
Abstract
Background
Mitochondrial DNA sequencing increasingly results in the recognition of genetically divergent, but morphologically cryptic lineages. Species delimitation approaches that rely on multiple lines of evidence in areas of co-occurrence are particularly powerful to infer their specific status. We investigated the species boundaries of two cryptic lineages of the land snail genus Trochulus in a contact zone, using mitochondrial and nuclear DNA marker as well as shell morphometrics.
Results
Both mitochondrial lineages have a distinct geographical distribution with a small zone of co-occurrence. In the same area, we detected two nuclear genotype clusters, each being highly significantly associated to one mitochondrial lineage. This association however had exceptions: a small number of individuals in the contact zone showed intermediate genotypes (4%) or cytonuclear disequilibrium (12%). Both mitochondrial lineage and nuclear cluster were statistically significant predictors for the shell shape indicating morphological divergence. Nevertheless, the lineage morphospaces largely overlapped (low posterior classification success rate of 69% and 78%, respectively): the two lineages are truly cryptic.
Conclusion
The integrative approach using multiple lines of evidence supported the hypothesis that the investigated Trochulus lineages are reproductively isolated species. In the small contact area, however, the lineages hybridise to a limited extent. This detection of a hybrid zone adds an instance to the rare reported cases of hybridisation in land snails.
Similar content being viewed by others


Background
Mitochondrial gene sequences have become a powerful tool to identify evolutionary lineages or species in animals [[1, 2], e.g. [3–5]]. Their application has often lead to the detection of divergent lineages within otherwise morphologically uniform recognised species [6]. Without any additional evidence from an explicit species delimitation approach, it is however rarely clear whether these lineages constitute reproductively isolated and/or ecologically different species or whether they are fully compatible, belonging to a single species [7, 8]. The study of populations within a contact zone is thus a useful approach to assess the species status of otherwise cryptic lineages. Indeed, these areas bring genetically distinct individuals in contact and offer them the possibility to interbreed: the resulting genetic signature observed in the contact zone provides information on the degree of reproductive isolation between the lineages.
Here, we evaluated the species status of two lineages of the Trochulus sericeus/hispidus complex (Gastropoda: Pulmonata: Helicoidea: Hygromiidae) in an area of co-occurrence. These divergent mitochondrial lineages had been previously identified in a phylogenetic study of character evolution on the origin and the function of the hair-like shell structures in the genus [9]. While some species (i.e. T. villosus, T. montanus or T. caelatus) clearly matched a distinct genetic lineages each, other morphologically described species (i.e. T. plebeius, T. striolatus, T. sericeus and T. hispidus) appeared to consist of several divergent mitochondrial lineages (9 to 11% sequence divergence); the taxonomic status of the latter remained undefined due to the lack of other (e.g. morphological) criteria of discrimination [9].
This came as no surprise as the genus Trochulus has triggered a remarkably long taxonomic debate on species numbers and limits [10]. The large morphological variability within Trochulus and the multitude of transition forms led some authors to describe dozens of species [Locard in [11]] while others, based on the same shell morphological and genital anatomical evidence, recognised only a single one [11, 12]. Species delimitation based on morphological features in land snails is often critical: shells have been shown in several cases to be strongly influenced by environmental conditions during growth [13, 14], whereas genital anatomic criteria, whose dependence on the environment has not yet been assessed, require minute dissections and often rely, such as for Trochulus spp., on subtle differences in size ratios [15].
Yet, a large mitochondrial sequence divergence per se does not necessarily warrant species status in land snails. Unusually high levels of genetic diversity within populations have been shown for some gastropod taxa [16–18] while other studies have demonstrated the presence of "good" cryptic species [[19], e.g. [20, 21]]. We used both mitochondrial and nuclear markers along with shell morphometrics to answer the following questions: (1) Is the mitochondrial divergence between the lineages reflected in the nuclear gene-pool, i.e. what is the degree of their reproductive isolation? (2) Are there consistent differences in shell shape among the identified lineages or are they truly cryptic? (3) Does the combination of these three datasets provide sufficient evidence to support the hypothesis of two distinct species?
