
Abstract
Background
The evolutionary history of the Asian colobines is less understood. Although monophyly of the odd-nosed monkeys was recently confirmed, the relationships among the langur genera Presbytis, Semnopithecus and Trachypithecus and their position among Asian colobines remained unclear. Moreover, in Trachypithecus various species groups are recognized, but their affiliations are still disputed. To address these issues, mitochondrial and Y chromosomal sequence data were phylogenetically related and combined with presence/absence analyses of retroposon integrations.
Results
The analysed 5 kb fragment of the mitochondrial genome allows no resolution of the phylogenetic relationships among langur genera, but five retroposon integrations were detected which link Trachypithecus and Semnopithecus. According to Y chromosomal data and a 573 bp fragment of the mitochondrial cytochrome b gene, a common origin of the species groups T. [cristatus], T. [obscurus] and T. [francoisi] and their reciprocal monophyly is supported, which is also underpinned by an orthologous retroposon insertion. T. [vetulus] clusters within Semnopithecus, which is confirmed by two retroposon integrations. Moreover, this species group is paraphyletic, with T. vetulus forming a clade with the Sri Lankan, and T. johnii with the South Indian form of S. entellus. Incongruence between gene trees was detected for T. [pileatus], in that Y chromosomal data link it with T. [cristatus], T. [obscurus] and T. [francoisi], whereas mitochondrial data affiliates it with the Semnopithecus clade.
Conclusion
Neither relationships among the three langur genera nor their position within Asian colobines can be settled with 5 kb mitochondrial sequence data, but retroposon integrations confirm at least a common origin of Semnopithecus and Trachypithecus. According to Y chromosomal and 573 bp mitochondrial sequence data, T. [cristatus], T. [obscurus] and T. [francoisi] represent true members of the genus Trachypithecus, whereas T. [vetulus] clusters within Semnopithecus. Due to paraphyly of T. [vetulus] and polyphyly of Semnopithecus, a split of the genus into three species groups (S. entellus - North India, S. entellus - South India + T. johnii, S. entellus - Sri Lanka + T. vetulus) seems to be appropriate. T. [pileatus] posses an intermediate position between both genera, indicating that the species group might be the result of ancestral hybridization.
Similar content being viewed by others


Background
The Old World monkeys are traditionally divided into the two subfamilies Cercopithecinae and Colobinae, which differ from each other by numerous morphological, behavioural and ecological characteristics [1–4]. While detailed information on the evolutionary history of cercopithecines (baboons, mangabeys, macaques and guenons) is at hand, knowledge on colobines is still scarce. Although some molecular studies [5–13] exist, they mainly focus on relationships within genera or species groups and not on the general phylogeny of the subfamily. Recently, a first mitochondrial phylogeny of colobine genera was established [14], which confirmed some previous assumptions, but also led to confusions, calling for further research to definitively elucidate their evolutionary history.
Based on distribution and morphology, the colobines are traditionally divided into an African and an Asian clade [3, 4], while Asian colobines are more diverse than their African relatives. Hence, the Asian forms are further split into the odd-nosed monkey (Pygathrix, Rhinopithecus, Nasalis, Simias) and langur (Presbytis, Trachypithecus, Semnopithecus) groups, which are both believed to be monophyletic. Accordingly, langurs were originally combined in the single genus Presbytis [15–17] or Semnopithecus [18], but based on neonatal colouration and cranial morphology, they were split into the three genera Semnopithecus, Trachypithecus and Presbytis [19], and a fourth genus (Kasi) was added [20]. Alternatively, Semnopithecus was separated from Presbytis, with Trachypithecus forming a subgenus of the former [21–23], but recent classifications use a subdivision of langurs into the three genera Presbytis, Trachypithecus and Semnopithecus [3, 4, 24–26].
Within the different langur genera, several species are recognized, which are lumped into species groups due to similar fur colouration, behaviour, ecology or distribution. With five species groups (T. [obscurus], T. [francoisi], T. [cristatus], T. [pileatus] and T. [vetulus]) [4], the genus Trachypithecus is the most diverse of all langurs and posses also the widest distribution, ranging from South India and Sri Lanka through mainland Southeast Asia to the Sundaland (Fig. 1, Fig. 2). Although all of them are morphologically similar, T. [vetulus] was sometimes separated in its own genus Kasi [20], and recent mitochondrial sequence data indicate a closer affiliation of T. [vetulus] and T. [pileatus] to Semnopithecus than to Trachypithecus [7, 9]. Accordingly, the two T. [vetulus] members were recognized as species of Semnopithecus [26]. In contrast to Trachypithecus, the genus Semnopithecus is restricted to the Indian subcontinent (Fig. 1) and traditionally regarded as monotypic with the only species S. entellus [3, 27], although recently several subspecies were elevated to species status [4]. The third langur genus, Presbytis, includes several species, which occur solely in the Sundaland, but are not lumped into distinct species groups [4].

