D* and Fst results.
Conclusion
Using extensive testing for selective neutrality, we confirmed that stochastic evolutionary factors have had a major role in shaping this polymorphic gene worldwide.
Similar content being viewed by others


Background
MBL (mannose-binding lectin) is an important component of innate immunity and a central recognition molecule of the lectin pathway of complement, which probably represents the most ancient pathway of complement activation [1]. It binds to an array of carbohydrates such as d- mannose and N- acetyl-d-glucosamine on the surface of pathogens and directly opsonizes the microorganism for phagocytosis or activates the complement system via interaction with MBL-associated serine proteases (MASP-1, -2, -3 and Map19). Complement activation kills the pathogen by the membrane-attack complex or by complement-mediated phagocytosis through increased deposition of opsonic C3 fragments. MBL is also able to recognize altered self structures present on apoptotic cells, promoting their clearance, and to modulate the release of various pro-inflammatory cytokines [2, 3].
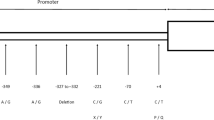
The MBL2 genetic polymorphism is responsible for the very common and widespread variation of circulating levels of MBL oligomers and of functional activity of the protein in the human species. This variation is mainly caused by three single nucleotide polymorphisms (SNPs) in the first exon of the gene: MBL2*D (Arg52Cys), *B (Gly54Asp) and *C (Gly57Glu). These mutations have a profound effect on the assembly and stability of the protein, which leads to an increase of low-molecular-mass MBL that has reduced capacity of activating complement and of ligand binding [4, 5]. The D, B and C SNPs have been collectively labeled O, whereas the major alleles at these loci have been called A. The concentration of the protein in serum is further modulated by at least three SNPs in the promoter region: MBL2*H,L (located 550 bp before the transcription start site), X, Y (located 221 bp before the transcription start site) and P, Q (non coding SNP located 4 bp after the transcription start site) [6, 7]. The combination of structural and promoter polymorphisms results in a dramatic variation in the concentration of high-order MBL oligomers in apparently healthy individuals of up to 1,000-fold (European: range <20-10,000 ng/ml) [8]. Linkage disequilibrium between the SNPs is responsible for only eight haplotypes (as opposed to the 64 theoretically possible) associated with increasingly lower MBL serum concentration: MBL2*HYPA = LYQA = LYPA > LXPA ≫ HYPD = LYPB = LYQC = LYPD [7, 9–13]. Using a haplotyping strategy developed by one of us, we recently defined 14 additional allelic haplotypes, most of them similar to LYQA or LYPA [2]. Genotypes carrying two copies of either HYPD, LYPB, LYQC or LYPD or one of them and LXPA are particularly associated with the susceptibility and severity of many diseases, as well as with protection against intracellular infections such as tuberculosis, leprosy and leishmaniasis [14–16].
In this work, we aimed to improve our former analysis by sequencing and haplotyping larger samples of European- and African-derived populations. In order to standardize and simplify comparisons between future association studies, we propose a nomenclature based on the evolutionary convergence of the identified MBL2 haplotypes [17]. We tested our samples for the hypothesis of selective neutrality and suggest that stochastic evolutionary factors have had a major role in shaping this polymorphism worldwide.
Results
To uncover the selective role diseases could have exerted on the MBL2 polymorphism, we evaluated the MBL2 promoter and exon 1 region from 856 chromosomes of Gabonese adults (this work) and children [2], as well as from 470 chromosomes belonging to individuals of European descent, and compared it with previously published data. Genotype frequencies were at Hardy and Weinberg equilibrium.