Results
Distribution of mitochondrial lineages
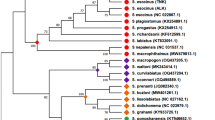
Two hundred and twenty four individuals were sequenced for a 16SrRNA gene fragment. The mitochondrial haplotype tree clustered the sampled individuals into 47 haplotypes (GQ253517–GQ253561) grouped into seven lineages (lineages 1 to 7; Figure 1), all of which were also identified in a previous study [9]. Neighbour-joining and Maximum parsimony methods revealed the same clusters (data not shown). Genetic divergence within lineage ranged between 0.3 and 0.4% and between lineages between 5.5 – 16.7%. Lineages 1 and 2 included most of the observed haplotypes (13 and 23 haplotypes respectively) and accounted for 89% of all individuals (39% and 50% of the total sample respectively) (Table 1). Lineages 1 and 2 diverged by 7.3%. Finally, the mitochondrial tree pointed out a series of lineages (i.e. [3–7]), largely distributed among recognised Trochulus species: these were individuals sampled as juveniles and misidentified (Figure 1); they were ruled out from the subsequent analyses.

Bayesian haplotype tree of Trochulus 16SrRNA gene fragments. Branches with a posterior probability lower than 50% are dashed. Perforatella incarnata and Trochulus lubomirskii were included as outgroups. The 44 haplotypes clustered into seven lineages (L1 to L7). Most of them had previously been identified (lineages A-I as in [9]).
Lineage 1 was predominant in the northern lower parts of the Sarine valley, while lineage 2 was preferentially found upstream in the South. There were, however, several sites in the middle of the valley where both lineages co-occurred (Figure 2).

Spatial distribution of nuclear clusters and haplotype lineages. The spatial distribution of the sampling sites in the Sarine valley (Switzerland) is given together with the cyto-nuclear composition of the individuals sampled. Each horizontal bar represents a single individual, the first column shows the haplotype lineage (white = lineage 1, black = lineage 2) and the second column shows the estimated nuclear composition of the multilocus microsatellite genotype (dark grey = cluster α, light grey = cluster β).
In addition, as evidenced by the mismatch distributions, lineage 1 contained a signal of spatial expansion, while lineage 2 did not (Figure 3).

Mismatch distributions of haplotype lineages. The lineage 1 shows a signal of population expansion, which is not the case of lineage 2. The observed distribution is in full stroke, the simulated distribution in a dashed line. For each lineage, expansion statistics are given.
Spatial nuclear clustering and cyto-nuclear association
The spatial clustering approach with K = 2 of microsatellite multilocus genotype data from 192 individuals revealed a clear pattern: one hundred and two individuals were attributed to one cluster (α), 83 to another (β); all with posterior probabilities larger than 0.9. Seven individuals had genotypes that appeared to be of mixed origin (i.e. with posterior probabilities for each cluster between 0.1 and 0.9). These results were identical for the best 15% of runs. Geographically, cluster α was largely distributed in the north, downstream of the Sarine valley, while cluster β was preferentially found upstream in the south (Figure 2). The contribution of alleles to these clusters were visualised with a factorial correspondence analysis (see additional file 1).
There was a highly significant association of nuclear clusters with mitochondrial haplotype lineages (N = 192, p < 0.001 exact rxc contingency test, Table 2); lineage 1 being associated with nuclear cluster α and lineage 2 with cluster β. This association remained significant when only mixed populations were considered (p < 0.001). However, the association was not categorical: 22 individuals with lineage 2 had cluster α and one individual vice versa. Finally, all seven individuals classified as nuclear intermediates had a lineage 2 mitochondrial genome (Table 2).
The sampling sites with exclusively 1/α and 2/β individuals were situated in the North and South margins of the sampling area, respectively (Figure 2). The sampling sites where both mitochondrial lineages and/or nuclear cluster co-occurred were all in the middle section of the valley (i.e. between sampling sites Enney and Rossens) and all were close to either the rivers or the reservoir lake (Figure 2).
Shell variation
A principle component analysis showed no tendency to group the individuals either according to mitochondrial lineage or nuclear cluster, and this along any of the significant axes (data not shown). Discriminant analyses, however, with grouping according to nuclear cluster or mitochondrial lineage were both significant (Figure 4). Grouping according to nuclear cluster differentiated the snails on variables such as HFW (standardised coefficient = 0.79), HSP (0.71), DOM (0.69) and AES (0.6) which tended to be slightly larger on cluster α individuals. This resulted in 78% correct posterior classifications. Grouping after mitochondrial lineage was less effective with only 69% of the cases correctly classified; the same variables were important, except DOM.

Discriminant analyses of shell morphometric data. A: Schematic representation of the 14 measurements taken on the shell from above, below and the front. Details on the measures are given in the text. Dotted lines represent orientation axes. B: Frequency plot of canonical scores on canonical root of discriminant analyses on morphometric variables grouped according to nuclear cluster membership (>90%). N = 171. Variables retained in the model: HFW (standardised coefficient = 0.79), HSP (0.71), DOM (0.69) and AES (0.67). Grouping resulted in 77.2% correct classifications. C: Grouped according to mitochondrial haplotype. N = 178. Variables retained in the model: HFW (0.86), HSP (0,83) and AES (0.80). Grouping resulted in 68.0% correct classifications.
