
Abstract
Background
This study addresses the apportionment of genetic diversity between Cycas revoluta and C. taitungensis, species that constitute the section Asiorientales and represent a unique, basal lineage of the Laurasian genus Cycas. Fossil evidence indicates divergence of the section from the rest of Cycas at least 30 million years ago. Geographically, C. taitungensis is limited to Taiwan whereas C. revoluta is found in the Ryukyu Archipelago and on mainland China.
Results
The phylogenies of ribosomal ITS region of mtDNA and the intergenic spacer between atpB and rbcL genes of cpDNA were reconstructed. Phylogenetic analyses revealed paraphyly of both loci in the two species and also in the section Asiorientales. The lack of reciprocal monophyly between these long isolated sections is likely due to persistent shared ancestral polymorphisms. Molecular dating estimated that mt- and cp DNA lineages coalesced to the most recent common ancestors (TMRCA) about 327 (mt) and 204 MYA (cp), corresponding with the divergence of cycad sections in the Mesozoic.
Conclusion
Fates of newly derived mutations of cycads follow Klopfstein et al.'s surfing model where the majority of new mutations do not spread geographically and remain at low frequencies or are eventually lost by genetic drift. Only successful 'surfing mutations' reach very high frequencies and occupy a large portion of a species range. These mutations exist as dominant cytotypes across populations and species. Geographical subdivision is lacking in both species, even though recurrent gene flow by both pollen and seed is severely limited. In total, the contrasting levels between historical and ongoing gene flow, large population sizes, a long lifespan, and slow mutation rates in both organelle DNAs have all likely contributed to the unusually long duration of paraphyly in cycads.
Similar content being viewed by others

Background
The analysis of geographical structure and phylogeographical pattern has been a major focus in population genetics and molecular ecology [1]. Populations descending from a common ancestral population are expected to differentiate from each other and eventually show reciprocal monophyly. Reciprocal monophyly is a result of the accumulated effects of genetic drift and selection when little gene exchange occurs after divergence. In contrast, populations may be genetically similar due to recent isolation, continuous gene exchange, or secondary contact [2]. Plant species on islands provide ideal subjects for teasing apart the contrasting effects of geographical isolation and gene flow on population differentiation. Island populations are often genetically isolated from each other and most island populations are small, thus facilitating random genetic drift.
Coalescence studies provide information on the genealogical processes (backwards in time) that have led to the current apportionment of genetic variation within populations or species. Coalescence-based phylogeography uses haplotype networks, which trace the mutational relationships among alleles within or among species in a geographical context [3], to infer processes such as migration, divergence, and isolation [4–6]. Noncoding spacer regions of organelle DNAs are often used as genetic markers because they are nearly neutral with low functional constraints and are free of the complexities associated with recombination for nuclear markers [6].
Cycads are a unique and ancient lineage of land plants that have attracted much attention from evolutionary biologists [7]. Cycas revoluta Thunb. and C. taitungensis Shen et al. constitute the section Asiorientales which is a basal lineage of this Laurasian genus [8]. Cycas revoluta occurs in a scattered distribution pattern from the southernmost part of Kyushu and throughout the Ryukyu Islands in Japan, and is also distributed in Fukien Province of China [9]; while C. taitungensis is the only extant and endemic species with two endangered populations along the eastern coast of Taiwan. The two species resemble each other morphologically; a few characters such as revolute vs. plane leaflet margins distinguish the species from each other. An Eocene fossil, C. fujiana Yokoyama, is much like C. revoluta; both the morphological similarity and the occurrence of both taxa in Japan suggest that these species are the same [10]. The divergence of the species in section Asiorientales is thought to have occurred some 30 million years before present [10]. Asiorientales is allopatric from other cycads.
Both species are agriculturally important. Each year thousands of seeds and plants have been exported for use in the nursery trade and in floral arrangements [11]. In the early 20th Century, cycad seeds and stems were used for making cake and soup. Cycad flour is toxic and causes neurological disorder [12]. Fermented seeds are used for producing alcoholic drinks [13]. Unfortunately, human overexploitation has led to the extinction of populations of C. revoluta in Fukien Province of China [9], and only two remaining populations of C. taitungensis occur in Taiwan [14, 15]. Large specimens of C. revoluta are prized by the nursery industry and natural populations of C. revoluta in mainland China have been decimated by poachers [9]. In contrast, populations of C. revoluta are numerous across the Ryukyu Archipelago and Kyushu [13, 16]. On the Island Amami-O-Shima, protected cycad forests contain thousands of plants [17]. Despite the high morphological similarity, the two species differ in habitat preference. Cycas revoluta usually grows along coasts, often subject to salt spray. In contrast, C. taitungensis grows on cliffs along riverbanks [18].
Island systems have long served as natural laboratories for evolutionary studies [e.g. [19, 20]]. Despite their often small geographic size, islands can contain a diversity of habitats suitable for the colonization of an array of plants and animals. Once an island is colonized, population differentiation between islands can be facilitated by isolation, vicariance events and local extinction-recolonization [21]. Taiwan and the Ryukyu islands lie at the boundary between the Eurasian and Pacific plates and are a constituent of the island-arc system along the western edge of the Pacific Ocean [22]. In contrast to sequentially emerging oceanic islands of volcanic origin, these continental islands emerged almost simultaneously via collision between the continental and oceanic tectonic plates and are geologically young, estimated at 5–6 million years old [23, 24]. Since emerging from the Pacific Ocean, these islands gradually acquired their floras and faunas from the Eurasian mainland [25, 26]. Most island species/populations thus have close phylogenetic links to their mainland relatives.
Today, islands of the Ryukyu archipelago are isolated from each other. In the past, sea level oscillated during periodic glacial cycles (Milankovitch cycles) [27–29]. During glacial maxima, the global sea level dropped by some 100–120 meters, exposing land bridges that connected islands [30] and thus providing migration corridors between today's isolated islands. Repeated stepwise migrations, plus occasional, long-distance colonization [cf. [31–33]], would allow various plants to fill newly available habitats on islands left by the expanding or retreating glaciers. The glacial cycles of the Quaternary played an important role in the evolution of species [cf. [34, 35]].
In the present study, phylogenies of the rITS region of mtDNA and the intergenic spacer between atpB and rbcL genes of cpDNA were used to trace phylogeographical relationships in C. revoluta and C. taitungensis. Organelle DNAs are maternally inherited in cycads; they disperse only via seeds [cf. [15]]. Most cycad seeds, including those of C. revoluta and C. taitungensis, are heavy and sink reducing the possibility of water/current dispersal over long distances [36, 37]. The fleshy outer seed coat of cycads attracts animals, mainly rodents, small fruit-eating bats, and birds which serve as dispersal agents [38]. In natural habitats, seeds drop and occasionally disperse for a limited distance via gravity. Migration between islands is thought to be very limited and such restricted dispersal has been documented in other island species [20, 39]. Ocean straits between large geographical regions (Fukien, Taiwan, and Ryukyu), and channels between islands make migration between islands unlikely in these cycads. Thus, genetic differentiation between populations for maternally inherited markers would be expected. Nevertheless, multiple colonization and recent isolation can counter forces leading to genetic differentiation between islands and monophyly [20]. Many plants distributed on the island arcs of the western Pacific Rim, including Cunninghamia [40], Michelia [41], Kandelia [42], Trema [43] and Lithocarpus [32, 44], show little differentiation. Coalescence in island populations can be a complicated process, determined by counteracting effects of isolation (i.e., historical connection), gene flow, drift, selection and repeated colonization.