MBL2 haplotypes identified in this study are listed in Table 1. They were named according to their evolutionary divergence [17] from a hypothetical ancient sequence probably related to LYQA and LYPA [11, 18]. According to the nomenclature system we adopted, the first clades to diverge are numbered with Arabic numerals. The 26 identified haplotypes are divided into two major phylogenetic branches by six polymorphisms (P1, Q1 or g.396A > C; P2, Q2 or g.474A > G; P3, Q3 or g.487A > G; P4, Q4 or g.495_500del6; P5, Q5 or g.753C > T, all in strong linkage disequilibrium with the commonly investigated P6, Q6 polymorphism or g.826C > T) (Figure 1). Clade *1 is represented by LYPA and other haplotypes with P variants. Clade *4 is represented by LYQA and other haplotypes with Q variants. Other clades are represented by the intermediate rare haplotypes previously found by our group in Gabon (2 and 3) [2]. Sublineages of each clade are subsequently designated with capital letters (e.g. LYQA-derived haplotypes = *4A and LYQC-derived haplotypes = *4F), and individual present-day haplotypes are given Arabic numerals (e.g. LYQA = *4A1), following the schema numerals/letters/numerals, if they diverge further (e.g. the LYQC-derived haplotype with the g.797C > A SNP, associated with severe malaria = *4F2A). This system is flexible enough for the accommodation of new haplotypes. For example, we added the LYPA-similar haplotypes H16 and H19 found by others exclusively in Pygmy populations [19] as *1K1 and *1L1, and added the HYPG haplotype described by us in another study [16], as 1B4. It is however not suited for recombinant haplotypes. In this case, we chose to call them by the names of the parental haplotypes, separated by a dot. LYPD for example is most probably the product of a recent intragenic recombination event between HYPD (*1B2) and LYPA (*1A1) or LYPB (*1F1) [20]. Since the recombination between HYPD and LYPB would have generated HYPB, which has not been found, we arbitrarily chose to call this haplotype *1A1.1B2 (equivalent to LYPA × HYPD). We also wished to incorporate reported associations of haplotypes with MBL concentration. In order to do this, we added a dash followed by small capitalized "h" or "l" letters, referring to "high" or "low" MBL levels in serum, respectively (e.g. LYQA = *4A1-h).

Maximum parsimony tree with suggested phylogenetic nomenclature (see text). The recombinant haplotype *1A1.1B2-l (LYPD) was excluded. Bootstrap values are given at nodes of the tree.
We identified in this and in other studies 14 haplotypes belonging to clade *1 and 9 haplotypes belonging to clade *4 and added data from others for comparison (Table 2). Eight of the first 14 and 6 of the last 9 haplotypes were polymorph in at least one population. Among the rare haplotypes, we found three previously unknown in the European population: *1B3, a rare HYPA- similar haplotype; *1C2, the only LXPA- similar haplotype; and *4C1-h, a LYQA-similar haplotype with a g.456G > T SNP found in three heterozygotes (the first two haplotypes were singletons). The g.456G > T SNP was assigned by others to an otherwise HYPA haplotype reconstructed from unphased genotypic data of one Sardinian heterozygote [19]. Maximum likelihood phasing of our own data with the EM and ELB algorithms generated 1-2% erroneously assembled haplotypes in the Gabonese and European samples. Only in the Gabonese, seven spurious "new" haplotypes were generated with the EM and eleven with the ELB algorithm (Table 3). To verify the effect of sample size in frequency estimates, we compared the haplotype distribution between some populations investigated by us and by others [19]. Although there were no significant differences with the exact population differentiation test, differences between individual haplotype frequencies were significant, even between samples with similar ancestry (Table 2).
With the exception of LYQA (*4A1-h), *4 haplotypes are well represented only in the African population. In contrast, HYPA (*1B1-h) and LYPB (*1F1-l) are among the *1 haplotypes that reach high frequencies in the European, Asian and Native American, but not in the African population (Figure 2). The uneven haplotype distribution around the world is reflected by the average Fst value among all segregating sites (0.1831, P < 0.00001), which indicate great genetic differentiation between the analysed populations. One of the lowest individual significant Fst values corresponded to the X/Y SNP, whereas the highest values corresponded to the H, L and P, Q segregating sites (Figure 3). The time to the most recent common ancestor of the MBL2 alleles was inferred at 73,251 years ago [95% CI 5,220 - 214,440]. The mean coalescence time implies that the ancestor of groups *1 and *4 alleles were separated before the modern human dispersal from Africa [21]. The TMRCA of groups *1 and *4 was estimated to be ca. 55,000 years ago, which also indicates that the presence of alleles of African populations in both clades is a result of an ancient ancestry.


Global Fst values distributed along the sequence. Fsts were calculated using the data of Table 2 and of [19]. Nucleotide positions corresponding to variant sites are shown on the x-axis. *** p < 0.001, ** p < 0.01.