Discussion
Mitochondrial lineages constitute good species
The use of a short 267 bp 16SrRNA gene fragment for identification purposes has shown the power of this approach [22]. A couple of individuals morphologically similar to the Trochulus complex or juveniles from other species were detected due to their genetic divergence. While it is possible that these lineages interact among each other, these individuals were excluded from further analysis to ensure a rigorous statistical analysis.
The highly significant association between mitochondrial lineage (1 or 2) and nuclear cluster (α or β) in the contact zone of these parapatric lineages argues for two distinct gene-pools (Table 2) and, hence, a specific distinctness of these evolutionary lineages, satisfying the criteria of most species concepts, in particular the unified species concept [23]. Such a picture might have been also produced by very recent contact. But even considering the low dispersal capacity of land snails, the lineages could have come in contact at any time in the Holocene after the retreat of the glacier from the valley, given the small geographic scale of the study. The chances are thus minimal that the contact zone formed only within the last few generations (Figure 3). We expect therefore that free interbreeding of the lineages for many generations would have resulted in completely intermixed genotypes in the contact zone and produced a deeper zone of reciprocal introgression. The rarity of such mixed genotypes and the spatially restricted area of introgression argue for some degree of reproductive isolation. This potential reproductive isolation does not allow equating the sampling sites with freely reproducing populations, which precludes their model dependent population genetic analysis.
If one applies the fastest mitochondrial divergence rate of 5% per million years ever suggested for land snails [16], a sequence divergence of more than 7% between the lineages suggests that they split at least 1.4 million years ago. Slower, more realistic rates would entail an even older divergence. This implies that the lineages were separated for at least most of the Pleistocene, giving them ample time to diverge and speciate.
This distinctness is also reflected in the shell morphology. Both mitochondrial and nuclear data appeared to be suitable statistical predictors of different morphological groups. The nuclear cluster performs slightly better, which we attribute to the few cases of mitochondrial introgression of lineage l2 haplotypes into nuclear background α, blurring the picture (see below). One could attribute the overlap between the two morphogroups to extensive mutual introgression at the relevant genes. This is however unlikely: even the populations furthest apart from each other (and thus least prone to extensive exchange) have overlapping morphologies (data not shown). Moreover, the possibility to statistically tell the two species apart and the approximately normally distributed factor scores (Figure 4B, 4C) suggest that at least some of the quantitative shell shape differences are governed by multiple divergently evolved genes [24]. The observed morphological shell differences are thus rather due to drift than different biological function.
However, a classification success rate of only 78% and 69% respectively, means that it is impossible to reliably differentiate the two species' shells based on extensive morphometric measurements (Figure 4), let alone to distinguish them visually. In practice, this means that the two species will remain cryptic unless molecular markers are used for identification. While the lineage 1/cluster α is yet unnamed, 2/β individuals may be attributed to Trochulus piccardi [25]. Indeed, the lineage 1 individuals, in addition of grouping with the type individual of that species on the mitochondrial phylogeny, have been sampled within the geographical range described for this species [25].
The distribution of T. piccardi that appears to be restricted to the upper Sarine valley and surrounding regions suggests in situ evolution [25]. Such a hypothesis requires the continuous presence of suitable habitat throughout the Pleistocene glaciation cycles, probably scattered over steep south-facing slopes. However, such refugia need not to be large for such poor dispersing, small animals: it has been shown that they can survive on very small areas, provided they are surrounded by other population patches [26].
The current distribution of the other Trochulus species (i.e. the lineage 1) lacks preciseness as this study's sampling scheme did not reach its northern limit. It can nevertheless be argued that it probably evolved in its own, not yet located Pleistocene refugium and got into secondary contact with T. piccardi in the course of a Holocene range expansion (Figure 3).
A pattern of speciation in isolation may turn out to be quite common within Trochulus: several species with very restricted ranges in mountainous regions have been described such as T. graminicola (FALKNER 1973), T. montanus (STUDER 1820), T. caelatus (STUDER 1820) or T. biconicus (EDER 1917). Generally speaking, it has been repeatedly put forward that alpine areas could provide pocket refugia for gastropods [27–30]. Even though a Pleistocene survival of land snails in alpine or other Northern refugia has not been traditionally considered [31], accumulating evidence suggests their importance for gastropod biodiversity [32–35].