Given the similar systems of mating and demography of both cycad species, non-coding regions of organelle DNA's should provide information on species/population histories and demographic processes as well as gene flow and colonization. In this study, we examine the genetic diversity within and between species and populations, determine geographical associations and analyze phylogenetic relationships to infer the historical processes that have lead to the current pattern of genetic structure. Several specific questions are addressed:
1. Given the long time that these species have been isolated, has reciprocal monophyly of sections and species been attained?
2. Are populations and geographical regions genetically differentiated?
3. Is the spatial apportionment of genetic variation consistent with a stepwise model of colonization?
4. Is erosion of genetic diversity associated with population decline? Are levels of genetic diversity in populations of Fukien and Taiwan lower than those of Ryukyu?
Results
Genetic diversity within Cycas revoluta and C. taitungensis
The rITS region of mtDNA varies in length from 411 to 547 bp with a consensus length of 598 bp. The sequence length of atpB-rbcL intergenic spacer of cpDNA varies from 762 to 830 bp with a 913 bp consensus length. In total, 105 and 97 haplotypes of cpDNA as well as 170 and 55 haplotypes of mtDNA were detected in populations of Cycas revoluta (307 individuals) and C. taitungensis (102 individuals), respectively (Table 1). Haplotype sequences of mt- and cpDNAs were deposited in the GenBank database with accession numbers of FN397910-FN397954 and FM999745-FM999775, respectively.
Higher levels of genetic diversity of mtDNA are detected in C. revoluta, φ = 0.0500, than in C. taitungensis, φ = 0.0038 (Table 2). Among the Ryukyu Islands, populations from Yonaguni and Ishigaki have the highest levels of mtDNA diversity while the diversity in populations on Okinawa and Amami-O-Shima is relatively low. Despite its extinct status in the wild, the Fukien sample contains much higher levels of mtDNA diversity than any of the Ryukyu populations. The existence of the phylogenetically most distant mitotype, G, that occurs only in the Fukien population contributes to the high level of genetic diversity (Fig. 1). In C. taitungensis the level of genetic diversity is mostly associated with population size. Genetic diversity of the Red-Leaf (RL) population is about twice that of the Coastal (C) population. In contrast, the levels of genetic diversity of cpDNA are much lower in C. revoluta, φ = 0.008, than in C. taitungensis, φ = 0.018 (Table 3). Fukien has the lowest level of diversity among all populations, while the populations of Ishigaki (φ = 0.055) and Iriomote (φ = 0.022) have higher levels of genetic diversity.

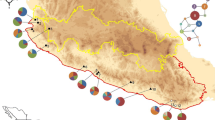
Map showing the spatial distribution of genetic polymorphisms of mtDNA in populations of Cycas revoluta (circles), and C. taitungensis (squares).
Phylogenies of organelle DNAs in C. revoluta and C. taitungensis
Knowledge of the process and pattern of nucleotide substitution is important for estimating the number of substitutional events between DNA sequences and phylogenetic reconstruction that incorporates models of DNA sequence evolution. Transition bias results in a substitution pattern that is characterized by transition/transversion (ti/tv) ratios typically ranging between 2 and 10 [45–47]. In contrast, higher rates of tranversional substitutions resulting in ti/tv ratios ~1 have often been equated with transitional saturation, in which the same nucleotide position undergoes multiple transitions, or considered indicative of high levels of homoplasy [48]. In this study, the transition/transversion ratios were detected to be 1.808 and 1.707 for mtDNA ITS and cpDNA, respectively, suggesting low likelihood of saturation, although relatively low. The phylogeny of the mtDNA ITS region was reconstructed by maximum-likelihood (ML) and maximum-parsimony (MP) analyses. Both analyses recover phylogenetic trees with consistent topologies. The gene tree identifies seven mitotypes, including haplotypes A to G (Fig. 2A). Reciprocal monophyly is not supported in either Cycas species. Geographically, mitotypes A and B are widespread (Fig. 1), while four mitotypes are restricted to single populations, including C (YO), D (AM), E (IS), and G (FU). Mitotypes C-G are absent in C. taitungensis while mitotype G does not occur in Ryukyus. Sequences from the two outgroups are mitotype A and do not form a monophyletic group, indicating that sections of Cycas are also paraphyletic for mtDNA (Fig. 2A and 3A). An ML tree based on nucleotide variation of the cpDNA atpB-rbcL intergenic spacer identifies six chlorotypes I-VI (Fig. 2B). Chlorotypes I, II, and IV are geographically widespread across islands and Fukien, while chlorotype II is restricted to C. taitungensis, and chlorotypes V and VI are found exclusively in Iriomote and Ishigaki (Fig. 4). The hypothesis of a molecular clock was not rejected at either locus based on the relative rate test (data not shown).

The maximum likelihood tree of haplotypes of Cycas revoluta and C. taitungensis with outgroup sequences (O). Numbers at nodes indicate the bootstrap values (ML/MP) in percentage. Symbols in parentheses indicate the geographical distribution of each mitotype. W = widespread. A) mtDNA. Clades A and B occur in both species, while clades D-G are restricted to C. revoluta. B) cpDNA. Clades I, III and IV occur in both species, while other are restricted to C. revoluta.

Network of haplotypes of Cycas revoluta and C. taitungensis. Numbers at arrows indicate the mutational changes between mitotypes. A) mtDNA, B) cpDNA, C) combined data.

Map showing the spatial distribution of genetic polymorphisms of cpDNA in populations of Cycas revoluta (circles), and C. taitungensis (suqares).
Cycas species are not monophyletic for cpDNA. Haplotypes of outgroups belong to chlorotype I and are also found in C. revoluta and C. taitungensis (Fig. 2B and 3B), again reflecting paraphyly of the section Asiorientales. In both phylogenies numerous mutations are located at tips of the network, and are derived from ancestral haplotypes at the interior nodes in the network. This pattern results in a star-like phylogeny. Furthermore, tip haplotypes are mostly confined to a single population while the interior haplotypes are geographically widespread.
Genetic differentiation and population demography
Almost all dominant cytotypes of both organelle genes are shared among cycad populations. For mtDNA, mitotype A (95.3%) is common and widely distributed across populations, while rare mitotypes C, D, E, and G occur in single populations (Fig. 1). Likewise, for the cpDNA intergenic spacer, chlorotype I (90.1%) is the most common and widespread in all populations, while rare chlorotypes II, V, and VI are restricted to single populations (Fig. 4). Despite the lack of physical linkage between the two organelle genomes, high levels of linkage disequilibrium are detected in both Cycas species (P < 0.05). All rare mitotypes (4.7%), including B-G, are associated with chlorotype I; while all rare chlorotypes II–VI (9.9%) are associated with the mitotypes A, in the pooled data reflecting a nonequilibrium state.
Since common haplotypes are shared among populations, cycads show little genetic differentiation at the two organelle DNA markers. Nm values obtained from Fst estimates in C. revoluta vary among pairwise comparisons between populations (Tables 4 and 5). High levels of differentiation between Japan and China populations are detected for all comparisons, e.g., Fst of 0.115 (P < 0.01) between OK and FU based on mtDNA, and Fst = 0.159 between IR and FU (P < 0.01) based on cpDNA (Tables 4 and 5). For example the estimated number of migrants per generation is high between populations C and R of C. taitungensis, Nm = 90.41 and 23.18 based on cp- and mtDNAs, respectively (60). The Nm values based on Fst estimates from mtDNA average 106.10 which is greater than the average from cpDNA data, 10.95 (Tables 4 and 5).