Distribution of the genus Semnopithecus and Trachypithecus [ obscurus ]. Genus affiliations and species groups after Groves [4].

Distribution of the Trachypithecus species groups T . [ vetulus ], T . [ pileatus ], T . [ francoisi ] and T . [ cristatus ]. Genus affiliations and species groups after Groves [4].
The phylogenetic relationships among the different Asian colobine genera are disputed. Although a common origin of the odd-nosed monkeys was recently confirmed [14], evidence for monophyly of its putative sister clade, the langur group, is still lacking. Moreover, available data depict Trachypithecus and Presbytis as sister taxa to the exclusion of Semnopithecus [14], which contradicts with traditional classifications, in which Trachypithecus and Semnopithecus are believed to form a clade to the exclusion of Presbytis [4, 21–23]. These findings raise the question of what positions the langur genera occupy among Asian colobines and whether the langurs form a monophyletic clade in general. Moreover, the affiliations of different Trachypithecus species groups, especially T. [vetulus] and T. [pileatus], are disputed, and hence, led to different classifications. Currently, only few genetic data are available [7, 9, 28], so that further information from other markers is required to definitively establish their relationships.
To address all these issues, mitochondrial and Y chromosomal sequence data were phylogenetically related and combined with presence/absence analysis of retroposon integrations. This approach was used to simultaneously analyse paternal-, maternal- and biparental-inherited markers, which allow the detection of incongruence between different gene trees indicating possible hybridization or introgression events between different lineages [29–31]. To determine the phylogenetic position of the langur genera among Asian colobines, a 5 kb fragment of the mitochondrial genome was sequenced from eight colobine genera, and combined with presence/absence analysis of retroposon integrations. Retroposon insertion events are nearly homoplasy-free and precise excision of elements is highly unlikely [32, 33]. Accordingly, retroposon insertions are powerful informative markers, which were already successfully applied to elucidate phylogenetic relationships in various primate lineages [34–38]. To study relationships among different langur species groups and their genus affiliations, a 573 bp fragment of the mitochondrial cytochrome b gene and a 777 bp portion of the SRY (sex-determining region, Y chromosome) gene was sequenced from at least one representative per species group, and complemented with retroposon analysis.
Results and Discussion
Genus level phylogeny
To elucidate the phylogenetic relationships among the different langur genera and their position among Asian colobines, mitochondrial sequence studies were combined with presence/absence analysis of retroposon insertions.
The herein analysed 5 kb fragment of the mitochondrial genome was assembled from sequences derived from 1–2 kb long and partly overlapping PCR products, whereby no inconsistencies in overlapping sequence fragments were detected. As template material, mainly DNA extracted from feces was used, which minimizes the amplification of nuclear pseudogenes [39], and comparisons of the data with sequences already deposited at GenBank revealed only intra-species or -generic variation, indicating that no nuclear pseudogenes were amplified.
To determine the phylogenetic relationships among analysed genera, tree reconstructions were conducted with different algorithms, which all led to the same tree topology (Fig. 3). Moreover, most relationships are significantly supported and congruent with previous classifications [1–4], indicating the reliability of the data set. In detail, the reconstructions confirm relationships among cercopithecine genera (Macaca, Papio, Chlorocebus), and reciprocal monophyly of cercopithecines and colobines, as well as of African and Asian colobines. Within the Asian clade, relationships are not resolved, although at least a common origin of the odd-nosed monkeys is depicted. Neither the assumed monophyly of the langur genera, nor the expected close affinity of Trachypithecus and Semnopithecus or the recently indicated sister grouping of Trachypithecus and Presbytis [14] can be verified with significance. Moreover, all alternative tree topologies, in which the three langur genera were regarded as monophyletic, or variously recognized as sister clades to each other, to the odd-nosed monkey clade, or even as basal among Asian colobines, were not rejected (P = 0.097 – 0.776). Accordingly, an unresolved polytomy among the three langur genera and the odd-nosed monkey clade may best reflect the relationships among Asian colobines based on the mitochondrial data.

Phylogenetic relationships among colobines and related genera based on mitochondrial data. Numbers on nodes indicate support values >80% (first: ML, second: NJ, third: MP).
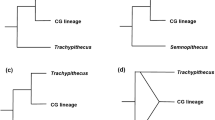
Several retroposon insertions were detected, which provide insights into the branching pattern of colobines (Fig. 4). Together with seven already published integrations [38], now 14 loci confirm monophyly of the Asian colobines to the exclusion of the African genus Colobus. Among Asian colobines, one insertion indicates a common origin of the odd-nosed monkeys, which is verified by three further loci [38]. Most prominent are the five retroposon integrations, which confirm a sister grouping of Trachypithecus and Semnopithecus. No loci were detected, which link Presbytis either with the other two langur genera or the odd-nosed monkeys.