Mitochondrial introgression in a hybrid zone
A few individuals with apparently admixed nuclear genotypes as well as some with the "wrong" mitochondrial genome could be detected (Figure 2). These intermediates suggest that, despite the relatively large evolutionary distance among the species, occasional hybridisation can take place (Figure 1, [9]).
Interestingly, the introgression of mitochondrial genomes seems to be directional: only a single lineage 1/cluster β individual was found which is little as compared to the 22 lineage l2/cluster α individuals; besides the admixed nuclear genotypes all carried the lineage 2. Even though the sample sizes of the admixed populations were too small to perform formal tests on cytonuclear disequilibrium [36], the apparent directional introgression of lineage 2 mitochondrial haplotypes into cluster α nuclear background begets the question whether this is due to intrinsic reproductive barriers and/or unidirectional gene-flow downstream the Sarine river. Differential mating preference, the usual explanation for the observed pattern, is unlikely in the present case: since these snails are reciprocally mating hermaphrodites (i.e. they both receive the sperm of the other individual to fertilise their eggs), the nuclear and mitochondrial genomes of both individuals are expected to be transmitted. Given the low dispersal capacity of land snails and their more or less isolated populations [37–40], it is anyway likely that we are dealing here with a mixed hybrid zone [36], where isolated hybrid populations may take different evolutionary trajectories. A hint in this direction was the somewhat peripheral sampling site Motelon (Figure 2), where nuclear cluster α is associated to lineage 2, contrary to all other sites: it may be due to one or a few introgression events, followed by drift in isolation.
Conclusion
The application of several lines of evidence allowed rejecting the hypothesis of a single species consisting of divergent lineages, despite occasional hybridisation in a contact zone. The detection of a putative hybrid zone in Trochulus adds to the surprisingly low number of cases of hybridisation in land snails [41–44], despite the ubiquitous presence of hybridisation in animal taxa [45]. This system – two cryptic hermaphrodite species with a small hybrid zone – seems excellently suited for further investigations on the evolutionary forces shaping species diversity in mountainous habitat rich regions, with a focus on the evolutionary consequences of hybridisation.
Methods
Sampling
Fourteen sampling sites in a previously identified area of co-occurrence of mitochondrial lineage F and Trochulus nov. spec. as defined in Pfenninger et al. 2005 (subsequently described as T. piccardi PFENNINGER & PFENNINGER[25] were sampled in the Swiss Western Prealps, along the Sarine valley (Figure 2). As the attribution of Trochulus individuals to a certain species based on shell morphology alone is difficult, all apparently adult Trochulus sericeus/hispidus individuals found were sampled. The collected snails were brought alive to the lab and frozen at -80°C. The individuals were then pulled out of their shell and stored at -80°C for genetic analyses; the shell was kept for morphometric measurements.
Sequencing and mitochondrial analyses
Total DNA was extracted from a piece of foot muscle following a modified salt/chloroform procedure by adding one step of chloroform-isoamylalcohol (24:1) [46]. The mitochondrial haplotypes were determined by sequencing a 267 bp fragment of the 16SrRNA gene using universal primers [47]. Reactions were carried out in a 50 μl volume containing 2 μl of template, 1.5 units of Taq DNA Polymerase (Qiagen), 1× of its buffer, 1000 μM of MgCl2 and 100 μM of dNTP and 0.4 μM of each primer. The PCR was run on a DNA Thermal Cycler (Perkin Elmer, Norwalk, CT) starting with an initial denaturation at 94°C for 3 minutes, followed by 10 cycles at 44°C annealing temperature (50 sec at 92°C, 30 sec at 44°C and 40 sec at 72°C) and 35 cycles at 48°C annealing temperature (30 sec at 92°C, 30 sec at 48°C and 40 sec at 72°C), and ended by a 3 minute final extension at 72°C. After amplification, all samples were controlled on a 1% agarose minigel stained with ethidium bromide and run in a 1× TBE buffer. The PCR products were then purified with a QIAQuick PCR Purification Kit (Qiagen) according to the manufacturer's instructions. The purified DNA was eluted in 30 μl of dH2O. Samples were sent to a sequencing company (Microsynth AG, Balgach, Switzerland). Sequences were edited, aligned on SEQUENCHER 3.0 (Gene Codes Corporation) and the alignment manually corrected using SEAVIEW[48].