The program IM defined six demographic parameters estimated by the MCMC (Markov chain Monte Carlo) multiple loci simulations and tested for an 'isolation with migration' model in cycads. Of these demographic parameters, the distribution of migration parameters reveals a peak at the lower limit of resolution in one direction from Japanese populations to the Chinese population in C. revoluta (Table 6). When the MLE (maximum likelihood estimates) for the migration parameters are transformed into the population migration rates, M1 = 2N1m1 = m1*(θ1/2) estimated from Japanese populations to the Chinese population in C. revoluta is higher than the estimate, M2 = 2N2m2 = m2*(θ2/2), from China to Japan, i.e., 8.0134 vs. 0.5431 based on the combined data (Table 6). The Neτ (the product of effective population size and the generation time in years) value is also assessed; a higher effective population size is detected for Japan's populations than China population, i.e., Neτ = 4.753*106 vs. 1.437*105 based on the combined data (Table 6).
In the study, the demographic scenarios for the two species and populations of Japan and China of C. revoluta are represented by the Bayesian skyline plot (Fig. 5 and 6). Based on the pattern of variation in mtDNA, a long history of constant population size of C. revoluta, followed by a slight decline (bottleneck) with subsequent demographic expansion, is recovered (Fig. 5A). At the population level, in C. revolute, the Japanese and Fukien populations also display similar patterns (Fig. 5C and 5D). In contrast, slow population growth is detected in C. taitungensis based on the Bayesian skyline plot (Fig. 5B). Based on cpDNA, a scenario of population expansion is suggested for both C. revoluta and C. taitungensis, while a weak bottleneck event may have occurred in C. revoluta (Fig. 6A and 6B). Likewise, Japan's populations of C. revoluta almost remain nearly constant before a recent population expansion, whereas, the Fukien's population did not undergo large fluctuations in population size until its recent extinction (Fig. 6C and 6D).

Bayesian skyline plot based on the mtDNA for the effective population size fluctuation throughout time. X axis: population size × generation time; Y axis: time (million years ago). A) Cycas revoluta; B) C. taitungensis; C) all Japanese populations of C. revoluta; D) the Fukien population of C. revoluta (black line, median estimations; area between gray lines, 95%

Bayesian skyline plot based on the cpDNA for the effective population size fluctuations throughout time. X axis: population size × generation time; Y axis: time (million years ago). A) Cycas revoluta; B) C. taitungensis; C) all Japanese populations of C. revoluta; D) the Fukien population of C. revoluta (black line, median estimations; area between gray lines, 95% confidence interval.
We also use mismatch distribution analyses to infer the long-term demographic history of populations. Mismatch distribution analyses (Fig. 7 and 8) show different patterns between the cp- and mtDNA genomes. In most cycad populations, cpDNA has a unimodal mismatch distribution (Fig. 8), while multimodal and very ragged distributions are detected in mtDNA within all populations (Fig. 7). Nevertheless, when sequences from all populations are pooled together, both cp- and mtDNAs have unimodal mismatch distributions in C. revoluta, while only cpDNA displays a unimodal mismatch distribution in C. taitungensis. Unimodal mismatch distributions are often the result of past demographic expansions [50, 51]; whereas, multimodal mismatch distributions usually indicate stationary population structure. Since the data used to produce mismatch distributions are not independent [52], Tajima's D, Fu and Li's D* and Fu's Fs statistics can be used to detect departure from population equilibrium. In this study, values of Fu and Li's D* based on cpDNA sequences for C. revoluta and C. taitungensis are significantly negative, except for populations IR, KO, and OK (Table 3); while Tajima's D statistics are all significantly negative, except for the population IR. Fu's Fs statistics are significantly negative, except for populations IS, IR, and OK (Table 3). Likewise, Fu and Li's D*, Tajima's D, and Fu's Fs statistics are significantly or marginally significantly negative based on mtDNA data in most populations (Table 2).

Mismatch distributions of mtDNA haplotypes based on pairwise sequence differences against the frequencies of occurrence for cycad species and populations.

Mismatch distributions of cpDNA haplotypes based on pairwise sequence differences against the frequencies of occurrence for cycad species and populations.
Coalescence of lineages and molecular dating
Bayesian estimates of the mutation rates and the age of the most recent common ancestor (TMRCA) of the cycad sequences were obtained using BEAST v. 1.3. For mtDNA, all the haplotypes coalesced at about 327.3 MYA, while all chlorotypes could be traced back to a common ancestor about 204.0 MYA (Fig. 9). In the mtDNA haplotype tree, clusters (A, B, C) vs. D split at some 74.1 MYA; and the clusters A and (B, C) coalesced at about 30.6 MYA. Likewise, in the cpDNA tree, clusters I and (II, V) split some 25.6 MYA, while cluster (I, II, V) diverged from its sisters (III, IV) an estimated 43.9 MYA. Almost all coalescences of the major lineages predate the formation of the section Asiorientales. Coalescent events occurred much earlier in mtDNA than in cpDNA. On the other hand, most tip haplotypes in both gene trees coalesced recently to their common ancestors.

Coalescence-based analyses of lineages. Time coalesced to the most recent common ancestor (TMRCA) of major lineages are indicated. A) mtDNA, B) cpDNA.
Discussion
Highly polymorphic organelle DNAs in Cycas revoluta and C. taitungensis
Genetic variation in the two cycad species is high, comparable to or higher than many tree species (such as Quercus [31, 49]; Fagus [50]; Lithocarpus [32, 44, 51]. Such high levels of genetic variation within mt- and cpDNAs suggest that these are ancient lineages with enough time for mutations to accumulate within lineages, given the slow rate of organelle evolution [52]. Evidence of the deep splits in both phylogenetic trees (Fig. 2) and the estimated dates of divergence support this scenario. Moreover, star-like phylogenies with a mixture of long and short branches, which represent recently evolved haplotypes, illustrate the high levels of variation.
Although the two cycad species have high levels of genetic diversity at both loci, the loci do not vary in concert. For example, the C. revoluta population YO possesses the highest level of mtDNA variation while having the lowest level of cpDNA diversity. Similarly, the Fukien China C. revoluta population is highly diverse for mtDNA haplotypes while containing lower than average of cpDNA haplotypes. The large native cycad population on the island of Amami-O-Shima (AM) has genetic diversity at both loci comparable to or even lower than most populations.
In this study, genetic diversity tends to be higher in the south than in the north. A cline-like distribution of genetic diversity from south to north occurs in many continental plants of the Northern Hemisphere and is thought to be related to colonization history over glacial cycles [cf. [34, 53]]. Considering the geological history of the Taiwan-Ryukyus Archipelago, one expects a similar pattern for these cycad species where colonization occurs in a stepwise manner from China northwards through the Archipelago. Genetic diversity should decrease to the north, assuming that a small number of individuals colonize each subsequent island in a stepwise manner. However, the occurrence of highly diverse organelle DNAs across the range of Cycas revoluta and C. taitungensis, and many other tree species of the Taiwan-Ryukyus Archipelago suggests that single stepwise colonization events may not be the sole determinant of the overall pattern of genetic diversity [cf. [33]].
Austerlitz et al. [51] suggest that the level of diversity in tree species that recolonize glaciated regions is affected by restricted gene flow but is also tempered by their demography. In tree populations, the genetic effects of founder events are reduced due to overlapping generations and delayed reproduction associated with long life spans. These are traits found in most long lived trees including these cycads, which are characterized by great longevity, overlapping generations, age structured populations and a juvenile phase of 30 to 50 years. During the juvenile period when cycads grow vegetatively, newly founded cycad potentially populations can increase in number for many years only through the arrival of new migrants, especially when migration corridors across islands for migration are available. This pattern of colonization and reproduction effectively acts to increase in the number of initial colonizers of a given population and in turn decreases any founder effects. Even given a high likelihood of stepwise colonization along the Taiwan-Ryukyu Archipelago, as observed in the Pinus luchuensis complex [52, 33], multiple colonization events could compensate for potential reduction in genetic diversity during each founder event.