*1B-derived haplotypes, even those found using maximum-likelihood phasing by others [19], seem to be restricted to Euroasiatic populations. Beyond those described in this work, we recently identified *1B4 in the Euro-Brazilian population. This haplotype is similar to HYPA but with a synonymous substitution in codon 44 (also called HYPG) [16]. To our knowledge, LXPA (*1C1-l) has only one rare similar haplotype (*1C2), identified in one European individual. We also found only one LYPB- similar haplotype (*1F2), but others cite another four [19]. Each occur with frequencies around 2% in Asian/Amerindian groups (Ashkenazi Jewish, Japanese, Chinese and Kaingang), but three were defined by SNPs upstream to the region analysed in this study. The *1H1-h haplotype has a similar global distribution as the commonly investigated haplotypes and is well represented in African, Asian and Amerindian(-derived) populations, being less frequent in European groups. We found a similar haplotype (*1H2-h) once in a Gabonese and once in a Euro-Brazilian individual. All other clade *1 haplotypes are concentrated in African groups. *1E1-h has a rare coding mutation found only once in the Gabonese, as *1G1-h [2]. The *1D1-h haplotype, which we found with 3% frequency in this population, was found by others with comparable frequencies (1.6 - 4.2%) in the Mbuti Pygmy, Nigerian Yoruba and Somali populations [19]. *1J1-h was also found with 1.6% and 0.8% frequencies in Tanzanian Chagga and in the Somali groups, respectively. *2A1-h and *3A1-h are intermediate between P and Q containing haplotypes and most probably reminiscent of the ancient original MBL2 haplotype [2]. The LYQA- similar *4B1-l haplotype carries a coding mutation and was found only once in the Gabonese, as the LYQC-similar haplotype *4F2B-l. In addition to the Gabonese, *4D1-h was found by others with 1.6% frequencies in the Tanzanian Chagga [19]. *4E1-h has a SNP within a glucocorticoid responsive element and seems to be well distributed in Africa, except in the Mbuti and Baka Pygmies [19]. 4F2A-l was formerly found associated with severe malaria [2] and has a similar distribution, except for the fact that it is also present in South-West Asian and European(-derived) groups with 11.9% (Ashkenazi Jewish [19]) to 0.5% (Germans, this work) frequencies. *4F3-l was also found in the Biaka Pygmy (2.1%), Nigerian Yoruba (1.6%) and Tanzanian Chagga (4.7%) groups [19], as well as in Afro-Americans [18] and in one individual of the Kaingang Amerindian population, known to be of mixed ancestry [22].
Tajima's D was significant in those regions containing five of the six P, Q segregating sites in the Gabonese population (Figure 4A). Yet Fu and Li's D* was significant in regions with rare SNPs: the LXPA-derived *1C2 haplotype in Europeans and the LYPA- derived *1E1-h haplotype in the Gabonese (also called LYPF due to a non-synonymous SNP in the exon 1 region) (Figure 4B). Highest nucleotide diversity was registered in the same windows with Tajima's D peaks (Figure 4C). None of the neutrality tests employed for the whole sequence or parts of it yielded significant results (Table 4).

Sliding window plot of (A) Tajima's D values, (B) Fu and Li's D* values and (C) nucleotide diversity for the entire sequenced region. Statistics were calculated for overlapping windows of 60 bp, placed at 15 bp intervals along the sequence. * P < 0.05.
Discussion
Both circulating levels of MBL oligomers and functional activity have been correlated with common MBL2 genetic variants. There are at least 28 segregating sites in the MBL2 promoter and exon 1 sequence [23], and 26 allelic haplotypes were physically defined in this study. Nucleotide diversity in Afro-derived populations reached 5 × the average value of chromosome 10 (8.25 × 10-4) [24], where the MBL2 gene resides (10q11.2→q21). This is still 2 × less than the lowest values found for polymorphic MHC regions (1%) [25], indicating that the MBL2 promoter-exon1 nucleotide diversity is intermediate among immune protein coding genes.
Several of the newly identified haplotypes are polymorph and of interest for disease association studies. Nevertheless beside the A/B/C/D system adopted for exon 1 alleles since 1991 [26] and of the H, L, X, Y and P, Q names for promoter SNPs since 1998, no other nomenclature was suggested. We adopted a phylogenetic approach that easily accommodates new haplotypes following a logical order, and suggested a way to call eventual recombinant haplotypes, incorporating knowledge about MBL serum levels.
Nevertheless haplotypes generated with EM and ELB haplotyping algorithms should be included with caution, especially when containing singletons. In our comparison, EM and ELB algorithms allowed for 1-2% errors in populations with high nucleotide diversity (π). The pseudo-Bayesian ELB performed worse in groups with very high π values, as Africans, generating more spurious "new" haplotypes. We did not find six of the haplotypes reconstructed by others using the Bayesian method implemented in PHASE software [19]. Two were recombinant (LYQB and HXPA), one presented a SNP that we haplotyped to LYQA and three were LYPA-similar haplotypes that seemed to be restricted to Pygmy populations, with SNPs presenting high Fst values. To avoid the inclusion of false haplotypes in the nomenclature system, we followed the approach of a group which only analysed haplotypes having a minimal frequency of 10% [27]. Two of the Pygmy haplotypes fulfilled this requirement, but all other haplotypes should ideally be phased by a physical haplotyping technique before inclusion.