Phylogenetic relationships among Asian colobine genera based on retroposon integrations. Dark dots represent new generated data, whereas light dots refer to already published data [37]. Numbers in dots indicate single integration events.
Although in general the mitochondrial data are suitable to elucidate relationships among the different genera, as indicated by the correct and significantly supported branching patterns among all other studied genera, the relationships among the langurs are unresolved, which is concordant with previous results [14]. In contrast, the presence/absence analysis of retroposon integrations provides evidence for a monophyletic odd-nosed monkey clade and a common origin of Trachypithecus and Semnopithecus, which is in agreement with morphological hypotheses [1, 3, 4, 21–23]. Regardless which markers were used, the phylogenetic position of Presbytis among Asian colobines and accordingly the unity of the langurs remains unclear and needs further investigations.
Species group phylogeny
In order to settle affiliations among the different Trachypithecus species groups and their members, mitochondrial and Y chromosomal sequence data were combined with information on retroposon integrations.
The mitochondrial phylogeny was established on the basis of 573 bp long cytochrome b gene sequences, generated from most species recognized in the genus and its sister genus Semnopithecus. In all tree reconstructions, identical relationships were obtained, with most branches being significantly supported (Fig. 5a). Accordingly, the different species groups are divided into two major groups, with one including solely groups of the genus Trachypithecus, whereas the second one includes representatives of Trachypithecus and Semnopithecus. In the mixed clade, T. [vetulus] and T. [pileatus] members are lumped together with S. entellus. Whereby T. [pileatus] is monophyletic, the members of T. [vetulus] are paraphyletic, with T. vetulus forming a sister clade to S. entellus from Sri Lanka, and T. johnii with S. entellus from South India. Furthermore, a fourth lineage in the mixed clade was detected, which is represented by S. entellus from North India. In contrast, the three species groups (T. [obscurus], T. [francoisi], T. [cristatus]) in the clade comprising solely Trachypithecus groups are all monophyletic. Alternative relationships, in which T. [vetulus] is recognized as monophyletic, either T. [vetulus] or T. [pileatus] belongs to Trachypithecus or even both are members of Trachypithecus, were tested, but all of them were rejected (P < 0.05).

Phylogenetic relationships among Semnopithecus and Trachypithecus species groups based on a) mitochondrial data, b) Y chromosomal data, and c) retroposon integrations. Numbers on nodes indicate support values (first: ML, second: NJ, third: MP), and boxed species belong to a species group, with species in bold giving the name of the group.
With some exceptions, the Y chromosomal data provide a similar picture (Fig. 5b), but due to the low number of polymorphic sites, support values are in general not as high as in the mitochondrial tree. According to the reconstructions, the species groups are divided into two major clades, with one comprising T. [obscurus], T. [cristatus], T. [francoisi] and T. [pileatus], and the other, T. [vetulus] and Semnopithecus. Relationships among the latter are not resolved. All alternative tree topologies, in which either T. [vetulus] belongs to Trachypithecus or T. pileatus groups with Semnopithecus, were rejected (P < 0.05).
Retroposon insertions further deepened our knowledge on the species group relationships. Altogether, three informative integrations were analysed (Fig. 5c), with one occurring in T. [obscurus], T. [cristatus] and T. [francoisi], and the other two in T. [vetulus] and Semnopithecus. Interestingly, all three integrations are absent in T. pileatus.
With the exception of the varying position of T. [pileatus], the affiliations of the remaining species groups are congruent among different gene trees. Accordingly, all analysed markers relate T. [vetulus] with Semnopithecus, indicating that this species group is a real member of the genus Semnopithecus and not of Trachypithecus as assumed by morphological similarities [4]. These similarities may be the results of adaptations to similar ecological conditions (Semnopithecus is semi-terrestrial and lives in deciduous forest, whereas Trachypithecus including T. [vetulus] is arboreal and occurs in wet evergreen forests). Although the Y chromosomal data allow no resolution within the Semnopithecus – T. [vetulus] clade, the mitochondrial data indicate paraphyly of the two T. [vetulus] species, with T. vetulus clustering with S. entellus from Sri Lanka and T. johnii with S. entellus from South India, which is concordant with their geographical distribution. These findings indicate paraphyly of S. entellus, whereby North Indian representatives form a further distinct lineage. Accordingly, the langurs of the Indian subcontinent should be split into three species groups, with one occurring solely on Sri Lanka, one in Southern India and a third one in Northern India, whereas the Gondavari river seems to be barrier between the latter two.