Secondary contact of ancestral lineages can be another factor that contributes to high genetic diversity. The coalescence-based analysis reveals that mitotypes F and G diverged some 327.3 and 295.3 million years ago, respectively. Today these haplotypes exist exclusively in the mainland Fukien (mitotype G), and in the Fukien and Ishigaki (mitotype F) populations. Mitotype F is common in the Fukien population, suggesting an origin most likely on the Asian mainland with subsequent dispersal northward onto Ishigaki. Such secondary contact of divergent lineages is consistent with a U-shaped site-frequency distribution according to Wakeley and Aliacar's simulations (Fig. 8) [54].
Isolation and lack of geographical subdivision
Current gene flow between islands is greatly restricted due to their geographical isolation and the low dispersability of cycad seeds in water [36]. Thus, one expects that island populations should be significantly differentiated from each other. But, like many other tree species of the Taiwan-Ryukyu Archipelago [33], no significant geographical differentiation is detected between cycad populations among islands. Fst values are small and all pairwise Nm values, deduced from Fst values, are much greater than one (Tables 4 and 5). The Nm values based on mtDNA data are about ten-fold greater than those based on cpDNA sequences, 106.10 vs. 10.95. While both organelles are transmitted only via seeds, the difference in Nm values determined from cp- and mtDNAs coupled with deviation from an isolation-by-distance model, and the lack of geographical subdivision and genetic differentiation, suggest that the two loci are responding to different evolutionary forces.
Clearly, Nm values cannot be interpreted as current gene flow between populations. These high Nm values likely represent either ancient migration events or shared ancestral polymorphisms [55, 56]. For example, the Nm values between Chinese and Japanese populations range from 3.87 to 7.31 (mtDNA) and 1.87 to 10.25 (cpDNA) (Tables 4 and 5). Testing of 'isolation with migration' (IM), nevertheless, reveals asymmetrical, historical gene flow with more migrants of C. revoluta from Japan to China than vice versa, 8.0134 vs. 0.5431 based on combined data. These results suggest long periods of isolation with limited genetic exchange between the two geographical regions. C. taitungensis shows a similar asymmetric pattern of ancient gene flow. Migration is estimated to be more frequent from population C to population RL (3.86445 vs. 41.10288, cpDNA, and 2.25430 vs. 48.99630, mtDNA), contributing to the lack of genetic differentiation between populations, as indicated by Fst values of 0.0056 (cpDNA) and 0.021 (mtDNA) [cf. [55]].
Like other island species, cycad populations were colonized via founders from neighboring islands or the mainland. Today, deep water channels hinder seed dispersal among islands and most seed dispersal occurs only within islands. However, during the glacial maxima [cf. [29]], sea levels were much lower and migration could have occurred among islands. The rise of sea levels during glacial retreats terminated between-island seed exchange and isolated cycad populations of the Ryukyu Archipelago from each other [57]. The time since isolation approximates 200–400 cycad generations. Given the very short time that populations have been isolated and the low rate of molecular evolution for both organelle loci, it is not surprising that populations show genetic coherence. A recent simulation study [51] indicates that tree populations can remain genetically similar even with an isolation-by-distance pattern of colonization and can yield a phylogeographical pattern with little differentiation, here for the cycads of the Taiwan-Ryukyu Archipelago. The high levels of nucleotide diversity in both organelle DNAs and the existence of the highest number of private cytotypes suggest that population IS of the south Ryukyu and population RL of Taiwan may have acted as glacial refugia for C. revoluta and C. taitungensis, respectively.
Paraphyly of loci in Cycas species and sections
Although cycad populations are undifferentiated from each other due to recent historical gene flow (Table 6), given that the generic sections of cycads sections have been isolated for very long periods of time, one would expect monophyly at the sectional level. Nevertheless, both mt- and cpDNAs are paraphyletic or polyphyletic between section Asiorientales and its sisters. Such lack of reciprocal monophyly between taxa may result from selection, e.g., the S locus of Brassica [58], and α-tubulin genes of Miscanthus [59], lineage sorting [60], or hybridization/introgression [61]. Genetic exchange among species via hybridization and introgression introduces foreign alleles into a species or population and can result in a genetic admixture. In contrast, lineage sorting occurs when populations or species are recently diverged and retain ancestral polymorphisms [cf. [5, 52]].
Hybridization and/or lineage sorting are likely causes of shared polymorphisms in Cycas given that these are neutral loci and thus not under strong selection. Discerning between hybridization and lineage sorting can be difficult [61–64], since most statistical analyses have limited power. However, in this study neither hypothesis can by itself explain the current patterns of genetic variation among sections. First, all four Cycas species are allopatric which greatly reduce the possibility of recent recurrent interspecific hybridization. Lineage sorting can result in shared polymorphism but requires a relatively short time of isolation. The fossil record indicates that the sections of Cycas have been diverged for 30 million years, making such a scenario unlikely [10].
The pattern of coalescing DNA segments provides information on the genealogical processes that occur within populations [4, 65, 66]. The star-like phylogeny of the organelle trees shows that recently derived haplotypes are numerous, external on the networks and are geographically restricted. Ancestral haplotypes are found at interior nodes, and are geographically more widespread than the recently derived tip haplotypes. This pattern is consistent with a birth-and-death model where most mutations go extinct with only very few ancestral polymorphisms surviving to become dominant [67]. If the dynamics of mutations and fixation do not follow a birth and death model, one expects topologies with deep splits for most haplotypes.
The pattern of mutations in cycads follows a 'surfing' model [68], which predicts that a majority of new mutations do not spread geographically and either remain at low frequencies or are lost by genetic drift. Only successful 'surfing mutations' can reach high frequencies and eventually occupy a large geographical region [69]. In this model both population size and migration rate determine the success of new mutations. Deme size not only affects the probability of a mutation to 'surf' but also determines its final frequency and spatial distribution. When the effective population size is small, a mutation within a population has a high probability of extinction due to genetic drift and it is able to spread only if it reaches a very high frequency by chance. In contrast, large effective population sizes counter genetic drift while a large number of exchanged migrants between neighboring demes prevents a mutation from reaching high local frequencies [68]. Accordingly, as most cycad populations were probably large (Fig. 5 and 6), one expects to observe numerous recently derived mutations that were derived from a single ancestral haplotype, and the observation that few haplotypes appear to survive over long periods (Fig. 2). Most of the new mutations appear to be ephemeral and can be lost from a population or species during population contraction (the glacial maximum). As a consequence, very few cytotypes, e.g., mitotypes A, and B, and chlorotypes I, and III, survived and became dominant across the cycad populations. Such a birth-and death process corresponding to demographic fluctuations may have occurred regularly in cycads over repeated glacial cycles. As a result, the existence of rare and geographically restricted cytotypes contributes to the high levels of genetic diversity within populations and within species, whereas, they played very minor roles in differentiating populations or species from each other due to their rarity.
While the surfing phenomenon explains the dominance of widespread haplotypes and low genetic differentiation between cycad populations, it does not explain shared ancestral polymorphisms between sections. Both genetic drift and changes in population size have most likely influenced polymorphism as well. Population contraction-expansion, which has occurred regularly in the Quaternary, provides an opportunity for rapid stochastic loss or fixation of alleles (haplotypes). Rapid population growth and expansion in cycads is suggested by the star-shape phylogenies found in most lineages of both organelle genomes, or the Bayesian skyline plot and by the observation that DNA sequences are saturated with singletons at both the population and species levels (Fig. 3 and 4) [cf. [70, 71]], as well as the Bayesian skyline plotting. A mixture of long and short branch lengths in the star-like phylogenies could also be caused by heterogeneous mutation rates. The significant values for Tajima's D and Fu and Li's D* statistics, nevertheless, favor a demographic expansion hypothesis [72].