We constructed a haplotype phylogeny to visualise haplotype clusters using MRBAYES v. 3.1 [49]. A GeneBank reference sequence for each of the nine previously identified lineages was added [Lineages A to I as in [9]], as well as sequences of Trochulus biconicus, T. caelatus, T. clandestinus, T. alpicolus, T. piccardi and T. lubomirskii. Perforatella incarnata was included as the outgroup species. We ran MODELTEST V.3.7[50] on this extended dataset and selected a GTR+R+I model, assuming a gamma-shaped rate variation and invariant sites. We ran two runs of four Metropolis coupled Monte Carlo Markov chains (MC3) for 10 million generations, sampling every 500th generation and discarding the first 100'000 trees as burn-in. Convergence was monitored by checking that the average standard deviation of the split frequencies (i.e. the differences in likelihood between the 2 runs) were below 0.1% (MRBAYES).
Episodes of population growth and decline may also leave characteristic signatures in the distribution of pairwise nucleotide differences of populations (i.e. mismatch distribution). The mismatch distributions under a sudden expansion model were computed for the two mitochondrial lineages (L1 and L2) with ARLEQUIN v.3.1 [51]. This model assumes that an initial population at equilibrium with θ = θ0 grows rapidly to a new size with θ = θ1, τ units of mutational time ago, where θ = N e u and τ = 2 ut (N e = effective population size, u = mutation rate and t = time since the expansion in generations). Goodness-of-fit tests [52] of the observed to the expected distribution were computed. The confidence intervals for τ were obtained from 1000 bootstrap replicates.
Genotyping and spatial genotype clustering
The individuals belonging to the two mitochondrial lineages in focus were scored at 6 microsatellite loci – TROA6, TROA111, TROB5, TROB108, TROB111 and TROB112 – following the published PCR conditions [53]. Amplifications were run on an ABI 3100 capillary sequencer (Applied Biosystems) and scored with GENEMAPPER v.3 (Applied Biosystems).
We applied a Bayesian spatial population genetics clustering algorithm implemented in TESS 1.2 to obtain individual posterior membership probabilities of nuclear gene-pools [54, 55]. The method is based on a hierarchical mixture model where the prior distribution of cluster labels is defined as a Hidden Markov Random Field (HMRF) on a spatial individual tessellation network. The program seeks genetic structure from individual multilocus genotypes sampled at different geographical locations without assuming predefined clusters. It returns the membership probabilities and cluster assignments of the individuals. In the initial field sampling, we did not record individual sampling points, as it is required by TESS. Therefore, we assigned a randomised sampling point to each individual, drawn from a square of 100 × 100 m around the recorded sampling location as given in Table 1. This proceeding actually models very well the sampling of land snails.
As the sampling sites are located along a river valley without a priori obstacle to dispersal, we left the neighbourhood diagram unmodified. We used the admixture model in a Monte-Carlo-Markov-Chain approach for the microsatellite data set, the interaction parameter set to 0.6 and the allele frequency model parameter to 1.0. We were not interested in intraspecific population structure but in gene pool membership; the maximum number of clusters was therefore set to two. We performed 200 runs with 50,000 sweeps per run with a burn-in of 12,000 sweeps, which was sufficient to reach stationarity. We used the program CLUMPP[56] to obtain averages of the 15% runs with the highest likelihoods. We performed a G-test to test for independence of mitochondrial lineage from nuclear cluster.
Shell morphometrics and discriminant analysis
A hundred and seventy eight adult individuals were selected for shell morphometric analyses according to three criteria: the individual (i) belonged to one of the two major mtDNA lineages and microsatellite data was available, (ii) had a shell with at least five complete whorls and (iii) had a shell undamaged as much as to allow all measures. Fourteen measurements were taken on calibrated digital pictures of the shells (Figure 4). From above, we measured the decimal number of whorls (NTW computed as the number of complete whorls plus the partial whorl (in degrees; divided by 360), the width of the apex (APX), the width of the first (SP1), second (SP2) and third whorl (SP3) and the diameter of the spire without the last whorl (DWL). From underneath, the diameter of the umbilicus (DOM) and the width of the shell without the opening (WWO) were taken. Finally, from a side perspective, we measured the height of the spire (HSP), the height of the first whorl (HFW), the diameter of the peristoma (DPR), the height of the peristoma (HPR), the diameter of the spire (DSP) and the shape, measured as the angle formed by the apex and the most external sutures (AES) (Figure 4). As first exploratory approach, a principle component analysis was performed. Two discriminant analyses were run on the morphometric variables: a first with nuclear cluster as grouping factor, a second with the mtDNA lineage. All analyses of the morphometric data were performed with STATISTICA 7 [57].
References
Hebert PDN, Cywinska A, Ball SL, DeWaard JR: Biological identifications through DNA barcodes. Proceedings of the Royal Society of London Series B-Biological Sciences. 2003, 270: 313-321. 10.1098/rspb.2002.2218.