Others used sample sizes at least four times smaller than ours [18, 19]. This caused discrepant frequency results especially for the most common haplotypes. Since rare variants are not easily detected in small population samples, we also found considerable differences between our Fu and Li's D* and F* and other's results [18]. Indeed, two singletons caused significant D* values in regions with very low nucleotide diversity levels specifically in our European and Gabonese samples.
We added data from other studies [2, 18, 19, 22, 28] to calculate the Fst statistic. This approach resulted in much higher Fst values for the whole gene (0.18), than those found previously by others (0.06 [18]) and by us using only the Amerindian and Chinese samples (0.12, [22]). The same was true for the H/L and P/Q SNPs (Fst values around 0.2-0.25, compared to published 0.1-0.15, [18]), which indicate that they are good markers for population differentiation. As opposed to these high Fst values, the X/Y SNP presented values lower than 0.05 in this and in another study [18], compatible with global balancing selection.
We previously discussed the origin and distribution of the LYPA (*1A1-h), HYPA (*1B1-h), HYPD (*1B2-l), LXPA (*1C1-l), LYPB (*1F1-l), LYPD (*1A1.1B2-l), LYQA (*4A1-h) and LYQC (*4F1-l) haplotypes [22]. In general, the most frequent clade *1 haplotypes are globally distributed, whereas clade *4 haplotypes are more restricted to the African continent. Four of the five most ancient haplotypes also belong to clade *1: *1A1-h, *1B1-h, *1C1-l and *1H1-h. Among them, only *1C1-l (with the X variant) is associated with low (although complement-activating) MBL production. This and the *4A1-h haplotype do not naturally occur in native Aboriginal, Greenlandic and Amerindian populations [11, 22, 29, 30], having probably been lost through bottleneck effects along the migration routes. The other eight polymorph haplotypes (with a frequency higher than 1%) have probably had a more recent origin, being geographically more restricted. Among them, only two are associated with high MBL levels: *1D1-h and *4E1-h. All others generate low MBL levels that, in addition, are greatly restricted in complement activation due to the B, C and D mutations, which occur in critical residues of the collagen-like region (*1B2-l, *1F1-l, *4F1-l, *4F2A-l, *4F3-l and *1F2) (Figure 5). Interestingly, the MBL1P1 pseudogene has been selectively turned off during evolution through the same molecular mechanisms causing the non-functional recent MBL2 haplotypes in man [31]. A more restricted distribution is obviously the case of all haplotypes containing singletons, as well as of *1J1-h, *4D1-h and *4F3-l in Africa, *1A1.1B2-l, *1B2-l and *4C1-h in Europe. They are therefore characteristic of different ethnic groups.

Distribution and origins of the MBL2 alleles in the world. Arrows denote the mutational steps between haplotypes (six between *1 and *4) and when dotted, the ancient migratory routes with their approximate ages [51]. The haplotypes which could have been lost by natural selection and/or genetic drift were denoted by '?'. In bold: haplotypes generated before human out-of-Africa migration. Squared: more recent haplotypes, with geographically restricted distribution. KYA thousand years ago.
The clades *1 and *4 are separated by six mutational steps (P, Q variants), which probably occurred before the human out-of-Africa migration (Figure 5). Of these six segregating sites, probably the most ancient is the g.487G > A variant and the most recent, the g.396A > C variant [2]. Q variants are less widely distributed than P variants, justifying their high Fst values. They are functionally associated with higher promoter activity [6, 32] and five of them presented positive, significant Tajima's D values in the Gabonese population. A significant positive value for Tajima's D test indicates an excess of intermediate-frequency variants, as compared with expected frequencies under neutrality, and constitutes evidence of balancing selection (mutations leading to higher MBL levels could have been selectively retained in the ancient human population) or population subdivision. Nevertheless the emergence of several recent mutations as well as genetic drift erased the selective signature at the long haplotype scale, leading to non-significant, although positive, Tajima's D values for the whole haplotype in this and in other studies (eg. Table 4), one of which included 1,166 chromosomes from 24 worldwide populations [18, 19, 22]. The patterns of MBL2 variation at the large temporal scale would thus have been shaped by stochastic evolutionary factors and therefore be compatible with neutral evolution.