Mismatch distribution analyses are also used to infer the long-term demographic history of populations. In this study, contrasting patterns of unimodal vs. multimodal mismatch are found for mt- and cpDNAs (Fig. 7 and 8). Usually, unimodal mismatch distributions are interpreted as the result of past demographic expansions [73, 74], whereas multimodal mismatch distributions indicate stationary population sizes. These contrasting patterns in cycads are difficult to interpret although a mismatch inconsistency between different data sets is not unprecedented. A well-known example of contrasting mismatch patterns occurs between expanding human Neolithic and hunter-gatherer populations [75]; the unimodal distribution for Neolithic populations is interpreted as the result of greater Nm values [76]. Even with a multimodal distribution, demographic expansion cannot be rejected, especially if the Nm values are small. In this study, the contrasting mismatch patterns between unlinked organelle genomes seem to be uncorrelated with Nm values. The ragged distributions in mtDNA are more likely attributable to the low τ (fig. 5). In cycads, a very recent population expansion about 200–400 generations, a slow evolutionary rate, and putative intramolecular recombination [data not shown; cf. [55]], may preclude mismatch distribution analyses from assessing recent population expansion. Nevertheless, Tajima's D, Fu and Li's D*, and Fu's Fs statistics provide independent estimates for spatial expansion, especially considering that the DNA sequences are from neutral noncoding spacer regions.
Coalescence-based analyses measure the time of coalescence to the most recent common ancestor (TMRCA). In both organelle DNAs, the divergence time is estimated at 327.3 MYA in mtDNA and 204.0 MYA in cpDNA, agreeing with the divergence of major sections of cycads in the Mesozoic [77]. Moreover, the split of the common haplotypes, mitotype B and chlorotype I, from their sister haplotypes occurred about 21.0 and 25.6 MYA, respectively in the Tertiary period when cycads were diversifying and when divergence of section Asiorientales (30 MYA) occurred. Since then the two dominant cytotypes have persisted in cycads [cf. [55]]. The sharing of common cytotypes in both genomes is consistent with range contraction-expansion during glacial cycles where common haplotypes could persist, while most rare, newly derived haplotypes were lost. Consequently, both phylogenies show topologies with few ancient coalescence events vs. numerous recent coalescence events.
The evolution of these two cycad species is complex, affected by range expansion and contraction, fluctuations in population size, widespread historical and current restricted gene flow during complex glacial cycles as well as low rates of molecular evolution in organelle DNA. All of these processes result in a pattern of genetic similarity among populations, a lack of reciprocal monophyly, and long coalescent times.
Conclusion
In summary, paraphyly at both organelle DNAs has been long maintained in Cycas section Asiorientales ever since the split from its sisters some 30 million years ago. Large population sizes of the constituting species C. revoluta and C. taitungensis contributed to the lack of reciprocal monophyly betwen Cycas sections. Providing limited current seed dispersal across oceans, unexpected low levels of geographical subdivision are attributable to the sharing of common haplotypes among island populations. Demographic expansion and contraction with fluctuating population sizes, widespread historical vs. current restricted gene flow during complex glacial cycles, and low rates of molecular evolution in organelle DNA all have to be aroused to explain such population structuring.
Methods
Plant materials, PCR, and nucleotide sequencing
We assessed levels of sequence variation for cp and mt-DNAs among individuals and populations of Cycas revoluta and C. taitungensis in Taiwan, Fukien, and Ryukyus (Table 1, Fig. 1 and 4). A total of 307 and 102 plants of C. revoluta and C. taitungensis, respectively, were sampled. Cycas revoluta is extinct from the wild in Fukien; only old trees were collected from public gardens or nurseries to avoid sampling from recently introduced material. The samples of C. revoluta in China were collected from South China Botanic Garden, which had plants collected from the Fukien wild population before extinction, which located in the Lienchiahg County, Fukien. An additional six native island populations were collected in Ryukyus for C. revoluta and two populations of C. taitungensis. Two species, C. taiwaniana Carruthers (Section Stangerioides) and C. thouarsii R. Brown (Section Rumphiae) [cf. [8]], were chosen as outgroups. C. taiwaniana was obtained from the South China Botanic Garden, transplanted from a native population in Fukien, China, and C. thouarsii was sampled from the Taiwan Forestry Research Institute, transplanted from a native population in Pingtung, Taiwan. Young and healthy leaves were collected in the field, rinsed with tap water and dried in silica gel. All samples were stored at -70°C until they were processed. The voucher specimens (Hsu s.n.) of this research are deposited in the Herbarium, Endemic Species Research Institute, Nantou, Taiwan. Leaf tissue was ground to a powder in liquid nitrogen and stored at -70°C. Genomic DNA was extracted from the powdered tissue following a CTAB procedure [78]. The atpB-rbcL noncoding spacer of cpDNA and the rDNA internal transcribed spacer (rITS) of mtDNA were amplified and sequenced. PCR amplification was carried out in 100 μL reaction using 10 ng of template DNA, 10 μL of 10× reaction buffer, 10 μL MgCl2 (25 mM), 10 μL dNTP mix (8 mM), 10 pmole of each primer, 10 μL of 10% NP-40, and 2 U of Taq polymerase (Promega, Madison, USA). The reaction was programmed on a MJ Thermal Cycler (PTC 100) as one cycle of denaturation at 95°C for 4 min, 30 cycles of 45 s denaturation at 92°C, 1 min 15 s annealing at 52°C, and 1 min 30 s extension at 72°C, followed by 10 min extension at 72°C. A pair of universal primers for cpDNA atpB-rbcL spacer [79] or mtDNA rITS (mito 18 s F 5'-GTG,AAG,TCG,TAA,CAA,GGT,AGC-3'; mito 5 s R 5'-TCG,AGG,TCG,GAA,TGG,GAT,CGG-3') [cf. [80]], dNTP and Taq polymerase were added to the above ice-cold mix. The reaction was restarted at the first annealing at 52°C.
PCR products were purified by electrophoresis in 1.0% agarose gel using 1 × TAE buffer. The gel was stained with ethidium bromide and the desired DNA band was excised and eluted using QIAquick Gel Kit (QIAGEN). For all individuals, purified amplicons were ligated to a pGEM-T easy vector system (Promega). Five clones were randomly selected and purified using the Plasmid Mini Kit (QIAGEN). Purified cloned ampicons were sequenced in both directions using BigDye chemistry (Perkin Elmer) on ABI 377A automated sequencer (Applied Biosystems). Primers for sequence determination were T7-promoter and SP6-promoter located on p-GEM-T easy Vector termination site. As no within-individual variation was detected for either DNA marker, direct sequencing with eluted PCR products was also conducted to all individuals.
Data analysis
Sequence alignment and phylogenetic analyses
Nucleotide sequences were aligned with the program BlastAlign [81]. After the alignment, the indels were coded as missing data. Phylogenetic trees were reconstructed using maximum likelihood (ML) analyses of the nucleotide sequences with software PHYML v2.4.5 [82] and bootstrap consensus values calculated using 1,000 replicates. The general time reversible GTR + I + G model with 6 substitution categories was determined to be the most suitable model by Modeltest v3.6 [83] and was used for all subsequent nucleotide analyses. Maximally parsimonious (MP) trees were also sought using PAUP heuristic search strategies [84]. The length of the shortest trees was obtained by initiating 1,000 heuristic searches, each using random addition starting trees, with tree bisection-reconnection (TBR) branch swapping. The equally MP trees were then used as starting trees for TBR branch swapping. In all analyses, the maximum number of trees to be saved was set at 5000. Bootstrap values [85] were calculated from 1,000 replicate analyses using a heuristic search strategy, simple addition sequence of the taxa, and TBR branch swapping. The values of random number seed set as 64238 and 70189 for mtDNA and cpDNA data sets, respectively, were used to start the random number generator. Relationships among haplotypes were determined and displayed as nested phylogenies. In order to visualize the phylogeographical relationships, a network of each locus based on statistical parsimony was constructed with the computer program TCS [86]. A distance matrix was composed of all pairwise comparisons of haplotypes, for which the maximum number of mutations was determined that did not exceed the probability of parsimony by 0.95 [as defined in [87]]. All haplotypes satisfying this parsimony criterion were then connected to a single network, while those of which the probability exceeded 0.95 were resolved as a separate network. According to coalescent theory, tip nodes of a network are likely to represent descendents derived from ancestral, interior nodes [cf. [88, 89]].