Monophyly of each of T. [obscurus], T. [cristatus] and T. [francoisi] and their close affiliation is depicted in all gene trees, so that all of them can be regarded as true members of Trachypithecus. These findings confirm previous molecular studies [8–11, 13] and are in general agreement with recent classifications [4, 26].
The only discrepancies between different gene trees were obtained for T. [pileatus]. Whereas the mitochondrial data link the species group with Semnopithecus and T. [vetulus], the Y chromosomal data affiliates it with T. [obscurus], T. [cristatus] and T. [francoisi]. These findings might be explained by incomplete lineage sorting of ancestral mitochondrial or Y chromosomal haplotypes. Accordingly, the ancestor of Trachypithecus, Semnopithecus and T. [pileatus] carried multiple DNA lineages with one lineage being randomly fixed in two taxa, but not in the third. Alternatively, the varying position of T. [pileatus] in different gene trees might be explained by past hybridization between Semnopithecus and Trachypithecus. As depicted by the three retroposon insertions, this putative hybridization event would have occurred between ancestral forms of Semnopithecus and Trachypithecus, before both genera diverged into distinct species groups. The hybridization hypothesis is also supported by some intermediate morphological characteristics [4] and the distribution of T. [pileatus], which is sandwiched between those of Semnopithecus and other Trachypithecus species groups (Fig. 1, Fig. 2).
Conclusion
The present study provides detailed insights into the evolutionary history of Asian colobines and underpins the tremendous power of retroposon integrations as cladistic markers. Although mitochondrial data proved to be useful to elucidate and confirm several relationships among studied taxa, the data set failed to resolve the affiliations among the langur genera and to settle their position among Asian colobines. In contrast, retroposon insertions provided clear evidence for a sister grouping of Semnopithecus and Trachypithecus, but no integrations were detected, which link Presbytis either with the other two langur genera or with the odd-nosed monkeys, so that further research is required to solve this issue. Moreover, to definitively explain the evolutionary history of colobines, further molecular markers should be analysed, especially regarding possible discrepancies among gene trees due to hybridization or introgression, as such events are important speciation mechanisms in primates [31], and as it was possibly detected in the present study in the case of T. [pileatus].
Although further investigations are necessary to fully understand the evolution of colobines, the present study provides sufficient data to revise the current classification of the genera Trachypithecus and Semnopithecus (Table 1). Accordingly, within Trachypithecus, three reciprocal monophyletic species groups (T. [obscurus], T. [cristatus] and T. [francoisi]) should be recognized, whereas T. [vetulus] should be included in Semnopithecus. Moreover, due to their paraphyletic origin, T. vetulus and T. johnii should not be lumped into a single species group, and S. entellus should not be regarded as monotypic. A classification into three species groups with one occurring in Northern India, one in Southern India and one on Sri Lanka may best reflect the evolutionary relationships among the langurs of the Indian subcontinent. However, this arrangement is tentative, because further research is required to confirm their distinctiveness not only on mitochondrial, but also on nuclear DNA level. T. [pileatus] might be the result of an ancient hybridization event between Semnopithecus and Trachypithecus, and hence, its classification is difficult. While a separation in its own distinct genus may be appropriate, we provisionally accept its traditional recognition as member of Trachypithecus.
Methods
Sample collection, DNA extraction and preventing contaminations
The species analysed in this study are presented in Table 2. All study specimens were identified by fur colouration and other external characteristics. Hanuman langur (S. entellus) samples were collected only from founder animals, of which the area of capture was at least roughly known. Total genomic DNA was extracted from blood, tissue or feces using the DNeasy or Stool Mini Kits from Qiagen. When hair follicle cells were used, 1–3 hairs were directly implemented into the PCR reaction after they were washed with sterile water and 95% ethanol. To prevent contaminations, sample collection and laboratory procedures followed described standard protocols [40–43]. In detail, all fecal and hair samples were collected with gloves and stored in sterile tubes or plastic bags before further processing. DNA extraction, PCR, gel extraction and sequencing was performed in separate laboratories and repeated after several months, while always only one individual per species was tested. Moreover, from most specimens two different sample types were available, which both were used as template material. Sequences from independent analyses were identical. Finally, all PCR reactions were performed with negative (distilled water) controls.
