
Abstract
Background
Evolutionary genetics provides a rich theoretical framework for empirical studies of phylogeography. Investigations of intraspecific genetic variation can uncover new putative species while allowing inference into the evolutionary origin and history of extant populations. With a distribution on four continents ranging throughout most of the Old World, Lampides boeticus (Lepidoptera: Lycaenidae) is one of the most widely distributed species of butterfly. It is placed in a monotypic genus with no commonly accepted subspecies. Here, we investigate the demographic history and taxonomic status of this widespread species, and screen for the presence or absence of the bacterial endosymbiont Wolbachia.
Results
We performed phylogenetic, population genetic, and phylogeographic analyses using 1799 bp of mitochondrial sequence data from 57 specimens collected throughout the species' range. Most of the samples (>90%) were nearly genetically identical, with uncorrected pairwise sequence differences of 0 – 0.5% across geographic distances > 9,000 km. However, five samples from central Thailand, Madagascar, northern Australia and the Moluccas formed two divergent clades differing from the majority of samples by uncorrected pairwise distances ranging from 1.79 – 2.21%. Phylogenetic analyses suggest that L. boeticus is almost certainly monophyletic, with all sampled genes coalescing well after the divergence from three closely related taxa included for outgroup comparisons. Analyses of molecular diversity indicate that most L. boeticus individuals in extant populations are descended from one or two relatively recent population bottlenecks.
Conclusion
The combined analyses suggest a scenario in which the most recent common ancestor of L. boeticus and its sister taxon lived in the African region approximately 7 Mya; extant lineages of L. boeticus began spreading throughout the Old World at least 1.5 Mya. More recently, expansion after population bottlenecks approximately 1.4 Mya seem to have displaced most of the ancestral polymorphism throughout its range, though at least two early-branching lineages still persist. One of these lineages, in northern Australia and the Moluccas, may have experienced accelerated differentiation due to infection with the bacterial endosymbiont Wolbachia, which affects reproduction. Examination of a haplotype network suggests that Australia has been colonized by the species several times. While there is little evidence for the existence of morphologically cryptic species, these results suggest a complex history affected by repeated dispersal events.
Similar content being viewed by others


Background
The study of speciation lies at the nexus of micro- and macroevolution, i.e., phylogenetics and population genetics. Phylogeography, which incorporates both approaches in a geographical context, examines the role of different historical processes in population demography, differentiation and speciation [1]. The advent of rapid and affordable DNA sequencing over the past 15 years has catalyzed studies on the evolutionary dynamics of populations and the discovery of previously unrecognized morphologically cryptic species [2].
The pea blue butterfly, Lampides boeticus (L.) (Lepidoptera: Lycaenidae), is one of the most widely distributed butterflies in the world, and is currently found across the Palaearctic region from Britain to Japan, throughout suitable habitat in Africa, Madagascar, South East Asia, and Australia, extending eastwards to parts of Oceania including Hawaii. It occurs in temperate, subtropical, and tropical biomes in both lowland and montane localities, typically in open and/or disturbed areas.
Taxonomically, L. boeticus is the only species in its genus and has no commonly recognized subspecies, despite its wide distribution. The larval stages feed on plants in at least six families, although Leguminosae (particularly Papilionoideae) is the predominant host plant taxon [3]. Cultivated legumes, including broad beans (Vicia faba) and garden peas (Pisum sativum) are among its preferred host plants, and the butterfly is a crop pest in many parts of its range [4]. Lampides boeticus is among the approximately three-quarters of butterfly species in the family Lycaenidae that associate with ants as larvae and pupae [5]. The species is facultatively tended by a variety of ants throughout its range, including Camponotus spp., Iridomyrmex spp., and 'tramp' ant species including Tapinoma melanocephalum and the Argentine ant, Linepithema humile [6, 7].
We sampled 57 L. boeticus from 39 localities on four continents (Fig. 1) to test the hypothesis that this widespread species, as currently circumscribed, consists of more than one genetically distinct taxon. We also used nucleotide sequence data to further examine the genetic structure of this species and analyze the demographic history of the sampled populations.

Map of Lampides boeticus collection localities. Numbers refer to sample information in Table 1. Different colors distinguish labeled biogeographic regions.
Results
Phylogenetic analyses and node dating
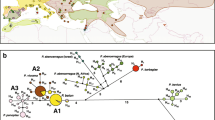
Bayesian, maximum likelihood and parsimony phylogenetic analyses arrived at similar phylogenetic hypotheses for the evolutionary history of L. boeticus that agreed on all major groupings (Fig. 2B). Cytochrome c oxidase subunit I (COI) had 48 variable sites and cytochrome b (cytB) had 28, of which 35 and 17 were parsimoniously informative, respectively. Thus, cytB was more variable – 5.35% of nucleotide sites were variable across all samples – than COI, in which 4.10% of nucleotide sites varied. The percentage of parsimoniously informative nucleotide sites was also higher in cytB (3.28% vs. 2.87%), as was the number of nucleotide sites with parsimoniously informative non-synonymous substitutions (3 vs. 0). The parsimony analysis resulted in 1,130 most parsimonious trees with a tree score of 336. The strict consensus of these trees differed with regard to two nodes when compared to the tree obtained in both Bayesian and maximum likelihood analyses (Fig. 2B). Lampides boeticus was monophyletic with regard to the three chosen outgroup species. In addition to the divergent genotypes in clades C and D (Fig. 2B), there were two other groups that were supported by Bayesian, maximum likelihood and parsimony analyses. Clade A contained all haplotypes from Africa, Madagascar, the eastern and western Palaearctic, Indo-Burma, and the Philippines not found in the divergent clades C and D. Grade B is a paraphyletic assemblage containing all of the haplotypes from the Sundaland, Wallacean, and Australian regions not found clades C and D. Only samples 26 and 56 from clade D were infected with Wolbachia as determined by PCR assay.