Conclusions
In this work, we evaluated the MBL2 promoter-exon 1 region using haplotype-specific sequencing in more than 700 chromosomes and found three new European haplotypes. We propose a phylogenetic nomenclature to standardize MBL2 studies and found two major phylogenetic branches due to six strongly linked polymorphisms associated with high MBL production. They present high Fst values and are imbedded in regions with high nucleotide diversity and significant Tajima's D values. Compared to others using small sample sizes and unphased genotypic data, we found differences in haplotyping, frequency estimation, Fu and Li's D* and Fst results. Using extensive testing for selective neutrality, we confirmed that stochastic evolutionary factors have had a major role in shaping this polymorphic gene worldwide.
Methods
Subjects and samples
We investigated 104 German Europeans, 131 Euro-Brazilians and 144 Gabonese adults. The German Europeans were healthy unrelated students and employees of the University of Tübingen, enrolled as controls in a genetic association study with type 2 diabetes, approved by the Ethics Committee of the University of Tübingen in Germany [33]. The Euro-Brazilians were healthy blood donors with mixed, however predominantly European ancestry, resident in Paraná state, South Brazil, sampled for different association studies, all approved by the Ethics Committee of Research in Humans of the Clinical Hospital, Federal University of Paraná, Brazil [16, 34, 35]. The Gabonese individuals took part in a large epidemiologic survey to detect the prevalence of asymptomatic Plasmodium falciparum infection in the villages around Lambaréné, Gabon, a study approved by the ethics committee of the International Foundation Albert Schweitzer Hospital [36]. All individuals signed an informed consent form prior to their inclusion in these studies.
MBL2 typing
DNA was collected with anticoagulant ethylenediaminetetraacetic acid and extracted from peripheral blood mononuclear cells through standard salting-out and phenol/chloroform/isoamyl alcohol methods. A fragment of 1059 nucleotides was amplified using the forward primers MBLfor (5'-ATGGGGCTAGGCTGCTGAG-3') and the reverse primer MBLrev (5'-CCAACACGTACCTGGTTCCC-3'). Sequence specific (SSP) PCR products were generated using the same reverse primer, combined to forward primers specific for variant H (Hf: 5'-GCTTACCCAGGCAAGCCTGTG-3') or for the variant L (Lf: 5'-GCTTACCCAGGCAAGCCTGTC-3'); for the variant X (Xf: 5'-CCATTTGTTCTCACTGCCACC-3') or for the variant Y (Yf: 5'-CCATTTGTTCTCACTGCCACG-3'). The PCR products with the primers Hf or Lf with MBLrev and Xf or Yf with MBLrev were 837 and 508 nucleotides in length, respectively. Hf and Lf were also combined to specific reverse primers for the variant P (Pr: 5'-CTCAGTTAATGAACACATATTTACCG-3') or for the variant Q (Qr: 5'-CTCAGTTAATGAACACATATTTACCA-3'), generating a product of 599 nucleotides. All fragments were sequenced with the amplification primers or with an internal exon 1 sequencing primer, MBLint (5'-GAGGCCAGGGATGGGTCATC-3'), using Big dye terminator version 1.1 chemistry (Applied Biosystems, Foster City, CA). Amplification conditions are described in detail elsewhere [20]. The reactions were purified with the Performa DTR V3 system (Edge BioSystems, Gaithersburg, MD) and analyzed on an automated sequencer (ABI Prism 3100 Genetic Analyzer, Applied Biosystems, Foster City, CA). New variants (singletons) were verified by reamplification and resequencing.
Statistical analyses
Genotype and haplotype frequencies were obtained by direct counting. We tested for deviations from Hardy-Weinberg proportions with the exact test of Guo and Thompson [37]. The haplotype frequency distributions of the populations examined by our group and by others were compared by applying the exact test of population differentiation of Raymond and Rousset [38]. Genetic differentiation among populations was estimated from haplotype frequencies using the Fst statistic, based on the analysis of molecular variance [39]. To verify the effect of other methods to infer haplotypes compared to physical haplotyping of SNPs, we simulated our own data using the (maximum-likelihood) EM algorithm or the (pseudo-Bayesian) ELB algorithm, with the settings recommended by the authors [40, 41]. These statistical analyses were done using the software package ARLEQUIN version 3.1 [42]. Fisher's exact tests were performed for differences between individual haplotype frequencies, using SISA software package http://home.clara.net/sisa.