The hypothesis of a molecular clock was tested by relative rate tests [90, 91]. The null hypothesis of a molecular clock suggests that the number of nucleotide substitutions between two lineages would be the same. Based on the assumption of a normal distribution of nucleotide substitutions [91], the hypothesis of a molecular clock will be rejected with 95% significance, when the difference of substitution rates between two lineages is greater than 1.96 times the standard error.
Population genetic analysis
Summary statistics such as the number of segregating sites (S), and the average number of pairwise difference (k) between haplotypes were determined. The level of genetic diversity within populations was quantified by measures of nucleotide divergence, θ [92] and φ [93], using DnaSP Version 4.10 [94]. In order to make inferences about demographic changes in the cycads, we employed both mismatch distributions and statistical tests of neutrality. We calculated Tajima's D [91], Fu and Li's D* statistic [95], and Fu's Fs statistics [96], which is powerful for detecting population growth [97], in the noncoding DNA fragments as indicators of demographic expansion in DnaSP. We also investigated the historical demographics of populations by plotting mismatch distributions [73] and comparing them to Poisson distributions. Mismatch distributions for each sample were used to distinguish between models that invoke past exponential growth and historical population stasis. Parameters of demographic expansion were estimated using the methods of Schneider and Excoffier [98]. The validity of the demographic expansion hypothesis was tested using a parametric bootstrap approach, in which the sum of squared deviation (SSD) between the observed distribution and the expected distribution was compared to the SSD between the simulated distributions and the expected distribution (Arlequin version 3.0 [99]). Patterns of geographical subdivision and genetic differentiation were estimated hierarchically in DnaSP. Statistical significance of Fst estimates was assessed using software Arlequin with 10,000 permutations.
Estimation of coalescence times of cp and mtDNA lineages
We estimated the coalescence time of the the sister lineages of C. taitungensis and C. revoluta split for both mt- and cpDNAs. To estimate divergence between lineages a well-supported rate of evolution is required. In seed plants, evolutionary rates are estimated at 1.01 × 10-9 and 4.50 × 10-10 substitutions per site per year for synonymous sites of cp- and mt-DNAs, respectively [cf. [52, 100]]. These values approximate the evolutionary rates of introns and noncoding spacers of organelle DNAs. Bayesian estimates of the mutation rates and the ages of the most recent common ancestor (TMRCA) of the cycad sequences were obtained using BEAST v. 1.4, available from http://beast.bio.ed.ac.uk[101]. We used the GTR+I+G model of nucleotide substitution with estimated base frequencies, gamma shape distribution (with six categories), proportion of invariant sites, and a strict molecular clock with uncorrelated log normal distribution of branch lengths. Posterior estimates of the mutation rate and age of the TMRCA were obtained by Markov chain Monte Carlo (MCMC) analysis, with samples drawn every 500 steps over a total of 25,000,000 steps. The BEAST program was also used to create a Bayesian skyline plot with 10 steps. The analysis was run for 107 iterations with a burn-in of 106 under the GTR+I+G model with estimated base frequencies, gamma shape distribution (with six categories), proportion of invariant sites, and a strict molecular clock with uncorrelated log normal distribution of branch lengths. Genealogies and model parameters were sampled every 1000 iterations. All operators were automatically optimized. Convergence of parameters and mixing of chains were followed by visual inspection of parameter trend lines and checking of ESS values by three pre-runs. The effective sampling size (ESS) parameter was found to exceed 100, which suggests acceptable mixing and sufficient sampling. Adequate sampling and convergence to the stationary distribution were checked using TRACER v. 1.3 [102]. Posterior estimates of parameters were all distinctly unimodal (although with wide 95% highest posterior densities), and all parameters were identifiable, despite the relatively low information content in the sequences and the small age range of the sequences.
Isolation by migration and estimating of ancestral population size
We used the simulation program IM [1, 103] to investigate isolation with a migration model. By applying coalescent simulations and Bayesian computation procedures, IM yielded six demographic parameters, including population-split time, effective population size for the ancestral and two current populations, and migration rates. The posterior probability densities of these parameters are generated by MCMC simulations, and simulations were run with individual simulations being updated 50 million times. Within each simulation, we used the procedure to swap among 10 heated chains (Metropolis coupling) and observed sufficient swapping rate while the simulation was running. These simulations were carried out using several independent runs, with each chain started at a different starting point and initiated with a burn-in period of 100,000 updates. Convergence upon the stationary distribution was assessed by estimating the effective sample size (ESS) for the parameters, based on the autocorrelation of parameter values measured over the course of the run. The analysis was considered to have converged upon a stationary distribution if the independent runs generated similar posterior distributions with a minimum ESS of 100. Each simulation yielded a marginal density histogram for the six parameters of interest. The peaks of the resulting distributions were considered as the maximum likelihood estimates (MLE) of the parameter with credibility intervals equaling the 95% highest posterior density (HPD) intervals.
References
Chiang TY, Schaal BA: Phylogeography of plants in Taiwan and the Ryukyu Archipelago. Taxon. 2006, 55: 3-41.
Hoelzer GA, Wallman J, Melnick DJ: The effects of social structure, geographical structure, and population size on the evolution of mitochondrial DNA: II. Molecular clocks and the lineage sorting period. J Mol Evol. 1998, 47: 21-31.
Schaal BA, Olsen K: Gene genealogies and population variation in plants. Proc Natl Acad Sci. 2000, 97: 7024-7029.
Donnelly P, Tavaré S: Coalescents and genealogical structure under neutrality. Ann Rev Genet. 1995, 29: 401-421.
Chiang TY: Lineage sorting accounting for the disassociations between chloroplast and mitochondrial lineages in oaks of southern France. Genome. 2000, 43: 1090-1094.
Matos JA, Schaal BA: Chloroplast evolution in the Pinus montezumae complex: a coalescent approach to hybridization between P. hartwegii and P. montezumae. Evolution. 2000, 54: 1218-1233.
Willis KJ, McElwain JC: The evolution of plants. 2002, Oxford: Oxford University Press
Hill KD: Cycas – an evolutionary perspective. Biology and conservation of cycads. Proceedings of the Fourth International Conference on Cycad Biology. Edited by: Chen CJ. 1999, Beijing: International Academic Publishers, 98-115.
Ye ZY: The investigation into the past and the current state of Cycas revoluta in Fujian. Biology and conservation of cycads. Proceedings of the Fourth International Conference on Cycad Biology. Edited by: Chen CJ. 1999, Beijing: International Academic Publishers, 73-74.
Pant DD: A study in contrasts: the present and past distribution and form of certain cycads. Biology and conservation of cycads. Proceedings of the Fourth International Conference on Cycad Biology. Edited by: Chen CJ. 1999, Beijing: International Academic Publishers, 1-23.
Zhou GS, Jian SX, Zhou W: Cycad cultivation and trade in Fujian of China. Biology and conservation of cycads. Proceedings of the Fourth International Conference on Cycad Biology. Edited by: Chen CJ. 1999, Beijing: International Academic Publishers, 368-370.