Mitochondrial sequence analysis
To determine the phylogenetic position of langur genera among Asian colobines, a ~5 kb fragment of the mitochondrial genome was sequenced from all colobine genera with the exception of Simias and Procolobus. This region spans the cytochrome b gene, the control region, the 12S rDNA and 16S rDNA, and the intermediate tRNAs. To exclude contaminations of the dataset with nuclear pseudogenes, mainly DNA extracted from feces was used as template material, and ~1–2 kb long and ~200–400 bp overlapping fragments were amplified. PCR products were generated via hot-start technique using a set of 24 primers (Table 3) and PCR conditions comprising a pre-denaturation step at 94°C for 2 min, followed by 40 cycles at 94°C for 1 min, 60°C for 1 min and 72°C for 1–2 min, and a final extension step at 72°C for 5 min. The results of the PCR amplifications were checked on agarose gels. PCR products were cleaned with the Qiagen PCR Purification Kit and subsequently sequenced on an ABI 3100-Avant sequencer using the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems). To obtain a comprehensive overview on the phylogeny of colobines, the dataset was expanded with further sequences from related taxa deposited at GenBank (Table 2). Accordingly, the final dataset comprised 15 taxa, including eight colobines, four cercopithecines, two hominoides and a New World monkey, which was used as outgroup taxon. Sequences were aligned with ClustalW [44] and subsequently checked by eye. Gaps and poorly aligned positions were removed with the G-blocks software [45], which reduced the final dataset to 4336 bp. Based on this alignment, phylogenetic trees were constructed with the maximum-parsimony (MP), neighbor-joining (NJ) and maximum-likelihood (ML) algorithms as implemented in PAUP 4.0b10 [46] and TREEPUZZLE 5.0 [47]. For MP analyses, all characters were treated as unordered and equally weighted throughout. A heuristic search was performed with the tree-bisection-reconnection (TBR) algorithm with random addition of sequences. The maximum number of trees was set to 100. NJ and ML trees were constructed with the GTR + I (= 0.2526) + Γ (= 0.4816) model of sequence evolution as it was selected as best-fitting model under the Akaike information criterion with MODELTEST 3.06 [48]. Relative support of internal nodes was performed by bootstrap analyses with 1,000 replications (MP, NJ), or by the quartet puzzling support values on the basis of 1,000 puzzling steps (ML). Finally, to evaluate the reliability of the depicted phylogenetic position of the langur genera among Asian colobines, alternative tree topologies were evaluated with the Kishino-Hasegawa [49] and Shimodaira-Hasegawa [50] tests in PAUP, both performed with full optimization and 1,000 bootstrap replications. Therefore, all three langur genera were regarded as monophyletic or variously recognized as sister clade to each other, to the odd-nosed monkey clade or even as sister clade to all other Asian colobines.
To determine phylogenetic affiliations of species groups, a 573 bp long fragment of the mitochondrial cytochrome b gene was analysed from all species of the different groups (Table 2). The generation of sequences followed laboratory methods as described [9, 11, 13]. The final alignment, which was easily generated by eye due to the absence of insertions or deletions, comprised 19 individuals including the outgroup taxon Presbytis melalophos. Phylogenetic trees were constructed as described above. As best-fitting model, MODELTEST selected the TIM + I (= 0.5977) + Γ (= 2.3137) model, which was applied for NJ and ML reconstructions. As for the 5 kb fragment, several alternative tree topologies, in which T. [vetulus] is recognized as monophyletic, either T. [vetulus] or T. [pileatus] belongs to Trachypithecus, or even both are members of Trachypithecus, were tested.
Y chromosomal sequence analysis
The SRY gene was selected as it represents a single-copy gene and as it is proved to be informative in reconstructing the Y chromosomal evolutionary history of macaques [51]. PCR conditions and primers were applied as described [51]. To amplify the SRY gene from fecal material, two overlapping fragments were generated with published primers [51] and the newly generated internal primers 5'-TGGGCGGAGTTGAGAGGGGT-3' and 5'-TAGCGGTCCCGTTGCTGCGG-3'. The final alignment of 777 bp comprised 15 taxa representing all species groups of the genera Semnopithecus and Trachypithecus as well as Presbytis melalophos, which was used as outgroup. To reconstruct NJ and ML trees, the K80 model of sequence evolution, determined with MODELTEST, was used. MP trees were generated as described above. The reliability of the depicted position of T. [vetulus] and T. [pileatus] was tested in PAUP by using alternative tree topologies, in which either T. [vetulus] belongs to Trachypithecus or T. pileatus groups with Semnopithecus.
Retroposon analysis
Due to their high copy number and relative small size (~300 bp), the primate specific Alu elements were selected as cladistic markers. The presence or absence of Alu elements in different colobines at specific loci was tested via PCR using primers occupying the flanking region of the insertion site. Details on analysed loci, primers and studied species are listed in Table 4.