We calculated the following summary statistics of genetic diversity: the number of polymorphic sites (S), the nucleotide diversity over loci (π) and Watterson's θ, defined as 4Neμ, where Ne is the effective population size and μ, the estimated mutation rate. We examined deviation from neutrality-equilibrium conditions using Tajima's D statistic [43], Fu and Li's D and Fu and Li's F without an outgroup (also known as D* and F*) [44] and Fay and Wu's H [45] tests. Significance was assessed by comparing the observed values to 104 coalescent simulations, conditional on the observed sample size and on the value of S or on the value of θ, assuming a standard neutral model with no recombination. Deletions were excluded from all analyses. To see if deviation from selective neutrality can be found in specific regions of the gene, we also tested the 5' upstream regulatory region (which includes the non-coding P, Q SNP) and the exon 1 coding region separately. The heterogeneity in π values and Tajima's D statistic across the sequenced region was also examined by use of the sliding window feature of the DnaSP program. Statistics were calculated for overlapping windows of 60 bp, placed at 15 bp intervals along the sequence. Neutrality tests and sequence diversity parameters were calculated using the DnaSP version 4.10.1 software [46].
The Network 4.1.1.2 package http://www.fluxus-technology.com/sharenet.htm was used to construct the minimum-mutation network, which reflects the mutational relationships among the MBL2 haplotypes by means of the Median Joining (MJ) algorithm [47]. The MEGA 3.1 program was used to construct the phylogenetic maximum parsimony tree with bootstrap test http://www.megasoftware.net/. The time to the most recent common ancestor (TMRCA) of MBL2 was estimated using a relaxed molecular clock approach [48]. Evolutionary rate was modeled by the uncorrelated lognormal distribution and a coalescent prior (Bayesian skyline) was assigned to the tree. The average rate of molecular evolution of the MBL2 gene (1 × 10-7) was obtained using a theta per site value of 0.0039 calculated for human sequences in DnaSP [46] and the estimate of human effective population size of 10,000 [49]. A normal prior with mean 1 × 10-7 and standard deviation of 1 × 10-7 was used for the rate of evolution. Divergence time inference was conducted in BEAST 1.4.8 [50]. In order to obtain the posterior distribution of divergence times, the Markov chain was sampled 50,000 times and 10% of the states were discarded as burn-in.
References
Fujita T, Matsushita M, Endo Y: The lectin-complement pathway - its role in innate immunity and evolution. Immunol Rev. 2004, 198: 185-202. 10.1111/j.0105-2896.2004.0123.x.
Boldt AB, Luty A, Grobusch MP, Dietz K, Dzeing A, Kombila M, et al: Association of a new mannose-binding lectin variant with severe malaria in Gabonese children. Genes Immun. 2006, 7: 393-400. 10.1038/sj.gene.6364312.
Gadjeva M, Takahashi K, Thiel S: Mannan-binding lectin--a soluble pattern recognition molecule. Mol Immunol. 2004, 41: 113-121. 10.1016/j.molimm.2004.03.015.
Garred P, Larsen F, Madsen HO, Koch C: Mannose-binding lectin deficiency--revisited. Mol Immunol. 2003, 40: 73-84. 10.1016/S0161-5890(03)00104-4.
Larsen F, Madsen HO, Sim RB, Koch C, Garred P: Disease-associated mutations in human mannose-binding lectin compromise oligomerization and activity of the final protein. J Biol Chem. 2004, 279: 21302-21311. 10.1074/jbc.M400520200.
Juliger S, Luckner D, Mordmuller B, May J, Weierich A, Lell B, et al: Promoter variants of the human mannose-binding lectin gene show different binding. Biochem Biophys Res Commun. 2000, 275: 617-622. 10.1006/bbrc.2000.3343.
Madsen HO, Garred P, Thiel S, Kurtzhals JA, Lamm LU, Ryder LP, et al: Interplay between promoter and structural gene variants control basal serum level of mannan-binding protein. J Immunol. 1995, 155: 3013-3020.
Dommett RM, Klein N, Turner MW: Mannose-binding lectin in innate immunity: past, present and future. Tissue Antigens. 2006, 68: 193-209. 10.1111/j.1399-0039.2006.00649.x.
Ip WK, To YF, Cheng SK, Lau YL: Serum mannose-binding lectin levels and mbl2 gene polymorphisms in different age and gender groups of southern Chinese adults. Scand J Immunol. 2004, 59: 310-314. 10.1111/j.0300-9475.2004.01392.x.
Lee SG, Yum JS, Moon HM, Kim HJ, Yang YJ, Kim HL, et al: Analysis of mannose-binding lectin 2 (MBL2) genotype and the serum protein levels in the Korean population. Mol Immunol. 2005, 42: 969-977. 10.1016/j.molimm.2004.09.036.
Madsen HO, Satz ML, Hogh B, Svejgaard A, Garred P: Different molecular events result in low protein levels of mannan-binding lectin in populations from southeast Africa and South America. J Immunol. 1998, 161: 3169-3175.
