
Abstract
Background
The origin of functional innovation is among the key questions in biology. Recently, it has been shown that new genes could arise from non-coding DNA and that such novel genes are often involved in male reproduction.
Results
With the aim of identifying novel genes, we used the technique "generation of longer cDNA fragments from serial analysis of gene expression (SAGE) tags for gene identification (GLGI)" to extend 84 sex-biased 3'end SAGE tags that previously could not be mapped to the D. pseudoobscura transcriptome. Eleven male-biased and 33 female-biased GLGI fragments were obtained, of which 5 male-biased and 3 female-biased tags corresponded to putatively novel genes. This excess of novel genes with a male-biased gene expression pattern is consistent with previous results, which found novel genes to be primarily expressed in male reproductive tissues. 5' RACE analysis indicated that these novel transcripts are very short in length and could contain introns. Interspecies comparisons revealed that most novel transcripts show evidence for purifying selection.
Conclusion
Overall, our data indicate that among sex-biased genes a considerable number of novel genes (~2–4%) exist in D. pseudoobscura, which could not be predicted based on D. melanogaster gene models.
Similar content being viewed by others

Background
Understanding functional innovation is one of the most interesting questions in biology. One important mechanism of functional innovation involves changes in gene expression [1] caused by cis-regulatory mutations [2]. While structural mutations within existing genes are an alternative mechanism to generate new functions [3], another possibility is the emergence of new genes. Several possible mechanisms are known to be involved in creating novel genes [4]. The best described origins of novel genes are gene duplication [5] and exon shuffling [6, 7]. Recently it has been shown that novel genes could also originate de novo from non-coding regions [8]. Comparative genome analyses permit the identification of previously uncharacterized genes through sequence conservation, but the identification of rapidly evolving genes or genes of very recent origin is frequently restricted to in silico predictions. As novel genes are typically short [8, 9], these may be easily missed. Alternatively, gene expression could serve as a good indicator for the presence of a gene. Hence, either Expressed Sequence Tag (EST) databases or reverse SAGE [10, 11] could be used to identify novel transcripts.
Drosophila served as model for the identification of novel genes since the 1990s. One of first novel genes in this genus was jingwei in D. melanogaster [12], which is a fusion of two genes, a retroposed copy of the alcohol dehydrogenase (Adh) gene and a duplicated copy of the yellow emperor (ymp) gene [13]. Since then several studies applied phylogenetic methods to the growing databases aiming for the identification of novel genes. The majority of the novel genes have a sex-biased gene expression and some reports suggested that sex-biased genes change their expression pattern more rapidly than unbiased genes [14, 15]. Furthermore, male-biased genes were shown to have a higher rate of protein evolution than unbiased genes [16–18]. In a recent report comparing the pattern of gene expression in D. melanogaster and D. pseudoobscura we failed to find evidence for an unconditionally faster rate of sequence evolution of male-biased genes. Rather, only genes with a male-biased gene expression in D. melanogaster were found to evolve faster. Genes with a male-biased gene expression in D. pseudoobscura only were evolving at a similar rate as unbiased genes [19]. As a large proportion of the sex-biased tags could not be mapped to the corresponding genes in D. pseudoobscura, the analysis of these tags should shed further light onto the pattern of protein evolution of sex-biased genes in D. pseudoobscura.
In this study, we identified eight novel genes with sex-biased gene expression in D. pseudoobscura using GLGI (Generation of longer cDNA fragments from serial analysis of gene expression tags for gene identification). Consistent, with previous results [8, 9], we observed significantly more novel genes with a male bias than with a female bias in gene expression. Interestingly, we found no significant excess of X-linked novel genes, as has been reported in the previous studies [8, 9].