Relationships among COI+cytB mitochondrial haplotypes of Lampides boeticus. Numbers refer to sample information in Table 1; colors denote biogeographic regions designated in Fig. 1. A. Most parsimonious haplotype network of L. boeticus constructed with 99% connection limit. Black circles indicate extinct or unsampled haplotypes that differ by one nucleotide substitution from the adjoining haplotype. B. Bayesian consensus tree of L. boeticus haplotypes. Numbers above braches indicate Bayesian posterior probability and maximum likelihood bootstrap support, respectively; numbers below indicate parsimony bootstrap symmetric resampling and jackknife support, respectively, for parsimony analyses that resulted in a topologically similar tree. Estimated node ages: I, 6.9 ± 0.6 My; II, 1.5 ± 0.2 My; III, 1.4 ± 0.2 My
Divergence of Lampides from its putatively closest relative, Cacyreus, occurred in the Miocene approximately 6.9 ± 0.6 Mya (node I; Fig. 2B). Divergence of clade D, containing haplotypes from north Queensland and the Moluccas (node II), occurred in the Pleistocene approximately 1.5 ± 0.2 Mya, and clade A and grade B, containing the majority of haplotypes (node III), diverged approximately 1.4 ± 0.2 Mya (Fig. 2B). However, given the relative paucity of genetic variation and the small magnitude of the difference of the inferred ages of nodes (both of which are likely to increase error), these age estimates should be regarded as approximations.
Pairwise distances among L. boeticus COI+cytB haplotypes ranged from 0–2.36% (Table 1), while distances between L. boeticus and the outgroup taxa ranged from 7.09–10.78% (data not shown). The relatively low levels of intraspecific sequence divergence among populations are consistent with the hypothesis that L. boeticus is a single species with pairwise genetic distances well below the upper ranges of intraspecific divergence estimates found in other lepidopteran species [8, 9].
Translated amino acid sequences were invariant within COI, but 16 changes at 8 sites were observed in cytB. McDonald and Kreitman tests found no evidence of natural selection acting on these mitochondrial genes (P > 0.20 in all possible pairwise tests).
Perhaps the most striking pattern in the data was the paucity of genetic variation across vast geographic distances. Our analyses showed that Lampides boeticus is a widely distributed and apparently panmictic species with little population differentiation. The most common COI+cytB haplotype was shared by specimens from Spain, Turkey, Kenya, Namibia, Madagascar, Laos, and Vietnam, spanning a distance of over 9,000 km or 100 longitudinal degrees on three continents (Figs. 1, 2A, Table 1). Coalescent theory predicts that internal nodes in a gene genealogy will be more common than tip nodes, as these represent older haplotypes. Mutations at different sites within these ancestral haplotypes result in descendent haplotypes that are younger and less common, and appear as multiple 'tips' emanating from the more abundant haplotypes of the internal node [10]. This pattern was evident in our haplotype tree (Fig. 2A). However, several samples were highly divergent from the majority of genetically similar, yet widely distributed haplotypes. These samples could not be connected to the others with a 90% parsimony connection limit in the COI+cytB network (the lowest parsimony value allowed by TCS 1.21; Fig. 2A). These haplotypes, corresponding to clades C and D in the phylogenetic analysis (Fig. 2B), were joined to very different sister haplotypes in the networks of COI and cytB, and with lower parsimony connection limits [see Additional file 1]. In these networks for individual genes, the divergent samples were on relatively long braches, with a haplotype from central Thailand closely related to a sample from Madagascar in clade C. Clade D contained a single sample from north Queensland, Australia, and differed at only two nucleotide sites from a haplotype shared by two samples from the Wallacean islands of Buton and Tomea in the Moluccas to the east of Sulawesi. Interestingly, other samples collected from the same sites in Tomea and Madagascar grouped with the bulk of genetically similar samples (e.g., samples 14, 15, and 25 in clade A and grade B, Fig. 2, Additional file 1), indicating substantial genetic diversity within these populations (e.g., 2.04% within Madagascar). In the phylogenetic analyses, these lineages appear to have diverged earlier than the more common genotypes (Fig. 2B). It is unlikely that these haplotypes are nuclear copies of the mitochondrial genes (numts), since all sequences could be translated into amino acids with no stop codons. In addition, both genes from the five specimens in clades C and D were amplified and sequenced twice to minimize the probability of human error.
Demographic and population genetic analyses
Indices of molecular diversity, results of Tajima's D and Fu's F tests, and output from the mismatch distribution analysis including estimated time since population bottlenecks are provided in Table 2. Grant and Bowen [11] suggested that comparison of h and π values within clades can provide information about patterns of past demographic expansion and/or constriction. They categorized numerical values of h and π as either high or low, and described situations that may have lead to each of four possible scenarios. In our data set, h and π values of COI and cytB from clade A and grade B considered separately or together all fall into category 2, with high h (> 0.5) and low π (< 0.005), indicating rapid expansion after a period of low effective population size. All values of Fu's F statistic revealed significantly negative deviations from mutation-drift equilibrium (note that, in Fu's F analysis, P = 0.02 is the threshold value corresponding to α = 0.05) [12]. In addition, Tajima's D statistic was significantly negative for COI data from clade A and marginally non-significant for cytB data in the same clade, indicating deviation from neutral evolution and suggestive of demographic expansion.
