
Abstract
Background
Due to its history, with a high number of migration events, the Mediterranean basin represents a challenging area for population genetic studies. A large number of genetic studies have been carried out in the Mediterranean area using different markers but no consensus has been reached on the genetic landscape of the Mediterranean populations. In order to further investigate the genetics of the human Mediterranean populations, we typed 894 individuals from 11 Mediterranean populations with 25 single-nucleotide polymorphisms (SNPs) located on the X-chromosome.
Results
A high overall homogeneity was found among the Mediterranean populations except for the population from Morocco, which seemed to differ genetically from the rest of the populations in the Mediterranean area. A very low genetic distance was found between populations in the Middle East and most of the western part of the Mediterranean Sea.
A higher migration rate in females versus males was observed by comparing data from X-chromosome, mt-DNA and Y-chromosome SNPs both in the Mediterranean and a wider geographic area.
Multilocus association was observed among the 25 SNPs on the X-chromosome in the populations from Ibiza and Cosenza.
Conclusion
Our results support both the hypothesis of (1) a reduced impact of the Neolithic Wave and more recent migration movements in NW-Africa, and (2) the importance of the Strait of Gibraltar as a geographic barrier. In contrast, the high genetic homogeneity observed in the Mediterranean area could be interpreted as the result of the Neolithic wave caused by a large demic diffusion and/or more recent migration events. A differentiated contribution of males and females to the genetic landscape of the Mediterranean area was observed with a higher migration rate in females than in males. A certain level of background linkage disequilibrium in populations in Ibiza and Cosenza could be attributed to their demographic background.
Similar content being viewed by others


Background
The X-chromosome has features that make it a good source of information for population genetic studies. The X-chromosome is present in a single copy in males, which makes it possible to determine the X-chromosome haplotypes in men. Compared with autosomes, the X-chromosome has lower recombination rate, lower mutation rate and smaller effective population size resulting in a faster genetic drift. In consequence, both linkage disequilibrium (LD) and population structure in the X chromosome are expected to be stronger than those in autosomes [1]. Two-thirds of the X-chromosome history has been spent in females. Thus, X chromosome polymorphisms mainly reflect the history of females. Due to recombination, X-chromosome markers in females provide a multilocus system, while the mtDNA and Y-chromosome are linked haplotypes. Thus, X-chromosome markers are valuable for population genetic studies [1].
Due to its history with constant migration movements, the Mediterranean area constitutes an challenging region for population genetic studies, both considering the whole area [2–4] and particular populations [5, 6]. Only very little genetic structure has been found among populations living on the northern and eastern shores of the Mediterranean Sea by analyses of Y-chromosome STRs [3] and mtDNA [7]. The pattern may be a consequence of the Neolithic demic diffusion in this region (around 9,000 YBP) and/or a high level of gene flow in the area. The presence of a strong genetic boundary between the northern and southern Mediterranean populations, especially in the western side of the sea has been suggested [3, 8–10]. Studies focused on smaller areas have also given interesting results, especially those regarding physical and/or cultural isolates such as Corsican [11] and Sardinian sub-isolates [12], Balearic populations [13–15] and Calabrian populations [16].
In order to further investigate the genetic characteristics of the populations in the Mediterranean basin, we analyzed 25 SNPs located on the X-chromosome in eleven populations from the Mediterranean area. In addition, two populations from Northern Europe and East Africa were analyzed. We carried out a wider analysis using data from the HapMap website on 21 out of the 25 SNPs used in this study (see Methods for details).
We wanted to use the X-chromosome SNPs as multilocus markers. We selected polymorphic X chromosome SNPs with a physical distance of at least 600 kb. Population history such as genetic drift and admixture has an important consequence on the degree of population LD [17]. The level of LD on the X-chromosome is expected to be higher than on autosomes. We also wanted to use these markers to study the pairwise linkage disequilibrium and multilocus association in order to obtain knowledge of the long-term and background LD in each population, because this information can be useful for studies of population genetics and the genetics of complex diseases.
We studied the informativeness of the markers selected and the sex-biased migration rate and compared the information to that of non-recombinant markers in mtDNA and the Y-chromosome.
Results
Intrapopulation variability
A total of 1,078 different haplotypes were found in the 13 populations analyzed by means of 25 X-chromosome SNPs. No single haplotype was shared among individuals within or between populations.
All the markers were polymorphic in all the populations studied. The minor allele frequency varied between a minimum percentage of 10.3% (X159 marker in the Moroccan population) and a maximum value of around 50.0% (widely found). The mean gene diversity (d) of the 25 markers analyzed was very homogeneous in the 13 populations sampled. They ranged from 0.437 in the Somali population to 0.461 in Iraqis and Tunisians. Taking into account that the maximum level of expected heterozygosity for biallelic markers is 0.5, a high degree of gene diversity was found for the 25 SNPs selected in all the populations analyzed.
Among the 300 pairwise comparisons between the 25 SNP markers in the populations, between 10 (Somalia) and 23 pairwise comparisons (Ibiza) showed significant p-values (p < 0.05). However, after Holm-Sidak correction, only two pairwise LD values were significant (X085-X159 in Cosenza, p < 0.001, and X018-X029 in Sicily, p = 0.02). Nevertheless, when multilocus tests of associations were performed using all the 25 markers, the null hypotheses of linkage equilibrium were rejected in two populations: Ibiza (p < 0.01) and Cosenza (p < 0.05). Both populations present some peculiarities that may explain the existence of a higher degree of background linkage disequilibrium (see Discussion).
Interpopulation differentiation
AMOVA and pairwise Fst values
Table 1 shows the results of AMOVA analyses in 11 Mediterranean populations based on 25 X-chromosome SNPs. A total of 99.61% of the global variation was due to the diversity within populations while only 0.39% of the variation was due to interpopulation variability. The Fst was 0.004 (p = 0.001). When the Moroccans were excluded, the significance level increased to p = 0.05. We also performed a similar statistical analysis in the nine "worldwide" populations (see Methods) by using 21 out of the 25 X-chromosome SNPs (Table 2). A ten times higher Fst value (0.045) was observed (p < 0.001). No significant difference was found among the three European populations or between the Chinese and the Japanese population, while a high differentiation was observed among the three African populations, especially between Moroccans and Sub-Saharans.