Minchinton RM, Dean MM, Clark TR, Heatley S, Mullighan CG: Analysis of the relationship between mannose-binding lectin (MBL) genotype, MBL levels and function in an Australian blood donor population. Scand J Immunol. 2002, 56: 630-641. 10.1046/j.1365-3083.2002.01167.x.
Steffensen R, Thiel S, Varming K, Jersild C, Jensenius JC: Detection of structural gene mutations and promoter polymorphisms in the mannan-binding lectin (MBL) gene by polymerase chain reaction with sequence-specific primers. J Immunol Methods. 2000, 241: 33-42. 10.1016/S0022-1759(00)00198-8.
Alonso DP, Ferreira AF, Ribolla PE, de MSI, do Socorro Pires e Cruz, Aecio dC, et al: Genotypes of the mannan-binding lectin gene and susceptibility to visceral leishmaniasis and clinical complications. J Infect Dis. 2007, 195: 1212-1217. 10.1086/512683.
Garred P, Harboe M, Oettinger T, Koch C, Svejgaard A: Dual role of mannan-binding protein in infections: another case of heterosis?. Eur J Immunogenet. 1994, 21: 125-131. 10.1111/j.1744-313X.1994.tb00183.x.
Messias-Reason IJ, Boldt AB, Moraes Braga AC, Rosen Seeling SE, Dornelles L, Pereira-Ferrari L, et al: The association between mannan-binding lectin gene polymorphism and clinical leprosy: new insight into an old paradigm. J Infect Dis. 2007, 196: 1379-1385. 10.1086/521627.
Nebert DW: Proposal for an allele nomenclature system based on the evolutionary divergence of haplotypes. Hum Mutat. 2002, 20: 463-472. 10.1002/humu.10143.
Bernig T, Taylor JG, Foster CB, Staats B, Yeager M, Chanock SJ: Sequence analysis of the mannose-binding lectin (MBL2) gene reveals a high degree of heterozygosity with evidence of selection. Genes Immun. 2004, 5: 461-476. 10.1038/sj.gene.6364116.
Verdu P, Barreiro LB, Patin E, Gessain A, Cassar O, Kidd JR, et al: Evolutionary insights into the high worldwide prevalence of MBL2 deficiency alleles. Hum Mol Genet. 2006, 15: 2650-2658. 10.1093/hmg/ddl193.
Boldt AB, Petzl-Erler ML: A new strategy for mannose-binding lectin gene haplotyping. Hum Mutat. 2002, 19: 296-306. 10.1002/humu.10051.
Cavalli-Sforza LL, Piazza A, Menozzi P, Mountain J: Reconstruction of human evolution: bringing together genetic, archaeological, and linguistic data. Proc Natl Acad Sci USA. 1988, 85: 6002-6006. 10.1073/pnas.85.16.6002.
Boldt AB, Culpi L, Tsuneto LT, de Souza IR, Kun JF, Petzl-Erler ML: Diversity of the MBL2 gene in various Brazilian populations and the case of selection at the mannose-binding lectin locus. Hum Immunol. 2006, 67: 722-734. 10.1016/j.humimm.2006.05.009.
Messias-Reason I, Boldt AB: Mannose-Binding Lectin, A Multifunctional Molecule of the Innate Immune System: Biology, Genetics and Disease Association. Genetic Predisposition to Disease. Edited by: Torres SL, Marin MS. 2008, New York: Nova Science, 1
Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, et al: A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001, 409: 928-933. 10.1038/35057149.
Gaudieri S, Dawkins RL, Habara K, Kulski JK, Gojobori T: SNP profile within the human major histocompatibility complex reveals an extreme and interrupted level of nucleotide diversity. Genome Res. 2000, 10: 1579-1586. 10.1101/gr.127200.
Sumiya M, Super M, Tabona P, Levinsky RJ, Arai T, Turner MW, et al: Molecular basis of opsonic defect in immunodeficient children. Lancet. 1991, 337: 1569-1570. 10.1016/0140-6736(91)93263-9.
Hummelshoj T, Munthe-Fog L, Madsen HO, Garred P: Functional SNPs in the human ficolin (FCN) genes reveal distinct geographical patterns. Mol Immunol. 2008, 45: 2508-2520. 10.1016/j.molimm.2008.01.003.
Zhang H, Zhou G, Zhi L, Yang H, Zhai Y, Dong X, et al: Association between mannose-binding lectin gene polymorphisms and susceptibility to severe acute respiratory syndrome coronavirus infection. J Infect Dis. 2005, 192: 1355-1361. 10.1086/491479.