Mismatch distributions are frequency distributions of the number of nucleotide differences in all pairwise comparisons. A population that has experienced sudden exponential growth from an initially small population is expected to have a unimodal mismatch distribution resulting from coalescence of haplotypes to the same bottleneck event [13, 14]. In clade A and grade B, deviations of the observed distributions of nucleotide frequencies were not significantly different from those expected under a model of stepwise expansion (Table 2), and visual inspection of the mismatch distributions (Fig. 3) indicate that the unimodal distributions of both genes in clade A are highly suggestive of a bottleneck, while the bimodal distributions of both genes in grade B suggest that the initial population size was larger before the expansion. This difference between clade A and grade B is reflected in differences between the pre- and post-expansion values of θ or the COI data, though this difference is not evident in the θ values calculated from the shorter cytB sequence fragment (Table 2). The estimated age of the bottleneck in clade A is more recent than that of grade B (Table 2), reflecting the more recent divergence of clade A in the estimated phylogeny. The estimated age of the bottleneck for the pair of clades, 2.30 My, predates the estimated divergence time of all extant L. boeticus 1.5 Mya (Fig. 2B), suggesting methodological discrepancy between the methods used to date divergence times and those used to estimate time since expansion. These differing estimates are no doubt affected by the inflated estimate of time since expansion due to the lack of a value for μ expressed in generations rather than years (see Methods).

Mismatch distributions of Lampides boeticus lineages clade A and grade B. Bars indicate observed mismatch differences and lines represent the expected distribution under a sudden population expansion model.
Beerli [15] provided evidence from analyses of simulated data that Bayesian methods of population parameter estimation are more likely to provide accurate estimates than maximum likelihood methods. However, our estimations of the parameters θ and g using both methods are remarkably concordant. The analyses suggest that the populations constituting grade B are larger than those composing clade A, and that grade B has a higher exponential growth rate than clade A (Table 3).
With the exceptions of L. boeticus and the species Cacyreus marshalli, which feeds on geraniums and recently underwent a range extension as an invasive species into southern Europe from Africa [16], all species in each of the genera closely related to Lampides are confined to the African region, with some species also present in Madagascar. Tracing the most parsimonious reconstruction of biogeographic regions on the given tree indicates that the most recent common ancestor of L. boeticus and its sister taxon lived in the African region. However, the optimization for the L. boeticus node is ambiguous, with an African origin being only one of several most parsimonious optimizations. The genus Harpendyreus was placed in the same section as Cacyreus by Eliot [17], implying a close relationship with Lampides, but was not sampled in the phylogenetic reconstruction of the Polyommatini. However, Harpendyreus is wholly African, and its absence does not affect our inferences.
Discussion
The phylogeographic structure of Lampides boeticus was remarkable in two respects. While most sampled populations had little or no genetic variation across half the planet, a small number of specimens from widely spaced locales were highly divergent from the most common haplotypes, yet relatively similar to each other.
As currently circumscribed, L. boeticus appears to be monophyletic with respect to the outgroups included in this analyses, and is nested within a wholly African clade. While we acknowledge the possibility that some extant species may have relictual distributions, it seems plausible – given the number of exclusively African close relatives – that the most recent common ancestor from which Lampides and its sister taxon evolved originated in Africa and spread eastward across the Old World. Descendants from early expansion events (clades C and D) appear to have been largely replaced throughout much of the range by descendants from expansion events after one or more recent bottlenecks (clade A and grade B).
The low genetic divergence among most members of this species could be the result of either recent and rapid population expansion after a population bottleneck or selection acting on some portion of the mitochondrial genome. These explanations are not mutually exclusive, but the weight of evidence suggests that most extant populations of L. boeticus have descended from expansion after one or more bottlenecks. Results from Fu's F and Tajima's D tests, analyses of mismatch distributions, and comparison of h and π values all suggest rapid expansion after a population bottleneck, with populations in clade A from Africa, Madagascar, the Palaearctic, and the Philippines resulting from a more severe population constriction than populations in grade B from Sundaland, Wallacea, and Australia. Moreover, since sampling was random with respect to genotype, the larger number of post-bottleneck haplotypes in clade A and grade B suggests that these individuals have a selective advantage over individuals from clades C and D, perhaps because they displaced most of the individuals from these basal lineages or because they are more effective dispersers or colonizers. Our data, however, cannot rule out the possibility that genetic drift is responsible for the predominance of haplotypes in clade A and grade B. Since L. boeticus is a polyphagous crop pest, human agriculture or commerce may have aided the rapid movement of the species around the globe by inadvertently transporting it with crop plants or produce. However, we suspect this factor has been minimal or geographically restricted, as no individuals from clade A were found in the areas inhabited by individuals from grade B, and vice versa.
Inspection of haplotype networks from individual and concatenated genes (Fig. 2A, Additional file 1) further suggest that Australia has been colonized by L. boeticus at least three times: once by an ancestor of the genetically divergent individual in clade D (specimen 56), once from a lineage originating in Wallacea (specimens 55 and 57), and at least once by a lineage from Sundaland (specimen 53).
A selective sweep acting on some portion of the mitochondrial genome might also explain the paucity of genetic variation in clade A and grade B. Zink [18] has shown that natural selection on a transmembrane portion of the mitochondrial ND2 gene is responsible for the shallow and unstructured haplotype trees of Parus montanus (Aves: Paridae), and similar selective forces on one or more mitochondrial gene(s) could account for the pattern observed in L. boeticus. However, MacDonald and Kreitman tests reveal no evidence for selection acting on the genes sequenced for this study.