When a locus by locus AMOVA was performed in the Mediterranean group, only three out of 25 markers showed significant (p < 0.05) variations among the Mediterranean populations (X029, X062, and X121), while 17 out of 21 markers were important for the differentiation of the 9 "worldwide" populations: X018, X029, X036, X046, X047, X056, X059, X062, X076, X085, X109, X121, X122, X131, X135, X143 and X165. The markers X004, X142, X159 and X175 were not included in these analyses due to the lack of information in the populations obtained from the HapMap Project. No correlation (data not shown) was found between the mean gene diversity and the level of differentiation among populations, as has been suggested by others [18].
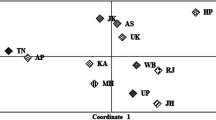
In order to obtain a graphical representation of the genetic structure of the populations studied, a principal coordinate analysis (PCoA) was made from the pairwise Reynold's Fst matrix obtained using 25 or 21 X-chromosome SNPs in the Mediterranean (Figure 1a) and the 17 populations (Figure 1b), respectively.

Principal coordinate analyses plotted from pairwise Reynold's F st values. a) The Mediterranean group analyzed by using 25 X-chromosome SNPs. b) All the 17 populations were represented, including those from the HapMap collection. Twenty-three X-chromosome SNPs were used. CEU: Utah residents with ancestry from western and northern Europe, DEN: Denmark, CHB: China, JPT: Japan, SOM: Somalia, YRI: Nigeria, CAT: Catanzaro, CSZ: Cosenza, IBZ: Ibiza, IRQ: Iraq, MAJ: Majorca, MOR: Morocco, RCA: Reggio di Calabria, SIC: Sicily, TUN: Tunisia, TKY: Turkey, VAL: Valencia
In Figure 1a, the first two axes accounted for 39% of the total variability (first axis: 25%, second axis: 14%). Thus, the Mediterranean populations are quite homogeneous. Equivalently to the results of the AMOVA analysis, the Moroccan population was considerably displaced in the first axis, and the Majorcans were placed in the opposite direction. In the second axis, the differentiation was less, but it is interesting that the Cosenza and Ibiza populations were located opposite to their neighbouring populations (i.e. Catanzaro and Valencia, respectively). Figure 1b shows the relative position of the Mediterranean populations in relation to a wider geographical area. The first two axes accounted for 51% of the global variability (first axis: 38%, second axis: 13%). The first axis displayed East Asian populations (Japanese and Chinese populations, which did not show a significant pairwise Fst value) opposite to the others (Middle East, European and African populations). In the second axis, the Mediterranean populations plus the CEU population (Utah residents with ancestry from northern and western Europe) formed a group between the North-European and African populations. Only the Moroccans showed a slight displacement from the others showing significant Fst values in most of the pairwise comparisons inside the Mediterranean group. The CEU population showed an intermediate position between the North of Europe, represented by the Danish population, and the Mediterranean group. None of the pairwise comparisons between the CEU population and the Mediterranean or Danish populations showed a significant Fst value. For the grouping proposed in Figure 1b, 4.58% of the variation could be explained by differences between groups (p < 0.001). A more detailed hierarchical AMOVA analysis showed that 8.86% of the global variation could be explained by the East Asian populations (Japan and China; p < 0.001), 3.49% of the variation was explained by Somalis and Sub-Saharans (in this case the significance level raised up to p < 0.05 due to the high heterogeneity between these two populations). The Mediterranean group (which included the CEU population) contributed to 1.33% of the global variation, a value that went down to 0.72% when the Moroccan population was excluded from the group (p < 0.001 in both cases). Finally, the North-West European populations (represented by CEU and Danish populations) only explained 0.77% of the global variation (no significant p-value).
Structure analysis
Additionally to the AMOVA analysis, a cluster analysis was performed using the STRUCTURE [19] software in order to assign individuals to a number of K populations allowing admixture. We estimated the posterior probability (P(X|K), where X represents the genotypes) for K between 1 and 6 for 17 populations (13 analyzed in this study and 4 from the HapMap project) using 21 X-chromosome SNPs. We observed that the proportions of the individuals assigned to each K subpopulation were balanced in all cases indicating that the X-chromosome SNPs did not identify to which subpopulation an individual belongs. According to the authors of the programme [19], this could be interpreted as a lack of population structure as a consequence of a high level of admixture.
Due to the statistical model implemented in STRUCTURE, it is possible to find significant Fst values without finding significant differences using STRUCTURE when the populations analyzed are closely related [19]. This is not the case of some of the populations included in the analysis. As it was discussed by other authors [20], it could be that the high migration rate of females could have resulted in a small genetic structure in X-linked loci. On the other hand, a lack of resolution of the investigated SNP markers could also be the cause of the low level of genetic structure observed.
Isolation by distance
We did not find any significant correlation between genetic differentiation (calculated as Fst/(1-Fst)) and the natural logarithm of the geographical distance (in kilometres) in the Mediterranean area based on the study of 25 X-chromosome SNPs (Figure 2a). According to Rousset's isolation-by-distance model [21], some demographic information can be obtained from the model drawn (Figure 2a). An indirect estimation of 4D μσ2, also known as the "Neighbourhood size" (where D is population density and σ2 the dispersal variance), could be calculated from the slope value (0.001). Considering the 25 biallelic markers analyzed in the present work, the estimated value of 4D μσ2 would be around 935 individuals, which seems to be an underestimated value according to the same author. This data can be interpreted as the result of a high number of migration events in the recent history of the human populations in the Mediterranean basin.

Pairwise differentiation versus the natural logarithm of the distances (in km) following Rousset's two-dimensional isolation by distance model. a) Plot based on the analyses of 25 X-chromosome SNPs in 11 Mediterranean populations: F st /(1 - F st ) = 0.001*ln(dist.) - 0.004. b) Analysis carried out on 9 worldwide populations and 21 SNPs: F st /(1 - F st ) = 0.062*ln(dist.) - 0.470.