To detect new loci, a subtractive hybridization approach [52] was performed with some modifications. As tracer and driver, different colobine genera were selected. Genomic DNA of tracer and driver was digested with RSAI (Fermentas), and subsequently, the adapters AdapA1/AdapAA1 (5'-TGTAGCGTGAAGACGACAGAAAGGGCGTGGTGCGGAGGGCGGT-3'/5'-ACCGCCCTCCG-3') and AdapA2/AdapAA2 (5'-TGTAGCGTGAAGACCTGTCTTAGGGCGTGGTGGCCAGGGCCGT-3'/5'-ACGGCCCTGGC-3') were ligated to the tracer fragments. Each of ~15 ng tracerA1 and tracerA2 were hybridized with ~1,500 ng driver for 20 h at 60°C. 2 μl of the hybridization result was used as template to amplify solely tracer fragments using the adapter primers A1 (5'-TGTAGCGTGAAGACGACAGAA-3') and A2 (5'-TGTAGCGTGAAGACCTGTCTT-3'). The PCR program consisted of a pre-extension step at 72°C for 6 min to fill in adaptor ends, followed by 25 cycles, each with a denaturation step at 95°C for 1 min, annealing at 60°C for 1 min and extension at 72°C for 2 min. To enrich fragments with Alu insertions, a semi-nested PCR was added using either primer A1 or A2 and the Alu-specific AluY (5'-GGAGAATGGCGTGAACCCGGGA-3') oligonueclotide. The PCR products were separated on agarose gels and fragments over 500 bp were excised from the gel. After purification, the fragments were cloned into the pGEMTeasy vector (Promega) and transformed into electro-competent TOP10 cells (Invitrogen). Bacterial clones were collected in 96-well microtiter plates and re-screened via PCR with the primers A1 or A2 and AluY. Positive clones were sequenced and analysed with REPEATMASKER and BLAST as implemented in NCBI and EMBL. Based on the generated alignments, locus-specific primers were constructed, with the forward primer occupying a region 5'-end upstream of the insertion site, which is conserved among the colobine and human or chimp sequences. Due to the absence of the 3'-end downstream sequence of the tested colobine species, reverse primers were constructed solely on the basis of human or chimp sequences. Subsequently, the presence or absence of respective Alu insertions in different colobine species was tested via PCR. The orthology of insertions was confirmed by sequencing of at least one species per genus or species group. Sequences were deposited at GenBank and are available under the accession numbers EU004484–EU004537 and EU006662–EU006692.
Abbreviations
Species names in square brackets ([]) indicate species groups.
References
Napier JR, Napier PH: Old World monkeys. 1970, New York: Academic Press
Szalay FS, Delson E: Evolutionary history of the primates. 1979, New York: Academic Press
Davies AG, Oates JF: Colobine monkeys: their ecology, behaviour, and evolution. 1994, Cambridge: Cambridge University Press
Groves CP: Primate Taxonomy. 2001, Washington: Smithsonian Institution Press
Rosenblum LL, Supriatna J, Hasan MN, Melnick DJ: High mitochondrial DNA diversity with little structure within and among leaf monkey populations (Trachypithecus cristatus and Trachypithecus auratus). Int J Primatol. 1997, 18: 1005-1028. 10.1023/A:1026304415648.
Wang W, Forstner MRJ, Zhang YP, Liu ZM, Wei Y, Huang HQ, Hu HG, Xie YX, Wu DH, Melnick DJ: Phylogeny of Chinese leaf monkeys using mitochondrial ND3–ND4 gene sequences. Int J Primatol. 1997, 18: 305-320. 10.1023/A:1026378215222.
Zhang YP, Ryder OA: Mitochondrial cytochrome b gene sequences of Old World monkeys: with special reference on evolution of Asian colobines. Primates. 1998, 39: 39-49. 10.1007/BF02557742.
Roos C: 2.3 Molecular phylogeny and systematics of Vietnamese leaf monkeys. Vietnam Primate Conservation Status Review 2002-Part 2: Leaf Monkeys. Edited by: Nadler T, Momberg F, Nguyen Xuan Dang, Lormee N. 2003, Hanoi: Fauna & Flora International and Frankfurt Zoological Society, 19-23.
Geissmann T, Groves CP, Roos C: The Tenasserim Lutung, Trachypithecus barbei (Blyth, 1847) (Primates: Cercopithecidae): Description, of a live specimen, and a reassessment of phylogenetic affinities, taxonomic history, and distribution. Contrib Zool. 2004, 73: 271-282.
Roos C: Molecular Evolution and Systematics of Vietnamese Primates. Conservation of Primates in Vietnam. Edited by: Nadler T, Streicher U, Ha Thang Long. 2004, Hanoi: Haki Publishing, 23-28.
Nadler T, Walter L, Roos C: Molecular evolution, systematics and distribution of the taxa within the silvered langur species group (Trachypithecus [cristatus]) in Southeast Asia. Zool Garten (NF). 2005, 75: 238-247.
Whittaker DJ, Ting N, Melnick DJ: Molecular phylogenetic affinities of the simakobu monkey Simias concolor. Mol Phylogenet Evol. 2006, 39: 887-892. 10.1016/j.ympev.2005.12.013.