Miller G: From cycad flour, a new emerges. Science. 2006, 313: 431-
Whitelock LM: The cycads. 2002, Portland: Timber Press
Osborne R, Stevenson DW, Hill KD: The world list of cycads. Biology and conservation of cycads. Proceedings of the Fourth International Conference on Cycad Biology. Edited by: Chen CJ. 1999, Beijing: International Academic Publishers, 224-239.
Huang S, Hsieh HT, Fang K, Chiang YC: Patterns of genetic variation and demography of Cycas taitungensis in Taiwan. Bot Rev. 2004, 70: 86-92.
Shimabuku K: Checklist of vascular flora of the the Ryukyu Islands. 1997, Kyushu University Press
Uehara K: Illustrations of trees. Cycadaceae. 1970, Tokyo: Erushima Press, 1:
Hsieh HT: Studies on population ecology and genetic variability of Cycas taitungensis. Master Thesis. 1999, Taipei: Department of Biology, National Taiwan Normal University
Darwin C: On the Origin of Species. 1859, London: John Murray
Emerson BC: Evolution on oceanic islands: molecular phylogenetic approaches to understanding pattern and process. Mol Ecol. 2002, 11: 951-966.
Juan II, Emerson BC, Orom II, Hewitt GM: Colonization and diversification: towards a phylogeographic synthesis for the Canary Islands. Trends Ecol Evol. 2000, 15: 104-109.
Vita-Finzi C: Deformation and seismicity of Taiwan. Proc Natl Acad Sci. 2000, 97: 11176-80.
Sibuet JC, Hsu SK: Geodynamics of the Taiwan arc-arc collision. Tectonophysics. 1997, 274: 221-251.
Sibuet JC, Hsu SK: How was Taiwan created?. Tectonophysics. 2004, 379: 159-181.
Wang JM: The Fenwei rift and its recent periodic activity. Tectonophysics. 1987, 133: 257-275.
Hikida T, Ota H: Biogeography of reptiles in the subtropical East Asian Islands. The Symposium on the Phylogeny, Biogeography and Conservation of Fauna and Flora of East Region. 1997, Taipei, Taiwan: National Taiwan Normal University, 11-18.
Bennett KD: Milankovitch cycles and their effects on species in ecological and evolutionary time. Paleobiology. 1990, 16: 11-21.
Lambeck K, Chappell J: Sea level change through the last glacial cycle. Science. 2001, 292: 679-685.
Hewitt GM: A climate for colonization. Heredity. 2004, 92: 1-2.
Kizaki K, Oshiro I: Paleogeography of the Ryukyu Islands. Marine Sci Month. 1977, 542-549.
Petit E, Balloux F, Goudet J: Sex-biased dispersal in a migratory bat: a characterization using sex-specific demographic parameters. Evolution. 2001, 55: 635-640.
Hung KH, Hsu TW, Schaal BA, Chiang TY: Loss of genetic diversity and erroneous phylogeographical inferences in Lithocarpus konishii (Fagaceae) of Taiwan caused by the Chi-Chi Earthquake: Implications for Conservation. Ann Missouri Bot Gard. 2005, 92: 52-65.
Chiang TY, Schaal BA: Phylogeography of plants in Taiwan and the Ryukyu Archipelago. Taxon. 2006, 55: 31-41.
Hewitt GM: The genetic legacy of the Quaternary ice ages. Nature. 2000, 405: 907-913.
Zachos J, Pagani M, Sloan L, Thomas E, Billups K: Trends, rhythms, and aberrations in global climate 65 Ma to present. Science. 2001, 292: 686-693.
Dehgan B, Yuen CKH: Seed morphology in relation to dispersal, evolution, and propagation of Cycas. Bot Gaz. 1983, 144: 412-418.
Hill KD: Evolution and biogeography of the genus Cycas. Proceedings of the 3rd International Conference of Cycas Biology. Edited by: Vorster P. 1996, Stellenbosch: The Cycad Society of South Africa
Tang W: Seed dispersal in the cycad Zamia pumila in Florida. Can J Bot. 1989, 67: 2066-2070.
Keppel G, Lee SW, Hodgskiss PD: Evidence for the long isolation among populations of a Pacific cycad: genetic diversity and differentiation in Cycas seemannii A. Br. (Cycadaceae). J Heredity. 2002, 93: 133-139.
Lu SY, Peng CI, Cheng YP, Hong KH, Chiang TY: Chloroplast DNA phylogeography of Cunninghamia konishii (Cupressaceae), an endemic conifer of Taiwan. Genome. 2001, 44: 797-807.
Lu SY, Hong KH, Liu SL, Cheng YP, Wu WL, Chiang TY: Genetic variation and population differentiation of Michelia formosana (Magnoliaceae) based on cpDNA variation and RAPD fingerprints: relevance to post-Pleistocene recolonization. J Plant Res. 2002, 115: 203-216.
Chiang TY, Chiang YC, Chen YJ, Chou CH, Havanond S, Hong TN, Huang S: Phylogeography of Kandelia candel in East Asiatic mangroves based on nucleotide variation of chloroplast and mitochondrial DNAs. Mol Ecol. 2001, 10: 2697-2710.
Chiang YC, Schaal BA, Ge XJ, Chiang TY: Range expansion leading to departures from neutrality in the nonsymbiotic hemoglobin gene and the cp DNA trn L-trnF intergenic spacer in Trema dielsiana (Ulmaceae). Mol Phyl Evol. 2004, 31: 929-942.
Chiang TY, Hung KH, Hsu TW, Wu WL: Lineage sorting and phylogeography in Lithocarpus formosanus and L. dodonaeifolius (Fagaceae) from Taiwan. Ann Missouri Bot Gard. 2004, 91: 207-222.
Purvis A, Bromham L: Estimating the transition/transversion ratio from independent pairwise compaisons with an assumed phylogeny. J Mol Evol. 1997, 44 (1): 112-119.
Ina Y: Estimation of the transition/transversion ratio. J Mol Evol. 1998, 46 (5): 521-533.
Bakker FT, Culham A, Gomez-Martinez R, Carvalho J, Compton J, Dawtrey R, Gibby M: Patterns of nucleotide substitution in angiosperm cpDNA trnL (UAA)-trnF (GAA) regions. Mol Biol Evol. 2000, 17: 1146-1155.
Hillis DM, Allard MW, Miyamoto MM: Analysis of DNA sequence data: phylogenetic inference. Methods Enzymol. 1993, 224: 456-490.
Dumolin-Lapeue S, Pemonge MH, Petit RJ: An enlarged set of consensus primers for the study of organelle DNA in plants. Mol Ecol. 1997, 6: 393-397.
Demesure B, Comps B, Petit RJ: Chloroplast DNA phylogeography of the common beech (Fagus sylvatica L.) in Europe. Evolution. 1996, 50: 2515-2520.
Austerlitz F, Mariette S, Machon N, Gouyon PH, Godelle B: Effects of colonization processes on genetic diversity: differences between annual plants and tree species. Genetics. 2000, 154: 1309-1321.
Chiang YC, Hung KH, Schaal BA, Ge XJ, Hsu TW, Chiang TY: Contrasting phylogeographical patterns between mainland and island taxa of the Pinus luchuensis complex. Mol Ecol. 2006, 765-779.
Petit RJ, Duminil J, Funeschi S, Hampe A, Salvini DA, Vendramin GG: Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol Ecol. 2005, 14: 689-701.
Wakeley J, Aliacar M: Gene genealogies in a metapopulation. Genetics. 2001, 159: 893-905.
Huang S, Chiang YC, Schaal BA, Chou CH, Chiang TY: Organelle DNA phylogeography of Cycas taitungensis, a relict species in Taiwna. Mol Ecol. 2001, 10: 2669-2681.
Whitlock MC, McCauley DE: Indirect measures of gene flow and migration: FST not equal to 1/(4Nm + 1). Heredity. 1999, 82: 117-125.