Figure 2a reflects a strong impact of gene flow versus genetic drift in the Mediterranean region (according to Koizumi et al [22]). Nevertheless, it is worth to pay attention to specific populations where other demographic scenarios such as geographic isolation, genetic drift or population admixture have probably played an important role in their current genetic constitution. As we have commented above, relative high genetic distances between neighbouring populations were found between Cosenza and (1) Catanzaro and (2) Reggio di Calabria and between Ibiza and (1) Majorca and (2) Valencia. The results support the hypothesis that the genetic differentiation of the Ibiza and Cosenza populations is a result of their particular demographic histories [15, 16, 23]. The Moroccan population showed a high level of differentiation in relation to the other populations, even to those that are geographically close to Morocco (Western side of the Mediterranean basin).
The number of estimated migrants was dramatically reduced when a much wider area was considered (Figure 2b, where 9 populations were taken into account: 3 African populations (from Morocco, Nigeria and Somalia), 3 Asian populations (from Iraq, China and Japan) and 3 European populations (from Denmark, CEU – Utah residents with ancestry from northern and western Europe- and South Italy). The analyses of populations belonging to an extended area are more prone to reflect more ancient demographic processes [24]. If the Neolithic transition occurred around 9,000 YBP, and human dispersal was close to 30 km per generation, pre-agricultural migration patterns could be observed at distances over 1,000 km [24]. Taking this information into account and the fact that the minimum geographic distance between two populations in Figure 2b was 954 km, pre-agricultural migration events should be reflected there. On the basis of 25 X-chromosome SNPs, an indirect estimate of 16 migrants between subpopulations was calculated from the regression slope (R2 = 0.680, p < 0.001 Spearman rank correlation coefficient). In this study, isolation by distance operated better at large geographic scales than at short distances.
X-chromosome SNPs vs. non-recombinant systems (mtDNA HVRI and 35 Y-chromosome SNPs)
Mantel test
We performed two Mantel tests for each of the three sets of markers: 25 X-chromosome SNPs, HVRI mtDNA and 35 Y-chromosome SNPs (Table 3). First, we analyzed 11 of the 13 populations in this work for which results of all the markers were available. Then, only the Mediterranean populations were analyzed (nine populations). Mantel correlations were corrected for geographical distances by computing partial correlations between genetic distances at constant geographic distance (see Methods). All tests were significant (p < 0.05) except the test between X-chromosome and mtDNA in the Mediterranean area, where the correlation index was low (rY1_2 = 0.190) but still positive. The lack of significance was most likely due to the low degree of genetic differentiation of both X-chromosomes and mtDNA in the area. All X-chromosome and mtDNA markers showed a significant correlation (p < 0.05) with Y-chromosome SNPs both when 9 or 11 populations were analyzed. Except in particular cases (e.g. Ibiza [25]), male and female movements in the Mediterranean have apparently had the same overall tendency. An important difference in the intensity of the migration events of both sexes could also be seen in the level of significance of the different Mantel tests.
Isolation-by-distance analyses by using different genetic systems
Figure 3 shows a plot of a two-dimensional isolation-by-distance model based on three sets of genetic markers in populations in the Mediterranean area (due to the lack of mtDNA data of the population from Catanzaro and Cosenza, these populations were not included in the analyses).

Two-dimensional isolation-by-distance plot. Representation of 9 populations from the Mediterranean region analyzed by three different markers: a) 25 X-chromosome SNPs (grey circles); b) HVRI mtDNA (black triangles) compared to X-chromosome; c) 35 Y-chromosome SNPs (empty rhombuses) compared to mtDNA and X-chromosome.
As expected from previous studies [3, 26], no significant correlation between genetic and geographic distances was found in any case. From a demographic point of view, the estimated number of migrants based on X-chromosome results would be approximately the same as calculated above (971 individuals), and around 32 were calculated from Y-chromosome SNP data. Surprisingly, a negative slope was obtained in the case of the mtDNA, reflecting a major effect of isolation and genetic drift versus migration in certain populations of the Mediterranean area.
When X-chromosome SNPs and mtDNA were analyzed in a wider geographic area (data not shown), a greater female migration rate compared to the male migration rate was still observed. As it has been commented above, the minimum geographic distance between two populations was approximately 954 km and mainly pre-agricultural migration events were then represented in this case.
Discussion
A considerable amount of information based on classical genetic markers [8, 27, 28], autosomal STRs [26], mtDNA [2, 7] and Y-chromosome haplotypes [3, 4, 10] has previously been reported concerning the genetics of populations in the Mediterranean region. A low level of genetic structure has been described due to the high degree of migration in the Mediterranean area [3]. Apparently, the surrounding lands of the Mediterranean Sea were settled by people from the Middle East approximately 40,000 years ago [5]. The migration events increased during the Neolithic era (around 9,000 YBP) with the transition to agriculture. The increase in migration events during the history constitutes the main cause of the genetic homogeneity in the area. Several authors [9, 10] have suggested the existence of a north-south genetic "barrier" in the western part, and no consensus on the current genetic landscape has been established [4, 29, 30]. Some isolated populations with deviating genetic differentiation have been described [5, 6, 16, 23].
Based on the study of 25 X-chromosome SNPs, we found a low value of population differentiation (Fst = 0.004) in 11 Mediterranean populations. The fixation index was significant (p = 0.001) but the p-value increased to p = 0.5 when the Moroccan population was excluded from the analysis. In contrast, a 10 times higher Fst value (p < 0.001) was observed when 9 populations from three continents (3 from Europe, 3 from Africa and 3 from Asia) were analyzed. Not surprisingly, the strongest genetic differentiation was found among the 3 African populations. It is well known that African populations have a higher level of genetic heterogeneity compared to European or Asian populations [31].
A certain bias of the results obtained could be due to the fact that the markers initially were selected for forensic purposes with a high level of polymorphism in different ethnic groups. A high level of intrapopulation variability will to some degree be associated with a low interpopulation variability [3]. Nevertheless, the significant Fst value found in the Mediterranean populations and the highly significant fixation index observed when distant populations were compared documents the suitability of these markers for population genetic studies.