Infection by Wolbachia spp., a group of rickettsial endocellular bacteria that manipulate host reproduction in a variety of ways, can spread rapidly throughout contiguous populations, purging genetic variation of the host species and causing a so-called 'Wolbachia sweep' [19] capable of reducing haplotype diversity within populations and whole species [20]. However, none of the samples in clade A, grade B, and clade C tested positive for Wolbachia infection, though two of the three samples (26 and 56) in clade D were infected. The uninfected specimen in this clade was from a dried specimen of unknown age purchased from a dealer who could not provide the collection date, and may also have been infected, but the Wolbachia DNA was too degraded to be amplified in the PCR test employed here.
Four lines of evidence suggest that all L. boeticus populations sampled in this study belong to a single, potentially interbreeding species: 1) even the highest L. boeticus intraspecific pairwise distance values are within the range of intraspecific variation recorded from other butterfly species; 2) monophyly of all sampled populations is well supported (100% by every measure of clade support); 3) the lack of external morphological differences among populations; and 4) estimated divergence time of all sampled L. boeticus genes (approximately 1.5 Mya using a dated node on the phylogeny or 2.3 Mya using coalescence time of COI) is more recent than the estimated divergence of Lampides from its putative sister genus Cacyreus 6.9 Mya. There are many caveats to using pairwise genetic distances and a molecular clock to infer the age of lineage splitting events [21]. These ages must thus remain speculative and, in the case of L. boeticus, cannot be confirmed by biogeographic or fossil evidence.
The haplotypes in clades C and D are most likely descended from basal lineages that diverged before the putative population bottleneck(s) that gave rise to clade A and grade B. Descendents of these bottlenecks seem to have largely displaced earlier lineages throughout the species' range. The relatively large number of synapomorphies that unite the haplotypes in clade C and clade D renders convergence unlikely, and the geographical distances between haplotypes in these clades – particularly between the Thai and Malagasy specimens in clade C – makes dispersal an unlikely explanation. The presence of these early-diverging clades, in conjunction with the inferred demographic history of clade A and grade B, suggest a demographic history profoundly affected by the species' propensity to undertake regional migrations [22–24].
The low genetic diversity of Lampides boeticus in clade A and grade B across its geographic range is similar to that of several other Old World lepidopteran crop pests. Like L. boeticus, the haplotype network of Helicoverpa armigera (Noctuidae) has a comparatively long branch, but the cluster of haplotypes at the end of this branch belong to a separate species, H. zea, which is morphologically distinguishable only by close examination of the genitalia [25]. The population parameters of θ estimated for clade A and grade B (MLEs of 0.00418 and 0.00711, respectively) overlap with θ values calculated for several other widespread pestiferous Lepidoptera, including H. armigera (0.0027 – 0.0099 within western Africa and France/Portugal, respectively) [26] and Ostrinia nubilalis (Crambidae; 0.00358 – 0.0315 for different loci within France). Values for the gypsy moth, Lymantria dispar (Lymantriidae), are notably lower (0.00129 – 0.00282 for Japan and mainland Asia, respectively) than for La. boeticus, but the demographic scenario inferred for Ly. dispar is remarkably similar to that of La. boeticus: divergent lineages in widespread locales (Japan and India) are thought to represent ancient splitting events from populations spreading throughout Eurasia, with current European populations harboring the most derived haplotypes [27].
Conclusion
Our analyses suggest that all populations of the butterfly Lampides boeticus (Lycaenidae) sampled in this study belong to a single, widespread species with a complex evolutionary history. Phylogenetic estimates suggest that the most recent common ancestor of L. boeticus and its sister taxon diverged in Africa in the Miocene approximately 6.9 Mya, but all extant populations coalesce to a most recent common ancestor that lived approximately 2.3 – 1.5 Mya, near the beginning of the Pleistocene. The majority of individuals are descended from expansion events after one or more population bottlenecks, though some populations harbor ancestral polymorphism predating these population constrictions. One lineage found in Northern Australia and the Moluccas may have experienced accelerated differentiation due to infection with the rickettsial endosymbiont Wolbachia, which infected at least 2 of 3 sampled individuals in this clade. The proclivity of L. boeticus to undergo regional migrations of unknown distance appears to have prevented genetic differentiation due to isolation by distance, and the species' apparent wanderlust has clearly shaped its demographic history, which is marked by expansion and long distance dispersal following periods of small population size.
Methods
DNA sequencing and selection of genetic markers
Genomic DNA was extracted from small pieces of abdominal tissue and legs using a CTAB phenol-chloroform extraction protocol, keeping wings and genitalia intact as morphological vouchers. Thirty-four samples were dried, papered specimens up to 11 years old; the bodies of all other specimens were stored in absolute ethanol immediately after collection for molecular study. We chose two rapidly evolving protein-coding mitochondrial genes for our analyses: cytochrome c oxidase subunit I (COI) and cytochrome b (cytB). The COI gene encompasses the 'Folmer region' used in DNA barcoding, and the cytB gene, located nearly opposite COI on the circular mitochondrial genome [28], is among the most variable protein-encoding genes in insects (R. Meier, unpublished data). Assuming the mitochondrial genome of L. boeticus is similar in size and organization to the lycaenid species Coreana raphaelis [28], the two genes represent two different regions and approximately 8.5% of the mitochondrial genome.