Pons J, Barraclough TG, Gomez-Zurita J, Cardoso A, Duran DP, Hazell S, Kamoun S, Sumlin WD, Vogler AP: Sequence based species delimitation for the DNA taxonomy of undescribed insects. Systematic Biology. 2006, 55: 595-609. 10.1080/10635150600852011.
Templeton AR: Using phylogeographic analyses of gene trees to test species status and processes. Molecular Ecology. 2001, 10 (3): 779-791. 10.1046/j.1365-294x.2001.01199.x.
Tautz D, Arctander P, Minelli A, Thomas RH, Vogler AP: A plea for DNA taxonomy. Trends in Ecology & Evolution. 2003, 18: 70-74. 10.1016/S0169-5347(02)00041-1.
Knowles LL, Carstens BC: Delimiting species without monophyletic gene trees. Systematic Biology. 2007, 56 (6): 887-895. 10.1080/10635150701701091.
Pfenninger M, Schwenk K: Cryptic animal species are homogeneously distributed among taxa and biogeographical regions. BMC Evolutionary Biology. 2007, 7: 121-10.1186/1471-2148-7-121.
Sites J, Jack W, Marshall JC: Delimiting species: a Renaissance issue in systematic biology. Trends in Ecology & Evolution. 2003, 18 (9): 462-470. 10.1016/S0169-5347(03)00184-8.
Wiens JJ: Species delimitation: new approaches for discovering diversity. Systematic Biology. 2007, 56 (6): 875-878. 10.1080/10635150701748506.
Pfenninger M, Hrabáková M, Steinke D, Dépraz A: Why do snails have hairs? A Bayesian inference of character evolution. BMC Evolutionary Biology. 2005, 5: 59-10.1186/1471-2148-5-59.
Falkner G: Zur Problematik der Gattung Trichia (Pulmonata, Helicidae) in Mitteleuropa. Mitteilungen der deutsche malakologische Gesellschaft. 1982, 3 (Supplement): 30-33.
Favre J: Les Mollusques post-glaciaires et actuels du bassin de Genève. Mémoires de la Societé de Physique et de l'Histoire Naturelle de Genève. 1927, 40 (3): 171-434.
Mermod G: Gastéropodes invertébrés de la Suisse, vol. Fasc. 18. Genève. 1930
Goodfriend GA: Variation in land snail shell form and size and its causes: a review. Systematic Zoology. 1986, 35 (5): 204-223. 10.2307/2413431.
Emberton KC: Partitioning a morphology among its controlling factors. Biological Journal Of the Linnean Society. 1994, 53 (4): 353-369. 10.1111/j.1095-8312.1994.tb01018.x.
Shileyko AA: On the systematics of Trichia s. lat. (Pulmonata: Helicoidea: Hygromiidae). Malacologia. 1978, 17 (1): 1-56.
Thomaz D, Guiller A, Clarke B: Extreme divergence of mitochondrial DNA within species of pulmonate land snails. Proceedings of the Royal Society of London Series B-Biological Sciences. 1996, 263 (1368): 363-368. 10.1098/rspb.1996.0056.
Uit de Weerd DR, Piel WH, Gittenberger E: Widespread polyphyly among Alopiinae snail genera: when phylogeny mirrors biogeography more closely than morphology. Molecular Phylogenetics and Evolution. 2004, 33 (3): 533-548. 10.1016/j.ympev.2004.07.010.
Pinceel J, Jordaens K, Backeljau T: Extreme mtDNA divergences in a terrestrial slug (Gastropoda, Pulmonata, Arionidae): accelerated evolution, allopatric divergence and secondary contact. Journal of Evolutionary Biology. 2005, 18 (5): 1264-1280. 10.1111/j.1420-9101.2005.00932.x.
Pinceel J, Jordaens K, Van Houtte N, De Winter AJ, Backeljau T: Molecular and morphological data reveal cryptic taxonomic diversity in the terrestrial slug complex Arion subfuscus/fuscus (Mollusca, Pulmonata, Arionidae) in continental north-west Europe. Biological Journal of the Linnean Society. 2004, 83 (1): 23-38. 10.1111/j.1095-8312.2004.00368.x.
Wilke T, Pfenninger M: Separating historic events from recurrent processes in cryptic species: phylogeography of mud snails (Hydrobia spp.). Molecular Ecology. 2002, 11 (8): 1439-1451. 10.1046/j.1365-294X.2002.01541.x.
Pfenninger M, Staubach S, Albrecht C, Streit B, Schwenk K: Ecological and morphological differentiation among cryptic evolutionary lineages in freshwater limpets of the nominal form-group Ancylus fluviatilis (O.F. Müller, 1774). Molecular Ecology. 2003, 12 (10): 2731-2745. 10.1046/j.1365-294X.2003.01943.x.