Geographic "barrier", genetic isolate and population admixture
The analysis of the Mediterranean populations in comparison to the "worldwide" populations showed a clear intermediate position between African and North-European populations with Morocco somewhat closer to the African populations than the other Mediterranean populations. When only the Mediterranean populations were analyzed in a principal coordinate plot, opposite positions of Moroccans and Majorcans were observed (Figure 1). This supports the existence of a north-south gene flow "barrier" in the western part of the Mediterranean area [9, 10]. It could be argued that the genetic differentiation of the Moroccan population was a result of the arab-berber sub-structure of the individuals sampled in this work, but this hypothesis does not seem likely. Several authors [26, 28] reported a high genetic homogeneity between berbers and arabs in NW Africa, so they suggested that the Arabisation of this area was probably a cultural phenomenon, which did not imply a replacement of the ancestry population. Our results give support the hypothesis of an early settlement of NW Africa [26]. The original berber population seem to have received a low genetic influx from the surrounding areas. Different hypothesis have been suggested to explain the genetic differentiation of the Moroccan population. An initial genetic drift [26, 30] could have caused differences in allele frequency distribution that have not been re-established due to a certain level of geographic isolation. The Strait of Gibraltar has been described by several authors [9, 10] as an important genetic barrier. Even a certain level of genetic exchange probably occurred between NW Africa and the South of the Iberian Peninsula [10, 27, 32, 33], sharp frequency changes have been described in this area [10, 33]. Also the Sahara desert has been suggested as responsible of the genetic isolation of NW African populations from Sub-Saharan populations [30]. There is no consensus about the impact of the Neolithic demic diffusion in the Mediterranean area [29, 30, 34]. According to our results, a low impact of the Neolithic expansions and/or later migration events on NW African populations would have occurred. Nevertheless, the high genetic homogeneity observed and especially the low level of genetic pairwise differentiations between the Iraqi and the Western Mediterranean populations (such as the East of Spain and South of Italy) supports the impact of several migration events [35] during the history in the genetic constitution of the Mediterranean area, where sailing at the Mediterranean Sea have been an important connection between various geographical areas and populations [33]. The high genetic homogeneity of X-chromosome markers contrasts with the genetic structure of Y-chromosome SNPs [33]. This highlights the differences between male and female migration rates, and stresses the importance of combining results obtained from different types of genetic markers.
Tunisians did not show a significant level of differentiation with northern populations as mentioned by others [3]. A discordant result may be due to the different nature of the markers used in the different works (e.g. mutation rate, recombination, sex specific inheritance). Also the heterogeneity described in Tunisian Berbers [36] could explain that studies carried out using different sets of Tunisian samples may end up showing different results.
More recent demographic events that took place in some particular populations in the area showed interesting exceptions to the general homogeneity. This was the case of the Cosenza and Ibiza populations that were placed relatively "far" from their neighbouring populations in the various statistical analyses performed.
Tagarelli et al [16] described the Calabria province as a collection of many "human genetic isolates". In contrast to Catanzaro and Reggio di Calabria, Cosenza did not suffer destructive telluric events that would have modified its population structure. So, the population heterogeneity inside Cosenza is more patent than in any of the other two populations. This is reflected by the heterogenic distribution of various genetic markers at the coast compared to the internal areas [16]. The results obtained with the 25 X-chromosome markers were in agreement with the previous studies. Cosenza showed not just a relatively high genetic differentiation compared to the populations from Catanzaro and Reggio di Calabria but also a certain displacement in relation to other Mediterranean populations. This was also the case of the Ibiza population. Ibiza, one of the three major islands of the Balearic archipelago, was reproductively isolated for centuries, their population effective size was reduced by infectious diseases and a high number of consanguineous marriages were reported in the 15th-17th centuries [25]. In 1970s, the tourist influx considerably increased the population of Ibiza [23]. Former studies have emphasized the genetic differentiation of this population as a consequence of the stochastic events that happened during its history [15, 23, 25]. Moreover, when a parametric multilocus association analysis was performed, significant values of associations were only observed in the Ibiza and Cosenza populations. In most human populations, high levels of linkage disequilibrium do not exist between markers separated >3 kb [37]. Taking the distance (0.6–145 Mb) between the markers selected into consideration, we did not expect to find significantly increased levels of LD in any of the populations studied. On the other hand, it is well known that different demographic scenarios could result in unexpected levels of LD over large genomic regions [38].
Sex-biased migration rate: a comparative study among different parts of the genome.
Sex-biased migration rates vary between populations as well as through population history. Matrilocality results in a mtDNA geographic structure while patrilocality results in a genetic structure in the non-recombinant region of the Y-chromosome [24]. The X-chromosome, which has spent 2/3 of its history in females, reflects matrilocality to a higher degree than patrilocality.
In the light of our findings it can be said that the migration rate was higher in women than in man, both when our results were analyzed with the Mantel test and under Rousset's isolation-by-distance model. The regression slope obtained from mtDNA (with a negative value) and X-chromosome markers were smaller than those obtained from Y-chromosome SNPs. The negative slope obtained from mtDNA data can be explained by the fact that populations geographically close to each other showed a relatively high genetic distance. In accordance with the findings of other authors [39], the female migration rate was greater than that for males (in this case, in the Mediterranean area). When a much wider area was included (which usually reflects ancient demographic events), the female migration rate was still larger than the male migration rate (data not shown). Thus, we did not find any indication of a change from matrilocality to patrilocality from pre- to post-agricultural societies as it has been suggested by others [24].
Conclusion
In summary, a general genetic homogeneity of the X-chromosome SNPs was observed in a number of populations in the Mediterranean area. The genetic distance between populations in the Middle East and the western part of the Mediterranean area was very low, most likely reflecting the effect of the Neolithic Wave and recent migration events. Only the Moroccan population showed a significant genetic distance from the remaining Mediterranean populations including populations that are geographically close to it, showing the importance of the Strait of Gibraltar as a geographic barrier and supporting the idea of a low impact of the Neolithic demic diffusion and more recent migrations in North-West Africa. In Ibiza and Cosenza populations, interesting differentiations from their neighbouring populations and significant multilocus associations were observed when all the 25 X-chromosome SNPs were analyzed reflecting the particular demographic histories of these populations. A deeper study of LD in these two populations could reveal interesting results for disease association studies.