Turner MW, Dinan L, Heatley S, Jack DL, Boettcher B, Lester S, et al: Restricted polymorphism of the mannose-binding lectin gene of indigenous Australians. Hum Mol Genet. 2000, 9: 1481-1486. 10.1093/hmg/9.10.1481.
Juliger S, Kremsner PG, Alpers MP, Reeder JC, Kun JF: Restricted polymorphisms of the mannose-binding lectin gene in a population of Papua New Guinea. Mutat Res. 2002, 505: 87-91.
Seyfarth J, Garred P, Madsen HO: The 'involution' of mannose-binding lectin. Hum Mol Genet. 2005, 14: 2859-2869. 10.1093/hmg/ddi318.
Naito H, Ikeda A, Hasegawa K, Oka S, Uemura K, Kawasaki N, et al: Characterization of human serum mannan-binding protein promoter. J Biochem (Tokyo). 1999, 126: 1004-1012.
Schwab M, Lupescu A, Mota M, Mota E, Frey A, Simon P, et al: Association of SGK1 gene polymorphisms with type 2 diabetes. Cell Physiol Biochem. 2008, 21: 151-160. 10.1159/000113757.
Alves Pedroso ML, Boldt AB, Pereira-Ferrari L, Steffensen R, Strauss E, Jensenius JC, et al: Mannan-binding lectin MBL2 gene polymorphism in chronic hepatitis C: association with the severity of liver fibrosis and response to interferon therapy. Clin Exp Immunol. 2008, 152: 258-264. 10.1111/j.1365-2249.2008.03614.x.
Messias RI, Schafranski MD, Jensenius JC, Steffensen R: The association between mannose-binding lectin gene polymorphism and rheumatic heart disease. Hum Immunol. 2006, 67: 991-998. 10.1016/j.humimm.2006.08.296.
Dal-Bianco MP, Koster KB, Kombila UD, Kun JF, Grobusch MP, Ngoma GM, et al: High prevalence of asymptomatic Plasmodium falciparum infection in Gabonese adults. Am J Trop Med Hyg. 2007, 77: 939-942.
Guo SW, Thompson EA: Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics. 1992, 48: 361-372. 10.2307/2532296.
Raymond M, Rousset F: An exact test for population differentiation. Evolution. 1995, 49: 1280-1283. 10.2307/2410454.
Excoffier L, Smouse PE, Quattro JM: Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992, 131: 479-491.
Excoffier L, Slatkin M: Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol. 1995, 12: 921-927.
Excoffier L, Laval G, Balding D: Gametic phase estimation over large genomic regions using an adaptive window approach. Hum Genomics. 2003, 1: 7-19.
Excoffier L, Laval G, Schneider S: Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol Bioinform Online. 2005, 1: 47-50.
Tajima F: Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989, 123: 585-595.
Fu YX, Li WH: Statistical tests of neutrality of mutations. Genetics. 1993, 133: 693-709.
Fay JC, Wu CI: Hitchhiking under positive Darwinian selection. Genetics. 2000, 155: 1405-1413.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R: DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003, 19: 2496-2497. 10.1093/bioinformatics/btg359.
Bandelt HJ, Forster P, Rohl A: Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999, 16: 37-48.
Drummond AJ, Ho SY, Phillips MJ, Rambaut A: Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4: e88-10.1371/journal.pbio.0040088.
Takahata N: Relaxed natural selection in human populations during the Pleistocene. Jpn J Genet. 1993, 68: 539-547. 10.1266/jjg.68.539.
Drummond AJ, Rambaut A: BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007, 7: 214-10.1186/1471-2148-7-214.
Stanyon R, Sazzini M, Luiselli D: Timing the first human migration into eastern Asia. J Biol. 2009, 8: 18-10.1186/jbiol115.
Acknowledgements
The subjects of this investigation were informed about the aims of the study and their consent to participate is gratefully acknowledged. We thank Andrea Weierich and Silvelia Grummes for excellent technical assistance. Financial support was provided by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
ABWB carried out the molecular biological studies and wrote the manuscript. IJM-R participated in the molecular work and conducted the recruitment of subjects. ABWB, DM, CGS, MLP-E and KD participated in statistical analyses. FL, BL, and PGK conducted the recruitment of individuals for the study. JFJK supervised the molecular work and finalised the manuscript.
All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Boldt, A.B., Messias-Reason, I.J., Meyer, D. et al. Phylogenetic nomenclature and evolution of mannose-binding lectin (MBL2) haplotypes. BMC Genet 11, 38 (2010). https://doi.org/10.1186/1471-2156-11-38
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2156-11-38