A 1,220 bp fragment of COI was amplified, typically in two overlapping fragments using LCO1490/Nancy and TN2126/Hobbes primer pairs (Table 4). However, six degraded samples had to be amplified in 3–4 fragments using the following primer pairs: LCO1490/ButterCOI-R2; DanausCOI-F3/ButterCOI-R3; ButterCOI-F4/ButterCOI-R4; and/or ButterCOI-F4/Hobbes (Table 4). The primer pair REVCBJ/REVCB2H successfully amplified a 579 bp region of the cytB gene from all samples. Each sample was also screened for the presence of Wolbachia, a maternally inherited bacterial endosymbiont. The primer pair 81F/691R has been used to amplify the Wolbachia surface protein (wsp) gene from Wolbachia infecting a variety of butterflies and other insects [29–31], and we used these primers in a PCR screen for the presence of Wolbachia. Each PCR reaction consisted of 2.5 μl 10× TaKaRa ExTaq buffer with 20 mM MgCl2, 1.2 μl of each 10 mM primer, 1 μl 100 mM dNTPs, 0.1 μl TaKaRa ExTaq polymerase, 17 μl H20, and 2 μl template DNA, for a total reaction volume of 25 μl, which was amplified with the following thermal cycler conditions: 3 min at 94°C followed by 35 cycles of 1 min at 94°C, 1 min at 52°C (COI), 54°C (cytB), or 55° (wsp) and 1.5 min at 72°C, and finally 5 min at 72°C. The resulting products were cleaned with Bioline SureClean, labelled with ABI BigDye Terminator 3.1, cleaned with Agencourt CleanSEQ, and sequenced in both directions on an ABI 3130xl DNA analyzer.
Phylogenetic analyses and node dating
Alignment of nucleotide sequences, which contained no indels, was performed with Sequencher 4.6 (Gene Codes Corp.), and data from both genes were concatenated with TaxonDNA 1.5 [32] for subsequent analyses. Replicate COI+cytB sequences were pruned from the dataset so that every haplotype in the phylogenetic analyses was unique. Cytochrome c oxidase subunit I (COI) sequences from three closely related butterflies [17] were used to root the ingroup taxa: Cacyreus marshalli, Uranothauma falkensteini, and Phlyaria cyara. Recent molecular phylogenetic investigation of the tribe Polyommatini has shown that these taxa are closely related to L. boeticus (N.E. Pierce et al., in prep.). Analyses performed with and without the inclusion of these sequences in the dataset showed that the absence of cytB sequence data for the outgroup species had no effect on the topology of the phylogenetic estimate for the ingroup.
Parsimony analyses were performed with TNT 1.1 [33]. After increasing the maximum number of saved trees to 3,000, a traditional TBR heuristic search was implemented, performing 1,000 replicates and saving 10 trees per replication, replacing existing trees. To assess confidence in the resulting phylogenetic estimate, the data were subjected to a bootstrap analysis using symmetric resampling [34] implementing a traditional search with 33% change probability (1,000 replicates). The results were summarized as absolute frequencies. In addition, the data were resampled with the jackknife technique using a traditional search with a 36% removal probability replicated 1,000 times.
Bayesian phylogenetic analyses were performed with MrBayes 3.1.2 [35]. MrModeltest 2.2 [36] selected the GTR+I+Γ [37] model for both COI and cytB partitions using Akaike's Information Criterion [38]. Parameter values for the substitution model were estimated from the data and allowed to vary independently between genes. Four Markov chains, one cold and three heated, were run simultaneously for 10 million generations. Trees were sampled every 100th generation. After completion of the analysis, the first 25,000 trees were discarded before a majority-rule consensus tree was calculated from the remaining 75,001 trees. Maximum likelihood analyses were performed with GARLI 0.951 [39] starting from a random tree using the GTR model with all model parameters estimated from the data. The analysis was automatically terminated after the search algorithm progressed 10,000 generations without improving the tree topology by a log likelihood of 0.01 or better. Maximum likelihood bootstrap values were obtained by repeating the analysis 100 times and constructing a majority-rule consensus tree with PAUP* 4.0b10 [40].
The most parsimonious haplotype network of L. boeticus was determined with TCS 1.21 [41]. Analyses were run multiple times with varying parsimony connection limits to ascertain the highest limit that would retain each connection. The haplotype networks of each gene and of the two concatenated genes were determined separately. Leading and trailing gaps resulting from declining quality at the ends of sequences and lack of overlap between forward and reverse strands were coded as missing data. These missing data caused only minor problems; only two sequences had missing values for three of the fifty-two parsimoniously variable sites. Anastomoses due to convergence of mutations at two or more nucleotide sites were pruned using the guidelines of Castelloe and Templeton [10] to produce strictly bi- or multifurcating topologies. Nested clade phylogeographic analysis was also performed to evaluate population histories of L. boeticus. The results of these analyses are presented in Additional file 1.
In the absence of a robust fossil record and/or vicariance events with which to calibrate divergence times, we estimated divergence times using a molecular clock (e.g. [42]). Cytochrome c oxidase subunit I (COI) exhibits the least rate homogeneity of any insect mitochondrial gene [43], and age estimations were based only on this gene, which shows an average mean uncorrected pairwise distance of 1.5% per million years (My) across a range of arthropod taxa [44]. Mean uncorrected pairwise distances between all samples on each branch of major bifurcating nodes were calculated with MEGA 4 [45] and divided by 0.015 (1.5%) to obtain a rough estimate of node age.