A higher migration rate was observed in females than in males in both the Mediterranean area and in a wider geographic area showing a greater genetic flow mediated by females than males probably both in pre-agricultural and post-agricultural societies. As it was suggested by other authors [24], patrilocality seems to have been common in post-agricultural populations. According to our results, patrilocality should have also been important in pre-agricultural societies. The negative slope obtained in Rousset's isolation-by-distance model using mtDNA data in the Mediterranean area emphasizes the effect of genetic drift and genetic isolation observed in females in some of the populations in the western part of the Mediterranean Sea.
Methods
Population sampled
We analyzed a total of 1,078 unrelated males from 13 populations (Figure 4). Eleven of these live around the Mediterranean basin: Catanzaro (61 males), Cosenza (37), Reggio di Calabria, (100) and Sicily (119) from the South of Italy; Valencia (60), Ibiza (108) and Majorca (100) from the East of Spain; Tunisia (100); Morocco (89); Turkey (57) and Iraq (63). The last two samples studied as out-groups were from Denmark (93) and Somalia (91). Nearly all DNA samples were purified from blood samples using QIAamp DNA blood mini kit (Qiagen). DNA from Tunisians was investigated with blood collected on FTA cards. The protocols were approved by the Danish local ethical committee (KF-01-037/03).

Geographical location of the populations included in this study. For abbreviators see Figure 1.
For further comparisons, data from males from the International HapMap Project were downloaded (Figure 4): 30 males from "Europe" (CEU: Utah residents with ancestry from Northern and Western Europe), 22 from China (CHB: Han Chinese in Beijing), 22 from Japan (JPT: Japanese in Tokyo) and 30 from Sub-Sahara Africa (YRI: Yoruba in Ibadan, Nigeria). We collected only data from males to make the comparison to our data easy. Depending on the population, data from between 21 and 23 out of the 25 X-chromosome SNPs analyzed in this work were obtained (data from markers X004 and X175 were not found for the CEU group, and results on X142 and X159 were not accessible for any of the four populations).
X-chromosome SNP selection and typing
Twenty-five X-chromosome SNP markers spaced across the chromosome were selected from the NCBI SNP database. The selected markers provide 300 pairwise comparisons over distances from 0.6 to 145 Mbp. Table 4 shows the location of the SNPs chosen. The criteria followed for marker selection were the following: i) High level of polymorphism in different ethnic groups, and ii) not in or close to a coding region, in order to minimize selection phenomena.
A 25-plex PCR reaction was developed in order to amplify all the DNA-fragments with the SNP markers in a single reaction, and a 25-plex minisequencing reaction was also developed. Table 5 shows the sequences of the PCR amplification primers, and Table 6 shows the sequences of the minisequencing primers. The primers were designed from GenBank sequences in the NCBI. The laboratory procedures have been previously described in detail [40]. Briefly, a 25-plex PCR amplification was followed by a multiplex SNaPshot reaction that extends a single base using fluorochrome-labelled ddNTPs. The resulting fluorescent fragments were analyzed by capillary electrophoresis using an ABI 3100 Genetic Analyzer (Applied Biosystems). PCR primers were designed to give amplicon lengths in the range of 62 to 120 bp.
Statistics
Allele and haplotype frequencies were estimated by direct gene counting. Intrapopulational genetic diversity parameters were computed with ARLEQUIN 3.1 [41]. Gene diversity (d), equivalent to the expected heterozygosity for diploid data, was calculated as: , where n is the number of gene copies, k the number of haplotypes and p i the sample frequency of the ith allele at the locus [42]. The significance of an association between pairs of loci was tested using an exact test of significance [41], with 100,000 steps in the Markov chain and 10,000 dememorization steps. The procedure is analogous to Fisher's exact test. The resulting p values were corrected with the step-down Holm-Sidak procedure: p s = 1 - (1 - pi)(n-i+1), where ps is the corrected p value, pi is ith p value of all the values sorted increasingly, and n is the number of pairwise comparisons [43]. The null hypothesis of linkage equilibrium from multilocus data was tested using a parametric method implemented in the LIAN 3.5 software [44]. In order to evaluate the amount of population genetic differentiation, the 13 populations selected for this study and the four populations from the HapMap collection were analyzed. For some of the analyses, the 17 populations were re-organized in two different groups: the "Mediterranean" group, which included 11 populations (Catanzaro, Cosenza, Ibiza, Iraqi, Majorcan, Moroccan, Calabrian, Sicilian, Tunisian, Turkish and Valencia), and the "worldwide" group with nine populations from three continents: Europe (Danes, CEU and Calabrian), Africa (Moroccans, YRI and Somalis) and Asia (Iraqis, CHB and JPT).
An AMOVA analysis with two hierarchical levels was performed using ARLEQUIN 3.1. In this case, Fst was calculated as , where σa2 is the covariance component due to differences among the populations, and σT2 is the total molecular variance calculated as the sum of the covariance components among haplotypes within a population, among haplotypes in different populations and the component originated by differences among populations. We also performed a three hierarchical level AMOVA using ARLEQUIN 3.1 when several groups of population were analyzed. Fst was tested by permuting haplotypes among populations among groups; Fsc was tested among populations within groups; and Fct was tested by permuting populations among groups. Pairwise Fst values [45] were obtained for all population pairs. A principal coordinate plot was drawn from the distance matrix obtained. Negative eigenvalues were corrected using Lingoes method [46] by means of DistPCoA program [47].
Additionally, genetic structure at individual level was investigated using STRUCTURE v. 2.2 [19]. We performed a cluster analysis using the admixture model. A burn-in time of 200,000 steps, followed by another 200,000 steps of the Markov Chain for data collection was used. We carried out five independent replicates for each value of K (predefined number of inferred populations).