Meusnier I, Singer GAC, Landry J-F, Hickey D, Hebert PD, Hajibabaei M: A universal DNA mini-barcode for biodiversity analysis. BMC Genomics. 2008, 9 (214):
De Queiroz K: Species concepts and species delimitation. Systematic Biology. 2007, 56 (6): 879-886. 10.1080/10635150701701083.
Boulding EG, Hay TK: Quantitative genetics of shell form of an intertidal snail: constraints on short-term reponse to selection. Evolution. 1993, 47 (2): 576-592. 10.2307/2410072.
Pfenninger M, Pfenninger A: A new Trochulus species from Switzerland (Gastropoda: Pulmonata: Hygromiidae). Archiv für Molluskenkunde. 2005, 134 (2): 261-269. 10.1127/arch.moll/0003-9284/134/261-269.
Pfenninger M, Bahl A: Influence of habitat size on the viability of spatially structured populations of the land snail Trochoidea geyeri. Verhandlungen der Gesellschaft für Ökologie. 1997, 27: 469-473.
Gittenberger E, Piel WH, Groenenberg DSJ: The Pleistocene glaciations and the evolutionary history of the polytypic snail species Arianta arbustorum (Gastropoda, Pulmonata, Helicidae). Molecular Phylogenetics and Evolution. 2004, 30 (1): 64-73. 10.1016/S1055-7903(03)00182-9.
Stehlik I, Schneller J, Bachmann K: Immigration and in situ glacial survival of the low-alpine Erinus alpinus (Scrophulariaceae). Biological Journal of the Linnean Society. 2002, 77: 87-103. 10.1046/j.1095-8312.2002.00094.x.
Schönswetter P, Stehlik I, Holderegger R, Tribsch A: Molecular evidence for glacial refugia of mountain plants in the European Alps. Molecular Ecology. 2005, 14 (11): 3547-3555. 10.1111/j.1365-294X.2005.02683.x.
Dépraz A, Cordellier M, Hausser J, Pfenninger M: Postglacial recolonisation at a snail's pace (Trochulus villosus): confronting competing refugia hypotheses using model selection. Molecular Ecology. 2008, 17: 2449-2462. 10.1111/j.1365-294X.2008.03760.x.
Ant H: Die Bedeutung der Eiszeiten für die rezente Verbreitung der europäischen Landgastropoden. Malacologia. 1966, 5: 61-62.
Haase M, Misof B, Wirth T, Baminger H, Baur B: Mitochondrial differentiation in a polymorphic land snail: evidence for Pleistocene survival within the boundaries of permafrost. Journal of Evolutionary Biology. 2003, 16 (3): 415-428. 10.1046/j.1420-9101.2003.00542.x.
Pfenninger M, Posada D, Magnin F: Evidence for survival of Pleistocene climatic changes in Northern refugia by the land snail Trochoidea geyeri (Soos 1926) (Helicellinae, Stylommatophora). BMC Evolutionary Biology. 2003, 3: 8-10.1186/1471-2148-3-8.
Pinceel J, Jordaens K, Pfenninger M, Backeljau T: Rangewide phylogeography of a terrestrial slug in Europe: evidence for Alpine refugia and rapid colonization after the Pleistocene glaciations. Molecular Ecology. 2005, 14: 1133-1150. 10.1111/j.1365-294X.2005.02479.x.
Wilke T, Duncan N: Phylogeographical patterns in the American Pacific Northwest: lessons from the arionid slug Prophysaon coeruleum. Molecular Ecology. 2004, 13: 2303-2315. 10.1111/j.1365-294X.2004.02234.x.
Arnold J: Cytonuclear disequilibria in hybrid zones. Annual Review in Ecology and Systematics. 1993, 24: 521-554. 10.1146/annurev.es.24.110193.002513.
Arnaud JF, Madec L, Guiller A, Bellido A: Spatial analysis of allozyme and microsatellite DNA polymorphisms in the land snail Helix aspersa (Gastropoda: Helicidae). Molecular Ecology. 2001, 10 (6): 1563-1576. 10.1046/j.1365-294X.2001.01292.x.
Schilthuizen M, Lombaerts M: Population structure and levels of gene flow in the Mediterranean land snail Albinaria corrugata (Pulmonata, Clausiliidae). Evolution. 1994, 48 (3): 577-586. 10.2307/2410470.
Pfenninger M: Relationship between microspatial population genetic structure and habitat heterogeneity in Pomatias elegans (OF Muller 1774) (Caenogastropoda, Pomatiasidae). Biological Journal of the Linnean Society. 2002, 76 (4): 565-575. 10.1046/j.1095-8312.2002.00080.x.