Demographic and population genetic analyses
To examine the evolutionary histories of sampled populations, we calculated several population genetic diversity indices that allow inference about demographic history. Identical sequences were not removed from the dataset for these analyses. Given the low levels of genetic variation and the paucity of samples at individual sampling sites, we grouped samples by clade (Fig. 2B) and provide separate analyses for clade A, grade B, and clade A + grade B, which, together, form a monophyletic group. We calculated the following population genetic indices using DnaSP [46]: number of haplotypes, number of variable nucleotide sites (S), haplotype diversity (h) and its standard deviation, nucleotide diversity (π) and its standard deviation, and the mean number of pairwise differences (k), along with its total variance (including components of stochastic and sampling variance) [47]. The haplotype diversity of a sample indicates the probability that two randomly chosen haplotypes within a sample will be identical [equation 8.5, [48]], while nucleotide diversity calculates the average proportion of nucleotide sites that differ in all pairwise comparisons [47]. In addition, the genetic imprint of rapid population expansion can be detected with Tajima's D test [49, 50], Fu's F-test [12], and by inspection of mismatch distributions, which plot the frequency distribution of observed pairwise differences [13].
Tajima's D test, Fu's F test, and calculation of observed and expected mismatch values were performed with Arlequin 3.11 [51], along with the moment estimators of the time to expansion (τ), and indices of population sizes before and after the expansion, θ0 and θ1, respectively [52], which are calculated with a generalized non-linear least-square approach with confidence intervals approximated with 1000 replicates of parametric bootstrapping [53]. To assess the validity of these estimates of demographic expansion, the probability of the sum of square deviations (SSD) between the observed mismatch values and values predicted by the model is approximated by determining the proportion of simulated SSDs that are larger than or equal to the observed SSD [51]. The time since expansion, t, is then calculated by substituting values for τ and μ in the equation τ = 2μt [51], where μ (the mutation rate per site per generation) is 0.75% between ancestor-descendent alleles (i.e., half of 1.5%, the average value for arthropod pairwise differences per million years) [44]. Note that τ is expressed in generations, while the value of μ used here is measured in years. Since L. boeticus passes through several generations per year, this method of estimating time since expansion is most likely an overestimate.
The relative effective population size parameter θ and exponential growth rate, g, as well as their 95% confidence intervals, were estimated using data from both COI and cytB and calculated with LAMARC 2.12b [54]. The two parameters and their confidence intervals were jointly estimated with separate Bayesian and maximum likelihood analyses. Each LAMARC analysis consisted of 3 simultaneous searches with heating temperature adjusted automatically with 15 initial chains sampled every 20 steps with a burn-in of 2000, followed by 6 final chains sampled every 20 steps with a burn-in of 2500. Final most likely estimates (MLEs) were calculated using parameter estimates from three replicated analyses.
To infer the geographical origin of L. boeticus, we connected our haplotype tree to a well-sampled, genus-level phylogenetic hypothesis for the tribe Polyommatini based on 4939 bp from seven nuclear and mitochondrial genes. Bayesian and maximum likelihood methods both recovered the following topology for L. boeticus and closely related genera: (((Lampides, Cacyreus) Actizera)(Phlyaria, Uranothauma)) (N.E. Pierce et al., in prep.). We coded the biogeographic distributions of each taxon in this phylogeny as a character and traced the most parsimonious reconstruction of biogeographic regions on the tree using MacClade 4.06 [55] to assess the probable region where L. boeticus and its most recent common ancestor diverged. MacClade was also used to translate DNA sequences to amino acids for tabulation of non-synonymous substitutions. PAUP* 4.0b10 was used to calculate mean pairwise distances among haplotypes. To determine whether non-synonymous changes might be the result of natural selection on the gene, McDonald and Kreitman tests [56] were performed with DnaSP.
Abbreviations
- CI:
-
confidence interval
- COI:
-
cytochrome c oxidase subunit I
- m:
-
meters
- MLE:
-
most likely estimate
- My:
-
million years
- Mya:
-
million years ago
- ND2:
-
nicotinamide adenine dinucleotide dehydrogenase subunit 2
- SSD:
-
sum of square deviations
- wsp:
-
Wolbachia surface protein
References
Avise JC: Molecular Markers, Natural History and Evolution. 2004, Sunderland, MA: Sinauer Associates, 2
Bickford D, Lohman DJ, Sodhi NS, Ng PKL, Meier R, Winker K, Ingram K, Das I: Cryptic species: a new window on diversity and conservation. Trends in Ecology and Evolution. 2006, 22: 148-155. 10.1016/j.tree.2006.11.004.
HOSTS – a Database of the World's Lepidopteran Hostplants. [http://www.nhm.ac.uk/research-curation/projects/hostplants/]
Mavi GS: A critical review on the distribution and host-range of pea blue butterfly, Lampides boeticus (Linn.). Journal of Insect Science. 1992, 5: 115-119.
Pierce NE, Braby MF, Heath A, Lohman DJ, Mathew J, Rand DB, Travassos MA: The ecology and evolution of ant association in the Lycaenidae (Lepidoptera). Annual Review of Entomology. 2002, 47: 733-771. 10.1146/annurev.ento.47.091201.145257.
Eastwood R, Fraser AM: Associations between lycaenid butterflies and ants in Australia. Australian Journal of Ecology. 1999, 24: 503-537. 10.1046/j.1440-169x.1999.01000.x.
Fiedler K: Ant associates of Palaearctic lycaenid butterfly larvae (Hymenoptera: Formicidae; Lepidoptera: Lycaenidae) – a review. Myrmecologische Nachrichten. 2006, 9: 77-87.
Hebert PDN, Cywinska A, Ball SL, deWaad JR: Biological identifications through DNA barcodes. Proceedings of the Royal Society (London) B. 2003, 270: 313-321. 10.1098/rspb.2002.2218.