A demographic analysis was performed using Rousset's two-dimensional isolation-by-distance model [21] with the ISOLDE program implemented in GENEPOP software version 3.4 [48]. The significance of the Spearman rank correlation coefficient was calculated with 106 Mantel's permutations.
Finally, we carried out a comparison between the results obtained from the 25 X-chromosome SNPs with results obtained on mainly the same populations by using non-recombinant markers (35 SNPs on the Y-chromosome and mtDNA HVRI sequences). We used the results obtained from the analysis of 35 biallelic Y-chromosome SNPs carried out on the same set of samples used for the present work (unpublished data and [49]). We used mtDNA data on HVRI sequences obtained from the literature: Italy [50], Sicily [51], Valencia [15], Ibiza [15], Majorca [15], Tunisia [2], Morocco [52], Turkey [53], Denmark [29], Germany [54], Iraq [29] and Somalia [55].
The sequences between the nucleotide positions 16,024 and 16,366 were analyzed [56]. Due to possible sequencing artefacts, the poly C tract (nucleotide positions from 16,184 to 16,191) was not taken into account for the analysis.
The levels of correlations between the different distance matrices obtained from the three groups of markers (X-chromosome, Y-chromosome and mtDNA) were calculated by means of the Mantel test (ARLEQUIN 3.1 software). Because the correlation between genetic distance matrices can be blurred by the geographic distance between populations [57], we calculated the partial correlations at constant geographical distances (r(set of markers 1, set of markers 2), geographic distance)). The sex-biased migration was studied by comparing the results obtained on the three data sets analyzed under Rousset's isolation-by-distance model [21] as suggested [24].
References
Schaffner SF: The X chromosome in population genetics. Nat Rev Genet. 2004, 5: 43-51. 10.1038/nrg1247.
Plaza S, Calafell F, Helal A, Bouzerna N, Lefranc G, Bertranpetit J, Comas D: Joining the pillars of Hercules: mtDNA sequences show multidirectional gene flow in the western Mediterranean. Ann Hum Genet. 2003, 67 (4): 312-328. 10.1046/j.1469-1809.2003.00039.x.
Quintana-Murci L, Veitia R, Fellous M, Semino O, Poloni ES: Genetic structure of Mediterranean populations revealed by Y-chromosome haplotypes analysis. Am J Phys Anthropol. 2003, 121: 157-171. 10.1002/ajpa.10187.
Capelli C, Redgead N, Romano V, Calì F, Lefranc G, Delague V, Megarbane A, Felice AE, Pascali VL, Neophytou PI, Poulli Z, Novelletto A, Malaspina P, Terrenato L, Berebbi A, Fellous M, Thomas MG, Goldstein DB: Population structure in the Mediterranean Basin: A Y chromosome perspective. Ann Hum Genet. 2006, 70: 207-225. 10.1111/j.1529-8817.2005.00224.x.
Falchi A, Giovannoni L, Calo CM, Piras IS, Moral P, Paoli G, Vona G, Varesi L: Genetic history of some western Mediterranean human isolates through mtDNA HVR1 polymorphisms. J Hum Genet. 2006, 51 (1): 9-14. 10.1007/s10038-005-0324-y.
Francalacci P, Morelli L, Underhill PA, Lillie AS, Passarino G, Useli A, Madeddu R, Paoli G, Tofanelli S, Calò CM, Ghiani ME, Varesi L, Memmi M, Vona G, Lin AA, Oefner P, Cavalli-Sforza LL: Peopling of three Mediterranean islands (Corsica, Sardinia, and Sicily) inferred by Y-chromosome biallelic markers. Am J Phys Anthropol. 2003, 121: 270-279. 10.1002/ajpa.10265.
Belledi M, Poloni ES, Casalotti R, Conterio F, Mikerezi I, Tagliavini J, Excoffier L: Maternal and paternal lineages in Albania and the genetic structure of Indo-European populations. Eur J Hum Genet. 2000, 8: 480-486. 10.1038/sj.ejhg.5200443.
Simoni L, Gueresi P, Pettener D, Barbujani G: Patterns of gene flow inferred from genetic distances in the Mediterranean region. Hum Biol. 1999, 71: 399-415.
Comas D, Calafell F, Benchemsi N, Helal A, Lefranc G, Stoneking M, Batzer MA, Bertranpetit J, Sajantila A: Alu insertion polymorphisms in NW Africa and the Iberian Peninsula: evidence for a strong genetic boundary through the Gibraltar Straits. Hum Genet. 2000, 107: 312-329. 10.1007/s004390000370.
Bosch E, Calafell F, Comas D, Oefner PJ, Underhill PA, Bertranpetit J: High-resolution analysis of human Y-chromosome variation shows a sharp discontinuity and limited gene flow between northwestern Africa and the Iberian Peninsula. Am J Hum Genet. 2001, 68: 1019-1029. 10.1086/319521.
Latini V, Sole G, Doratiotto S, Poddie D, Memmi M, Varesi L, Vona G, Cao A, Ristaldi MS: Genetic isolates in Corsica (France): linkage disequilibrium extension analysis on the Xq13 region. Eur J Hum Genet. 2004, 12: 613-619. 10.1038/sj.ejhg.5201205.
Angius A, Bebbere D, Petretto E, Falchi M, Forabosco P, Maestrale GB, Casu G, Persico I, Melis PM, Pirastu M: Not all isolates are the same: linkage disequilibrium analysis on Xq13.3 reveals different patterns in Sardinian subpopulations. Hum Genet. 2002, 111: 9-15. 10.1007/s00439-002-0753-z.
Jiménez G, Picornell A, Tomàs C, Castro JA, Ramon MM: Y-chromosome polymorphism data in Majorcan, Minorcan and Valencian populations (eastern Spain). Forensic Sci Int. 2001, 124 (2–3): 231-234. 10.1016/S0379-0738(01)00605-3.
Tomàs C, Picornell A, Castro JA, Ramon MM: Genetic analyses in five western Mediterranean populations: variation at five tetrameric short tandem repeat loci. Hum Biol. 2001, 73 (3): 349-363. 10.1353/hub.2001.0042.