Roos C, Thanh VN, Walter L, Nadler T: Molecular systematics of Indochinese primates. Vietn J Primatol. 2007, 1: 41-53.
Sterner KN, Raaum RL, Zhang YP, Stewart CB, Disotell TR: Mitochondrial data support an odd-nosed colobine clade. Mol Phylogenet Evol. 2006, 40: 1-7. 10.1016/j.ympev.2006.01.017.
Napier JR, Napier PH: A handbook of living primates. 1967, London: Academic Press
Delson E: Evolutionary history of the Cercopithecidae. Contrib Primatol. 1975, 5: 167-217.
Groves CP: The forgotten leaf-eaters and the phylogeny of Colobinae. Old World monkeys. Edited by: Napier JR, Napier PH. 1970, New York: Academic Press, 555-586.
Reichenbach HGL: Die vollständige Naturgeschichte der Affen. Die vollständige Naturgeschichte des In- und Auslandes. 1862, Dresden: Central-Atlas für Zoologische Gärten
Pocock RI: The monkeys of the genera Pithecus (or Presbytis) and Pygathrix found to the east of the Bay of Bengal. Proc Zool Sci London. 1935, 1934: 895-961.
Hill WCO: A monography on the purple-faced leaf-monkeys (Pithecus vetulus). Ceylon J Sci. 1934, 9: 23-88.
Brandon-Jones D: Colobus and leaf monkeys. Encylopedia of Mammals. Edited by: MacDonald ID. 1984, London: George Allen and Unwin, 398-408.
Strasser E, Delson E: Cladistic analysis of cercopithecid relationship. J Hum Evol. 1987, 16: 81-99. 10.1016/0047-2484(87)90061-3.
Brandon-Jones D: A revision of the Asian pied leaf monkeys (Mammalia: Cercopithecidae: Superspecies Semnopithecus auratus), with a description of a new subspecies. The Raffles Bulletin of Zoology. 1995, 43: 3-43.
Weitzel V, Yang CM, Groves CP: A catalogue of primates in the Singapore Zoological Reference Collection. The Raffles Bulletin of Zoology. 1988, 36: 1-166.
Groves CP: A theory of human and primate evolution. 1989, Oxford: Oxford University Press
Brandon-Jones D, Eudey AA, Geissmann T, Groves CP, Melnick DJ, Morales JC, Shekelle M, Stewart CB: Asian Primate Classification. Int J Primatol. 2004, 25: 97-164. 10.1023/B:IJOP.0000014647.18720.32.
Rowe N: The pictorial guide to the living primates. 1996, New York: Pogonias Press
Bigoni F, Stanyon R, Wimmer R, Schempp W: Chromosome painting shows that the proboscis monkey (Nasalis larvatus) has a derived karyotype and is phylogenetically nested within Asian colobines. Am J Primatol. 2003, 60: 85-93. 10.1002/ajp.10095.
Evans BJ, Supriatna J, Andayani N, Melnick DJ: Diversification of Sulawesi macaque monkeys: decoupled evolution of mitochondrial and autosomal DNA. Evol. 1999, 57 (8): 1931-1946.
Tosi AJ, Morales JC, Melnick DJ: Y-chromosome and mitochondrial markers in Macaca fascicularis indicate introgression with Indochinese M. mulatta and a biogeographic barrier in the Isthmus of Kra. Int J Primatol. 2002, 23: 161-178. 10.1023/A:1013258109954.
Arnold ML, Meyer A: Natural hybridization in primates: one evolutionary mechanism. Zoology. 2006, 109 (4): 261-276. 10.1016/j.zool.2006.03.006. Epub 2006 Aug 30.
Batzer MA, Deininger PL: Alu repeats and human genomic diversity. Nat Rev Genet. 2002, 3: 370-379. 10.1038/nrg798.
Shedlock AM, Okada N: SINE insertions: powerful tools for molecular systematics. Bioessays. 2000, 22: 148-160. 10.1002/(SICI)1521-1878(200002)22:2<148::AID-BIES6>3.0.CO;2-Z.
Schmitz J, Ohme M, Zischler H: SINE insertions in cladistic analyses and the phylogenetic affiliations of Tarsius bancanus to other primates. Genetics. 2001, 157: 777-784.
Roos C, Schmitz J, Zischler H: Primate jumping genes elucidate strepsirrhine phylogeny. Proc Natl Acad Sci USA. 2004, 101: 10650-10654. 10.1073/pnas.0403852101.
Ray DA, Xing JC, Hedges DJ, Hall MA, Laborde ME, Anders BA, White BR, Stoilova N, Fowlkes JD, Landry KE, Chemnick LG, Ryder OA, Batzer MA: Alu insertion loci and platyrrhine primate phylogeny. Mol Phylogenet Evol. 2005, 35: 117-126. 10.1016/j.ympev.2004.10.023.