Zakharov EV, Smith CR, Lees DC, Cameron A, Vane-Wright RI, Sperling FAH: Independent gene phylogenies and morphology demonstrate a Malagasy origin for a wide-ranging group of swallowtail butterflies. Evolution. 2004, 58: 2763-2782.
Castelloe J, Templeton AR: Root probabilities for intraspecific gene trees under neutral coalescent theory. Molecular Phylogenetics and Evolution. 1994, 3: 102-113. 10.1006/mpev.1994.1013.
Grant WAS, Bowen BW: Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. Journal of Heredity. 1998, 89: 415-426. 10.1093/jhered/89.5.415.
Fu YX: Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997, 147: 915-925.
Slatkin M, Hudson RR: Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics. 1991, 129: 555-562.
Rogers AR, Harpending H: Population growth makes waves in the distribution of pairwise genetic differences. Molecular Biology and Evolution. 1992, 9: 552-569.
Beerli P: Comparison of Bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics. 2006, 22: 341-345. 10.1093/bioinformatics/bti803.
Quacchia A, Ferracini C, Bonelli S, Balletto E, Alma A: Can the Geranium Bronze, Cacyreus marshalli, become a threat for European biodiversity?. Biodiversity and Conservation. 2008, 17: 1429-1437. 10.1007/s10531-008-9350-3.
Eliot JN: The higher classification of the Lycaenidae (Lepidoptera): a tentative arrangement. Bulletin of the British Museum of Natural History. 1973, 28: 373-505.
Zink RM: Natural selection on mitochondrial DNA in Parus and its relevance for phylogeographic studies. Proceedings of the Royal Society (London) B. 2005, 272: 71-78. 10.1098/rspb.2004.2908.
Werren JH: Biology of Wolbachia. Annual Review of Entomology. 1997, 42: 587-609. 10.1146/annurev.ento.42.1.587.
Narita S, Nomura M, Kato Y, Fukatsu T: Genetic structure of sibling butterfly species affected by Wolbachia infection sweep: evolutionary and biogeographical implications. Molecular Ecology. 2006, 15: 1095-1108. 10.1111/j.1365-294X.2006.02857.x.
Heads M: Dating nodes on molecular phylogenies: a critique of molecular biogeography. Cladistics. 2005, 21: 62-78. 10.1111/j.1096-0031.2005.00078.x.
de Vos R, Ellis WN: Migrating Lepidoptera and rarities in 2001 and recent adventive records. Entomologische Berichten. 2004, 64: 138-145.
Dingle H, Zalucki MP, Rochester WA: Season-specific directional movement in migratory Australian butterflies. Australian Journal of Entomology. 1999, 38: 323-329. 10.1046/j.1440-6055.1999.00117.x.
Corbet AS, Pendlebury HM, Eliot JN, D'Abrera B: Butterflies of the Malay Peninsula. 1994, Kuala Lumpur: Malayan Nature Society, 4
Behere GT, Tay WT, Russell DA, Heckel DG, Appleton BR, Kranthi KR, Batterham P: Mitochondrial DNA analysis of field populations of Helicoverpa armigera (Lepidoptera: Noctuidae) and of its relationship to H. zea. BMC Evolutionary Biology. 2007, 7: 117-10.1186/1471-2148-7-117.
Nibouche S, Buès R, Toubon J-F, Poitout S: Allozyme polymorphism in the cotton bollworm Helicoverpa armigera (Lepidoptera: Noctuidae): comparison of African and European populations. Heredity. 1998, 80: 438-445. 10.1046/j.1365-2540.1998.00273.x.
Bogdanowicz SM, Schaefer PW, Harrison RG: Mitochondrial DNA variation among worldwide populations of gypsy moths, Lymantria dispar. Molecular Phylogenetics and Evolution. 2000, 15: 487-495. 10.1006/mpev.1999.0744.
Kim I, Lee EM, Seol KY, Yun EY, Lee YB, Hwang JS, Jin BR: The mitochondrial genome of the Korean hairstreak, Coreana raphaelis (Lepidoptera: Lycaenidae). Insect Molecular Biology. 2006, 15: 217-225. 10.1111/j.1365-2583.2006.00630.x.
Tagami Y, Miura K: Distribution and prevalence of Wolbachia in Japanese populations of Lepidoptera. Insect Molecular Biology. 2004, 13: 359-364. 10.1111/j.0962-1075.2004.00492.x.
Jiggins FM, Bentley JK, Majerus MEN, Hurst GDD: How many species are infected with Wolbachia? Cryptic sex ratio distorters revealed to be common by intensive sampling. Proceedings of the Royal Society (London) B. 2001, 268: 1123-1126. 10.1098/rspb.2001.1632.
Hiroki M, Tagami Y, Miura K, Kato Y: Multiple infection with Wolbachia inducing different reproductive manipulations in the butterfly Eurema hecabe. Proceedings of the Royal Society (London) B. 2004, 271: 1751-1755. 10.1098/rspb.2004.2769.
Meier R, Shiyang K, Vaidya G, Ng PKL: DNA barcoding and taxonomy in Diptera: a tale of high intraspecific variability and low identification success. Systematic Biology. 2006, 55: 715-728. 10.1080/10635150600969864.
Goloboff PA, Farris JS, Nixon KC: TNT, a free program for phylogenetic analysis. Cladistics. 2008, 24: 774-786. 10.1111/j.1096-0031.2008.00217.x.
Goloboff P, Farris JS, Källersjö M, Oxelmann B, Ramirez M, Szumik C: Improvements to resampling measures of group support. Cladistics. 2003, 17: S26-S34. 10.1111/j.1096-0031.2001.tb00102.x.