Picornell A, Gómez-Barbeito L, Tomàs C, Castro JA, Ramon MM: Mitochondrial DNA HVRI variation in Balearic populations. Am J Phys Anthropol. 2005, 128: 119-130. 10.1002/ajpa.10423.
Tagarelli A, Piro A, Tagarelli G, Zinno F: Color-blindness in Calabria (Southern Italy): a North-South decreasing trend. Am J Hum Biol. 2000, 12: 17-24. 10.1002/(SICI)1520-6300(200001/02)12:1<17::AID-AJHB3>3.0.CO;2-D.
Ardlie KG, Kruglyak L, Seielstad M: Paterns of linkage disequilibrium in the human genome. Nat Rev Genet. 2002, 3: 299-309. 10.1038/nrg777.
Bowcock AM, Ruiz-Linares A, Tomfohrde J, Minch E, Kidd JR, Cavalli-Sforza LL: High resolution of human evolutionary trees with polymorphic microsatellites. Nature. 1994, 368 (6470): 455-457. 10.1038/368455a0.
Pritchard JK, Stephens M, Donnelly P: Inference of population structure using multilocus genotype data. Genetics. 2000, 155: 945-959.
Wilson JF, Weale ME, Smith AC, Gratix F, Fletcher B, Thomas MG, Bradman N, Goldstein DB: Population genetic structure of variable drug response. Nat Genet. 2001, 29: 265-269. 10.1038/ng761.
Rousset F: Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics. 1997, 145: 1219-1228.
Koizumi I, Yamamoto S, Maekawa K: Decomposed pairwise regression analysis of genetic and geographic distances reveals a metapopulation structure of stream-dwelling Dolly Varden charr. Mol Ecol. 2006, 15: 3175-3189. 10.1111/j.1365-294X.2006.03019.x.
Picornell A, Miguel A, Castro JA, Misericordia Ramon M, Arya R, Crawford MH: Genetic variation in the population of Ibiza (Spain): genetic structure, geography, and language. Hum Biol. 1996, 68 (6): 899-913.
Wilkins JF, Marlowe FW: Sex-biased migration in humans: what should we expect from genetic data?. BioEssays. 2006, 28: 290-300. 10.1002/bies.20378.
Tomàs C, Jiménez G, Picornell A, Castro JA, Ramon MM: Differential maternal and paternal contributions to the genetic pool of Ibiza Island, Balearic Archipelago. Am J Phys Anthropol. 2006, 129 (2): 268-278. 10.1002/ajpa.20273.
Bosch E, Calafell F, Pérez-Lezaun A, Clarimón J, Comas D, Mateu E, Martínez-Arias Rosa, Morera B, Brakez Z, Akhayat O, Sefiani A, Hariti G, Cambon-Thomsen A, Bertranpetit J: Genetic structure of north-west Africa revealed by STR analysis. Eur J Hum Genet. 2000, 8: 360-366. 10.1038/sj.ejhg.5200464.
Kandil M, Moral P, Esteban E, Autori L, Mameli GE, Zaoui D, Calo C, Luna F, Vacca L, Vona G: Red cell enzyme polymorphisms in Moroccans and Southern Spaniards: new data for the genetic history of the Western Mediterranean. Hum Biol. 1999, 71 (5): 791-802.
Harich N, Esteban E, Chafik A, López-Alomar A, Vona G, Moral P: Classical polymorphisms in Berbers from Moyen Atlas (Morocco): genetics, geography, and historical evidence in the Mediterranean peoples. Ann Hum Biol. 2002, 29 (5): 473-487. 10.1080/03014460110104393.
Richards M, Macaulay V, Hickey E, Vega E, Sykes B, Guida V, Rengo C, Sellitto D, Cruciani F, Kivisild T, Villems R, Thomas M, Rychkov S, Rychkov O, Rychkov Y, Gölge M, Dimitrov D, Hill E, Bradley D, Romano V, Calì F, Vona G, Demaine A, Papiha S, Triantaphyllidis C, Stefanescu G, Hatina J, Belledi M, Di Rienzo A, Novelletto A, Oppenheim A, Nørby S, Al-Zaheri N, Santachiara-Benerecetti S, Scozzari R, Torroni A, Bandelt HJ: Tracing European founder lineages in the Near Eastern mtDNA pool. Am J Hum Genet. 2000, 67: 1251-1276.
Arredi B, Poloni ES, Paracchini S, Zerjal T, Fathallah DM, Makrelouf M, Pascali VL, Novelletto A, Tyler-Smith C: A predominantly Neolithic origin for Y-chromosomal DNA variation in North Africa. Am J Hum Genet. 2004, 75: 338-345. 10.1086/423147.
Watson E, Forster P, Richards M, Bandelt HJ: Mitochondrial footprints of human expantions in Africa. Am J Hum Genet. 1997, 61 (3): 691-704. 10.1086/515503.
Scozzari R, Cruciani F, Pangrazio A, Santolamazza P, Vona G, Moral P, Latini V, Varesi L, Memmi MM, Romano V, De Leo G, Gennarelli M, Jaruzelska J, Villems R, Parik J, Macaulay V, Torroni A: Human Y-chromosome variation in the Western Mediterranean area: implications for the peopling of the region. Hum Immunol. 2001, 62: 871-884. 10.1016/S0198-8859(01)00286-5.
Malaspina P, Cruciani F, Santolamazza P, Torroni A, Pangrazio A, Akar N, Bakalli V, Brdicka R, Jaruzelska J, Kozlov A, Malyarchuk B, Mehdi SQ, Michalodimitrakis E, Varesi L, Memmi MM, Vona G, Villems R, Parik J, Romano V, Stefan M, Stenico M, Terrenato L, Novelletto A, Scozzari R: Patterns of male-specific inter-population divergence in Europe, West Asia and North Africa. Ann Hum Genet. 2000, 64: 395-412. 10.1046/j.1469-1809.2000.6450395.x.
Menozzi P, Piazza A, Cavalli-Sforza L: Synthetic maps of human frequencies in Europeans. Science. 1978, 204 (4358): 786-792. 10.1126/science.356262.