Lin CC: An outline of Taiwan's Quaternary geohistory with a special discussion of the relation between natural history and cultural history in Taiwan. Bull Dept Archaeol Anthropol. 1966, 23: 7-44.
Miege C, Ruffio-Chable V, Schierup MH, Cabrillac D, Dumas D, Gaude T, Cock JM: Intrahaplotype polymorphism at the Brassica S locus. Genetics. 2001, 159: 811-822.
Wu WL, Schaal BA, Hwang CY, Hwang MD, Chiang YC, Chiang TY: Characterization and adaptive evolution of α-tubulin genes in Miscanthus sinensis complex (Poaceae). Amer J Bot. 2003, 90: 1513-1521.
Jacob S, Blattner FR: A chloroplast genealogy of Hordeum (Poaceae): long-term persisting haplotypes, incomplete lineage sorting, regional extinction, and the consequences for phylogenetic inference. Mol Biol Evol. 2006, 23 (8): 1602-1612.
Chiang TY, Hung KH, Peng CI: Experimental hybridization reveals biased inheritance of the internal transcribed spacer of the nuclear ribosomal DNA in Begonia × taipeiensis. J Plant Res. 2001, 114: 343-351.
Donnelly MJ, Pinto J, Girod R, Besansky NJ, Lehmann T: Revisiting the role of introgression vs shared ancestral polymorphisms as key processes shaping genetic diversity in the recently separated sibling species of the Anopheles gambiae complex. Heredity. 2004, 92: 61-68.
Holder MT, Anderson JA, Holloway AK: Difficulties in detecting hybridization. Syst Biol. 2001, 50: 978-982.
Osada N, Wu CI: Inferring the mode of speciation from genomic data: a study of the great apes. Genetics. 2005, 169: 259-264.
Hudson RR: Island models and the coalescent process. Mol Ecol. 1998, 7: 413-418.
Templeton AR: Gene tree: A powerful tool for exploring the evolutionary biology species and speciation. Plant Species Biol. 2000, 15: 211-222.
Graham J, Thompson EA: A coalescent model of ancestry for a rare allele. Genetics. 2000, 156: 375-384.
Klopfstein S, Currat M, Excoffier L: The fate of mutations surfing on the wave of a range expansion. Mol Biol Evol. 2006, 23: 482-490.
Edmonds CA, Lillie AS, Cavalli-Sforza LL: Mutations arising in the wave front of an expanding population. Proc Natl Acad Sci. 2004, 101: 975-979.
Rosenberg NA, Hirsh AE: On the use of star-shaped genealogies in inference of coalescence times. Genetics. 2003, 164: 1677-1682.
Joy DA, Feng X, Mu J, Furuya T, Chotivanich K, Krettli AU, Ho M, Wang A, White NJ, Suh E, Beerli P, Su XZ: Early origin and recent expansion of Plasmodium falciparum. Science. 2003, 300: 318-321.
Aris-Brosou S, Excoffier L: The impact of population expansion and mutation rate heterogeneity on DNA sequence polymorphism. Mol Biol Evol. 1996, 13: 494-504.
Slatkin M, Hudson RR: Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics. 1991, 129: 555-562.
Rogers AR, Harpending H: Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol. 1992, 9: 552-569.
Excoffier L, Schneider S: Why hunter-gatherer populations do not show signs of Pleistocence demographic expansions. Proc Natl Acad Sci. 1999, 96: 10597-10602.
Ray N, Currat M, Excoffier L: Intra-deme molecular diversity in spatially expanding populations. Mol Biol Evol. 2003, 20: 76-86.
Treutlein J, Wink M: Molecular phylogeny of cycads inferred from rbc L sequences. Naturwissenschaften. 2002, 89: 221-225.
Murray MG, Thomposon WF: Rapid isolation of high molecular weight DNA. Nucleic Acids Res. 1980, 8: 4321-4325.
Chiang TY, Schaal BA, Peng CI: Universal primers for amplification and sequencing a noncoding spacer between atpB and rbcL genes of chloroplast DNA. Bot Bull Acad Sin. 1998, 39: 245-250.
Brennicke A, Möller S, Blanz PA: The 18S and 5S ribosomal RNA genes in Oenothera mitochondria: Sequence rearrangments in the 18S and 5S rRNA genes of higher plants. Mol Gen Gent. 1985, 198: 404-410.
Belshaw R, Katzourakis A: BlastAlign: a program that uses blast to align problematic nucleotide sequences. Bioinformatics. 2005, 21 (1): 122-123.
Guindon S, Gascuel O: A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, 52: 696-704.
Posada D, Crandall KA: MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998, 14 (9): 817-818.
Swofford DL: PAUP*: Phylogenetic Analysis Using Parsimony (and Other Methods), Version 4.0b10. 2002, Sinauer Associates, Inc, Sunderland, Massachusetts
Felsenstein J: Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985, 39: 783-791.
Clement M, Posada D, Crandall K: TCS, a computer program to estimate gene genealogies. Mol Ecol. 2000, 9: 1657-1660.
Templeton AR, Crandall KA, Sing CF: A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics. 1992, 132: 619-633.
Donnelly P, Tavare S: The ages of alleles and a coalescent. Adv Appl Probab. 1986, 18: 1-19.
Crandall KA, Templeton AR: Empirical tests of some predictions from coalescent theory with applications to intraspecific phylogeny reconstruction. Genetics. 1993, 134: 959-969.
Sarich VM, Wilson AC: Generation time and genomic evolution in primates. Science. 1973, 179: 1144-1147.
Wu CI, Li WH: Evidence for higher rates of nucleotide substitution in rodents than in man. Proc Natl Acad Sci USA. 1985, 82: 1741-1745.
Watterson GA: On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 1975, 7: 256-275.
Tajima F: Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989, 123: 585-595.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R: DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003, 19: 2496-24.
Fu YX, Li W: Statistical tests of neutrality of mutations. Genetics. 1993, 133: 693-709.
Fu YX: Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997, 147: 915-925.
Ramos-Onsins SE, Rozas J: Statistical properties of new neutrality tests against population growth. Mol Biol Evol. 2002, 19: 2092-2100.
Schneider S, Excoffier L: Estimation of past demographic parameters from the distribution of pairwise differences when mutation rates vary among sites: application to human mitochondrial DNA. Genetics. 1999, 152: 1079-1089.
Excoffier L, Laval G, Schneider S: Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evol Bioinform Online. 2005, 1: 47-50.
Graur D, Li WH: Fundamentals of molecular evolution. 2000, Sunderland: Sinauer Associates
Drummond AJ, Ho SY, Phillips MJ, Rambaut A: Relaxed phylogenetics and dating with confidence. PLOS Biology. 2006, 4: e88-
Rambaut A, Drummond A: Tracer – MCMC trace analysis tool. 2004, Oxford, UK: University of Oxford
Hey J, Nielsen R: Multilocus methods for estimating population sizes, migration rates, and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004, 167: 747-760.
Acknowledgements
This study was supported by the National Science Council (NSC) and Council of Agriculture of (COA) of Taiwan. We are grateful to the Tropical Research Centre of the University of Ryukyus for the assistance in the sample collecting.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Authors' contributions
YCC and KHH carried out the molecular genetic studies and the statistical analysis, and participated in the sequence alignment. TYC, TWH and BAS designed the research and drafted the manuscript. SJM, SH, and XJG took part in material sampling. All authors read and approved the final manuscript.
Yu-Chung Chiang, Kuo-Hsiang Hung contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chiang, YC., Hung, KH., Moore, SJ. et al. Paraphyly of organelle DNAs in Cycas Sect. Asiorientales due to ancient ancestral polymorphisms. BMC Evol Biol 9, 161 (2009). https://doi.org/10.1186/1471-2148-9-161
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-9-161