Schmitz J, Roos C, Zischler H: Primate phylogeny: molecular evidence from retroposons. Cytogenet Genome Res. 2005, 108: 26-37. 10.1159/000080799.
Xing J, Wang H, Han K, Ray DA, Huang CH, Chemnick LG, Stewart CB, Disotell TR, Ryder OA, Batzer MA: A mobile element based phylogeny of Old World monkeys. Mol Phylogenet Evol. 2005, 37: 872-880. 10.1016/j.ympev.2005.04.015.
Thalmann O, Hebler J, Poinar HN, Pääbo S, Vigilant L: Unreliable mtDNA data due to nuclear insertions: a cautionary tale from analysis of humans and great apes. Mol Ecol. 2004, 13: 321-335. 10.1046/j.1365-294X.2003.02070.x.
Nsubuga AM, Robbins MM, Roeder AD, Morin PA, Boesch C, Vigilant L: Factors affecting the amount of genomic DNA extracted from ape faeces and the identification of an improved sample storage method. Mol Ecol. 2004, 13: 2089-2094. 10.1111/j.1365-294X.2004.02207.x.
Goossens B, Chikhi L, Utami SS, De Ruiter JR, Bruford MW: A multi-samples, multi-extracts approach for microsatellite analysis of faecel samples in an arboreal ape. Conserv Genetics. 2000, 1: 157-162. 10.1023/A:1026535006318.
Taberlet P, Waits LP, Luikart G: Noninvasive genetic sampling: look before you leap. Trends Ecol Evol. 1999, 14: 323-327. 10.1016/S0169-5347(99)01637-7.
Karanth KP, Delefosse T, Rakotosamimanana B, Parsons TJ, Yoder AD: Ancient DNA from giant extinct lemurs confirms single origin of Malagasy primates. Proc Natl Acad Sci USA. 2005, 102: 5090-5095. 10.1073/pnas.0408354102.
Thompson JD, Higgins DG, Gibson TJ: Clustal W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22: 4673-4680. 10.1093/nar/22.22.4673.
Castresana J: Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000, 17: 540-552.
Swofford DL: PAUP*: Phylogenetic analysis using parsimony (*and other methods), Version 4.0b10. 2002, Sunderland: Sinauer Associates
Strimmer K, von Haeseler A: Quartet puzzling: a maximum likelihood method for reconstructing tree topologies. Mol Biol Evol. 1996, 13: 964-969.
Posada D, Crandall KA: Modeltest: testing the model of DNA substitutions. Bioinformatics. 1998, 14: 817-818. 10.1093/bioinformatics/14.9.817.
Kishino H, Hasegawa M: Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and their branching order of Hominoidea. J Mol Evol. 1989, 29: 170-179. 10.1007/BF02100115.
Shimodaira H, Hasegawa M: Multiple comparisons of log-likelihood with applications to phylogeneitc reference. Mol Biol Evol. 1999, 16: 1114-1116.
Tosi AJ, Morales JC, Melnick DJ: Comparison of Y chromosome and mtDNA phylogenies leads to unique inferences of macaque evolutionary history. Mol Phylogenet Evol. 2000, 17: 133-144. 10.1006/mpev.2000.0834.
Mamedov IZ, Arzumanyan ES, Amosova AL, Lebedev YB, Sverdlov ED: Whole-genome experimental identification of insertion/deletion polymorphisms of interspersed repeats by a new general approach. Nucleic Acid Res. 2005, 33: e16-10.1093/nar/gni018.
Acknowledgements
We are grateful to the staff of the zoos in Cologne, Dresden, Hannover, Krefeld, Erfurt, Stuttgart, Wuppertal, Howletts, Bristol and Singapore, and to Manfred Ade (Naturhistorisches Museum Berlin, Germany), Linda Vigilant (Max-Planck-Institute of Evolutionary Anthropology, Leipzig, Germany), Kai-Olaf Krüger (Angkor Centre for Conservation of Biodiversity, Cambodia) and Tilo Nadler (Endangered Primate Rescue Center, Vietnam) for samples. Finally, we thank Christiane Schwarz for her excellent technical assistance in the laboratory. The work was financially supported by the German Primate Center (DPZ).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
MO designed primers, performed parts of the laboratory work and wrote parts of the manuscript. LW wrote parts of the manuscript and performed parts of the statistical analysis. CR collected samples, designed primers, performed parts of the laboratory and statistical work and wrote parts of the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Osterholz, M., Walter, L. & Roos, C. Phylogenetic position of the langur genera Semnopithecus and Trachypithecus among Asian colobines, and genus affiliations of their species groups. BMC Evol Biol 8, 58 (2008). https://doi.org/10.1186/1471-2148-8-58
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-8-58