Pfenninger M, Bahl A, Streit B: Isolation by distance in a population of a small land snail Trochoidea geyeri: Evidence from direct and indirect methods. Proceedings of the Royal Society of London Series B-Biolological Sciences. 1996, 263 (1374): 1211-1217. 10.1098/rspb.1996.0178.
Chiba S: Appearance of morphological novelty in a hybrid zone between two species of land snail. Evolution. 2005, 59 (8): 1712-1720.
Teshima H, Davison A, Kuwahara Y, Yokoyama J, Chiba S, Fukuda T, Ogimura H, Kawata M: The evolution of extreme shell shape variation in the land snail Ainohelix editha: a phylogeny and hybrid zone analysis. Molecular Ecology. 2003, 12 (7): 1869-1878. 10.1046/j.1365-294X.2003.01862.x.
Giokas S, Mylonas M, Sotiropoulos K: Gene flow and differential mortality in a contact zone between two Albinaria species (Gastropoda; Clausiliidae). Biological Journal of the Linnean Society. 2000, 71 (4): 755-770. 10.1111/j.1095-8312.2000.tb01289.x.
Chiba S: Novel colour polymorphisms in a hybrid zone of Mandarina (Gastropoda: Pulmonata). Biological Journal Of the Linnean Society. 1997, 61 (3): 369-384.
Schwenk K, Brede N, Streit B: Introduction. Extent, processes and evolutionary impact of interspecific hybridization in animals. Philosophical Transactions of the Royal Society Series B-Biological Sciencesi. 2008, 363 (1505): 2805-2811. 10.1098/rstb.2008.0055.
Miller SA, Dykes DD, Polesky HF: A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research. 1988, 16 (3): 1215-1215. 10.1093/nar/16.3.1215.
Palumbi SR, Metz EC: Strong reproductive isolation between closely related tropical sea-urchins (genus Echinometra). Molecular Biology and Evolution. 1991, 8 (2): 227-239.
Galtier N, Gouy M, Gautier C: SEAVIEW and PHYLO_WIN: Two graphic tools for sequence alignment and molecular phylogeny. Computer Applications in the Biosciences. 1996, 12 (6): 543-548.
Ronquist F, Huelsenbeck JP: MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19 (12): 1572-1574. 10.1093/bioinformatics/btg180.
Posada D, Crandall KA: MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998, 14 (9): 817-818. 10.1093/bioinformatics/14.9.817.
Excoffier L, Laval G, Schneider S: Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online. 2005, 1: 47-50.
Schneider S, Excoffier L: Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates very among sites: Application to human mitochondrial DNA. Genetics. 1999, 152 (3): 1079-1089.
Dépraz A, Ratthey E, Hausser J: Characterisation of thirteen polymorphic microsatellite loci for two land snail species, Trochulus villosus and T. sericeus (Gastropoda: Pulmonata: Hygromiidae). Molecular Ecology Resources. 2008, 8: 704-706. 10.1111/j.1471-8286.2007.02055.x.
François O, Ancelet S, Guillot G: Bayesian clustering using hidden Markov random fields in spatial population genetics. Genetics. 2006, 174: 805-816. 10.1534/genetics.106.059923.
Chen C, Durand E, Forbes F, François O: Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Molecular Ecology Notes. 2007, 7: 747-756. 10.1111/j.1471-8286.2007.01769.x.
Jakobsson M, Rosenberg N: CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007, 23: 1801-1806. 10.1093/bioinformatics/btm233.
StatSoft I: STATISTICA for Windows. 1995, Tulsa: StatSoft, Inc, 5.5
Acknowledgements
We wish to thank Nicolas Juillet and Emilie Rathey for their help in the field and/or in the lab. We are also grateful to Lucie Büchi and Glenn Yannic for their invaluable advice and graphical expertise and to Patrick Basset and four anonymous reviewers for helpful comments on a previous draft. MP received support by the research funding programme "LOEWE – Landes-Offensive zur Entwicklung Wissenschaftlich-ökonomischer Exzellenz" of Hesse's Ministry of Higher Education, Research, and the Arts.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
AD designed the study, sampled the snails, performed the lab work and drafted the manuscript. JH took part in the design, performed the morphometric measurements and drafted the manuscript. MP analysed the data and drafted the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Dépraz, A., Hausser, J. & Pfenninger, M. A species delimitation approach in the Trochulus sericeus/hispiduscomplex reveals two cryptic species within a sharp contact zone. BMC Evol Biol 9, 171 (2009). https://doi.org/10.1186/1471-2148-9-171
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-9-171