Ronquist F, Huelsenbeck JP: MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19: 1572-1574. 10.1093/bioinformatics/btg180.
Nylander JAA: MrModeltest v.2.2. 2004, Evolutionary Biology Centre, Uppsala University: Program distributed by the author
Tavaré S: Some probabilistic and statistical problems in the analysis of DNA sequences. Some Mathematical Questions in Biology: DNA Sequence Analysis. Edited by: Miura RM. 1986, Providence, RI: American Mathematical Society, 57-86.
Akaike H: A new look at the statistical model identification. IEEE Transactions on Automatic Control. 1974, 19: 716-723. 10.1109/TAC.1974.1100705.
Zwickl DJ: Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. PhD Thesis. 2006, Austin: The University of Texas at Austin
Swofford DL: PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4.0b10. 2002, Sunderland, MA: Sinauer Associates
Clement M, Posada D, Crandall KA: TCS: a computer program to estimate gene geneologies. Molecular Ecology. 2000, 9: 1657-1659. 10.1046/j.1365-294x.2000.01020.x.
Quek S-P, Davies SJ, Ashton PS, Itino T, Pierce NE: The geography of diversification in mutualistic ants: a gene's-eye view into the Neogene history of Sundaland rain forests. Molecular Ecology. 2007, 16: 2045-2062. 10.1111/j.1365-294X.2007.03294.x.
Gaunt MW: An insect molecular clock dates the origin of the insects and accords with palaeontological and biogeographic landmarks. Molecular Biology and Evolution. 2002, 19: 748-761.
Quek S-P, Davies SJ, Itino T, Pierce NP: Codiversification in an ant-plant mutualism: stem texture and the evolution of host use in Crematogaster (Formicidae: Myrmicinae) inhabitants of Macaranga (Euphorbiaceae). Evolution. 2004, 58: 554-570.
Tamura K, Dudley J, Nei M, Kumar S: MEGA 4: Molecular Evolutionary genetics Analysis (MEGA) software version 4.0. Molecular Biology and Evolution. 2007, 24: 1596-1599. 10.1093/molbev/msm092.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R: DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003, 19: 2496-2497. 10.1093/bioinformatics/btg359.
Tajima F: Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983, 105: 437-460.
Nei M: Molecular Evolutionary Genetics. 1987, New York: Columbia University Press
Tajima F: Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989, 123: 585-595.
Tajima F: The effect of change in population size on DNA polymorphism. Genetics. 1989, 123: 597-601.
Excoffier L, Laval G, Schneider S: Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online. 2005, 1: 47-50.
Rogers AR: Genetic evidence for a Pleistocene population explosion. Evolution. 1995, 49: 608-615. 10.2307/2410314.
Schneider S, Excoffier L: Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics. 1999, 152: 1079-1089.
Kuhner MK: LAMARC 2.0: maximum likelihood and Bayesian estimation of population parameters. Bioinformatics. 2006, 22: 768-770. 10.1093/bioinformatics/btk051.
Maddison DR, Maddison W: MacClade 4.06. 2003, Sunderland, MA: Sinauer Associates
McDonald JH, Kreitman M: Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991, 351: 652-654. 10.1038/351652a0.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R: DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology. 1994, 3: 294-299.
Simon C, Frati F, Beckenbach AT, Crespi BJ, Liu H, Flook P: Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Annals of the Entomological Society of America. 1994, 87: 651-701.
Monteiro A, Pierce NE: Phylogeny of Bicyclus (Lepidoptera: Nymphalidae) inferred from COI, COII and EF-1alpha gene sequences. Molecular Phylogenetics and Evolution. 2001, 18: 264-281. 10.1006/mpev.2000.0872.
Simmons RB, Weller SJ: Utility and evolution of cytochrome b in insects. Molecular Phylogenetics and Evolution. 2001, 20: 196-210. 10.1006/mpev.2001.0958.
Zhou W, Rousset F, O'Neill S: Phylogeny and PCR-based classification of Wolbachia strains using wsp gene sequences. Proceedings of the Royal Society (London) B. 1998, 265: 509-515. 10.1098/rspb.1998.0324.
Acknowledgements
We thank Sujatha N. Kutty and Kathy Su for assistance in the laboratory, Rod Eastwood, Swee Peck Quek, Roger Vila, and four anonymous reviewers for comments on earlier versions of this manuscript, and the collectors who supplied specimens for this study. Navjot S. Sodhi provided helpful comments and logistical assistance for this study, and Dewi Prawiradilaga assisted with the coordination of fieldwork. DJL, DP, and RM were supported by grant R-154-000-270-112 from the Singapore Ministry of Education; NEP was supported by NSF grant DEB-0447242 and grants from the Putnam Expeditionary Fund and the Baker Fund.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
DJL coordinated fieldwork, collected specimens, performed the laboratory work and data analyses, and wrote the manuscript. DP assisted with fieldwork, contributed specimens, and helped write the manuscript. NEP assisted in the design of the study, contributed specimens, suggested additional analyses, and helped write the manuscript. RM conceived of the study, participated in the coordination of the study, suggested additional analyses, and helped write the manuscript.
Electronic supplementary material
12862_2008_870_MOESM1_ESM.pdf
Additional file 1: Additional information on the GeoDis analysis of our data including haplotype networks of individual and concatenated genes with step clades designated. (PDF 73 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lohman, D.J., Peggie, D., Pierce, N.E. et al. Phylogeography and genetic diversity of a widespread Old World butterfly, Lampides boeticus(Lepidoptera: Lycaenidae). BMC Evol Biol 8, 301 (2008). https://doi.org/10.1186/1471-2148-8-301
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-8-301