Semino O, Magri C, Benuzzi G, Lin AA, Al-Zahery N, Battaglia V, Maccioni L, Triantaphyllidis C, Shen P, Oefner PJ, Zhivotovsky LA, King R, Torroni A, Cavalli-Sforza LL, Underhill PA, Santachiara-Benerecetti S: Origin, diffusion, and differentiation of Y-chromosome haplogroups E and J: inferences on the Neolithization of Europe and later migratory events in the Mediterranean area. Am J Hum Genet. 2004, 74: 1023-1034. 10.1086/386295.
Fadhlaoui-Zid K, Plaza S, Calafell F, Ben Amor M, Comas D, Bennamar El, gaaied A: Mitochondrial DNA heterogeneity in Tunisian Berbers. Ann Hum Genet. 2004, 68: 222-233. 10.1046/j.1529-8817.2004.00096.x.
Kruglyak L: Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet. 1999, 22: 139-144. 10.1038/9642.
Wilson JF, Goldstein DB: Consistent long-range linkage disequilibrium generated by admixture in a Bantu-semitic hybrid population. Am J Hum Genet. 2000, 67: 926-935. 10.1086/303083.
Seielstad MT, Minch E, Cavalli.Sforza LL: Genetic evidence for a higher female migration rate in humans. Nat Genet. 1998, 20: 278-280. 10.1038/3088.
Sanchez JJ, Børsting C, Morling N: Typing of Y chromosome SNPs with multiplex PCR methods. Methods Mol Biol. 2005, 297: 209-228.
Excoffier L, Laval G, Schneider S: Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online. 2005, 1: 47-50.
Nei M: Molecular evolutionary genetics. 1987, Columbia University Press, New York, NY, USA
Sokal RR, Rohlf FJ: Biometry: the principle and practice of statistics in biological research. 1995, WH Freeman, New York
Haubold H, Hudson RR: LIAN 3.0: detecting linkage disequilibrium in multilocus data. Bioinformatics. 2000, 16: 847-848. 10.1093/bioinformatics/16.9.847.
Reynolds J, Weir BS, Cockerham CC: Estimation for the coancestry coefficient: a basis for short term genetic distance. Genetics. 1983, 105: 767-779.
Legendre P, Anderson MJ: Distance-based redundancy analysis: testing multi-species responses in multi-factorial ecological experiments. Ecol Monogr. 1999, 69 (1): 1-24.
Legendre P, Anderson MJ: Program DistPCoA. Département de sciences biologiques, Université de Montréal. 1998, 10 pages
Raymond M, Rousset F: GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Heredity. 1995, 86: 248-249.
Sanchez JJ, Hallenberg C, Børsting C, Hernandez A, Morling N: High frequencies of Y chromosome lineages characterized by E3b1, DYS19-11, DYS392-12 in Somali males. Eur J Hum Gent. 2005, 13: 856-866. 10.1038/sj.ejhg.5201390.
Tagliabracci A, Turchi C, Buscemi L, Sassaroli C: Polymorphism of the mitochondrial DNA control region in Italians. Int J Legal Med. 2001, 114: 224-228. 10.1007/s004140000168.
Calì F, Le Roux MG, D'Anna R, Flugy A, De Leo G, Chiavetta V, Ayala GF, Romano V: MtDNA control region and RFLP data for Sicily and France. Int J Legal Med. 2001, 114: 229-231. 10.1007/s004140000169.
Brakez Z, Bosch E, Izaabel H, Akhayat O, Comas D, Bertranpetit J, Calafell F: Human mitochondrial DNA sequence variation in the Moroccan population of the Souss area. Ann Hum Biol. 2001, 28: 295-307. 10.1080/030144601300119106.
Comas D, Calafell F, Mateu E, Pérez-Lezaun A, Bertranpetit J: Geographic variation in human mitochondrial DNA control region sequence: the population history of Turkey and its relationship to the European populations. Mol Biol Evol. 1996, 13 (8): 1067-1077.
Lutz S, Weisser HJ, Heizmann J, Pollak S: Location and frequency of polymorphic positions in the mtDNA control region of individuals from Germany. Int J Legal Med. 1998, 11: 67-77. 10.1007/s004140050117.
Watson E, Bauer K, Aman R, Weiss G, Haeseler A, Pääbo S: mtDNA sequence diversity in Africa. Am J Hum Genet. 1996, 59: 437-444.
Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG: Sequence and organization of the human mitochondrial genome. Nature. 1981, 290 (5806): 457-465. 10.1038/290457a0.
Kremer A, Kleinschmit J, Cottrell J, Cundall EP, Dean JD, Ducousso A, Köniig AO, Lowe AJ, Munro RC, Petit RJ, Stephan BR: Is there a correlation between chloroplastic and nuclear divergence, or what are the roles of history and selection on genetic diversity in European oaks?. For Ecol Manage. 2002, 156: 75-87. 10.1016/S0378-1127(01)00635-1.
Acknowledgements
Carmen Tomas is a postdoc at the Section of Forensic Genetics, Department of Forensic Medicine, Faculty of Health Sciences, University of Copenhagen, Denmark. The work was supported by grants from Ellen and Aage Andersen's Foundation and the European Commission (GROWTH Programme, SNPforID project, contract G6RD-CT-2002-00844).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
CT was primarily responsible for the design of the study, carried out the SNP typing, performed the statistical analysis and elaborated the manuscript. JJS participated in designing the study and the manuscript. AB participated in designing the study and the manuscript, and sent samples from South Italy and Sicily. CBC participated in designing the study and the manuscript, and sent samples from Tunisia. AH participated in designing the study and the manuscript, and sent samples from Morocco. MBD participated in designing the study and the manuscript, and sent samples from Tunisia. MR participated in designing the study and the manuscript, and sent samples from Spain. NM participated in designing the study, analyzing the results and preparing the manuscript. All authors read and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Tomas, C., Sanchez, J.J., Barbaro, A. et al. X-chromosome SNP analyses in 11 human Mediterranean populations show a high overall genetic homogeneity except in North-west Africans (Moroccans). BMC Evol Biol 8, 75 (2008). https://doi.org/10.1186/1471-2148-8-75
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-8-75