Results
GLGI analysis
We used recently published SAGE data to identify sex-biased tags in D. pseudoobscura [19]. Previous analysis showed a substantially higher efficiency of tag to gene mapping for male-biased tags than for female-biased tags [19]. As the D. pseudoobscura genome annotation is heavily based on D. melanogaster gene models, this may be due to a higher proportion of novel genes among the genes with a female-biased gene expression. To test this, we selected 20 male-biased and 64 female-biased tags that were previously not mapped, relatively highly expressed and showed significant difference in expression between the sexes (p < 0.001), for further analysis. Using the GLGI method, we successfully generated longer 3'cDNA fragments for 44 SAGE tags. This success rate is in agreement with a previous GLGI analysis [20]. The GLGI fragments include 11 male-biased and 33 female-biased tags (Table 1). Thirty female-biased (91%) tags were mapped close to putative orthologs of D. melanogaster in the 3' end while only six male-biased (55%) tags were in this category (Additional file 1, Tables 1a and 1b). The remaining eight tags are falling into genomic regions that show no sequence conservation between D. melanogaster and D. pseudoobscura and no genes are annotated in D. melanogaster. Furthermore, we could not identify these regions by either nucleotide or protein BLAST search in any other species whose genome is completely sequenced except for D. persimilis. This suggests that these tags potentially represent novel genes that arose in the obscura group and are absent in other Drosophila species. Alternatively, these genes may have diverged from their orthologs in other Drosophila species to such an extent that they could not be identified by BLAST algorithm. We identified five male-biased and three female-biased transcripts that are putatively novel. Hence, 46% of the male-biased and 9% of the female-biased tags are putatively novel in the genome of D. pseudoobscura. The difference between male- and female-biased tags is significant (p = 0.02), suggesting that the majority of the novel genes show male biased expression [8, 9].
Modified SAGE-GLGI approach
As the GLGI analysis of sex-biased genes indicated a high number of novel genes, we were interested if this applies only to highly sex-biased genes or is a more general phenomenon. We sequenced 126 clones using a cDNA library that mimics the generation of GLGI fragments from SAGE tags. Of the 126 clones sequenced, 89 were unique and the remaining clones were redundant. From these 89 clones, 46 clones could be matched to the SAGE expression data [19]. Out of 89, 35 clones could be mapped to the annotated genes of D. pseudoobscura. Thirty-two clones mapped close to predicted genes in the 3' end. Six clones fell unambiguously into genomic regions for which genes are predicted in D. melanogaster but not in D. pseudoobscura. For these clones we noted that the incomplete annotation in D. pseudoobscura is probably due to duplications of the genes in these regions. One clone was mapped to a mitochondrial rRNA gene that is not included in the annotation of D. pseudoobscura. Three clones were missing in the D. pseudoobscura genome assembly but exist in the trace archive database [21] and have homologous sequences in D. melanogaster. Four clones were too short to be mapped unambiguously and two clones were anti-sense transcripts. For four clones of the six remaining ones, no prediction exists in either of the species despite sequence conservation. Hence, these genes are not novel to D. pseudoobscura. The remaining two clones are putatively novel genes as these sequences are located in genomic regions with no gene predicted in D. pseudoobscura and no orthologous region could be identified in D. melanogaster by a BLAST search. Both of these clones map to D. pseudoobscura chromosome arm XR. One of them matched a SAGE tag that was present in the male and female SAGE library [19] and showed female-biased expression. This sequence is located 72-nucleotides away from a predicted protein coding gene GA23511 in D. pseudoobscura. However, TBLASTN search of this protein against other completely sequenced Drosophila genomes did not reveal any hit except in the sibling species D. persimilis. The other clone did not match to any of the SAGE tags and was located in a region of the genome where no gene is predicted.
Chromosomal distribution of novel sex-biased genes
Six of the eight GLGI sequences representing putatively novel genes are located on autosomes, while only two were located on the X-chromosome. Based on the number of predicted genes on X- and autosomes in the D. pseudoobscura annotation release 2.0, we tested for an overrepresentation of novel genes on the X-chromosome and did not find support for an enrichment of new genes on one chromosome (p = 0.72, Fisher's exact test). Even after accounting for the two novel genes identified by the modified GLGI approach, we still found no evidence for an enrichment of novel genes on the X-chromosome (p = 0.75, Fisher's exact test).
Structure of the novel genes
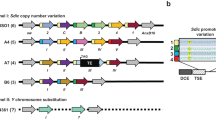
In order to obtain full-length transcripts of the novel genes identified by GLGI, we performed 5' RACE analysis. All the five male biased tags were successfully amplified with 5' RACE. Out of three female-biased clones, only one could be cloned and sequenced. Based on the 5' RACE analysis, we could show that the transcripts of these novel genes are very short, ranging from 138 base pairs to 442 base pairs, yielding very short coding sequences ranging from 29 amino acids to 95 amino acids (Figure 1). These transcripts have start and stop codons at the same position in the orthologous regions of the closely relatives D. persimilis and D. miranda and the complete ORF of these transcripts is conserved in the three species. Interestingly, the gene containing the shortest ORF was found to possess an intron. Nevertheless, further experiments are required to determine whether or not these short ORFs are translated into peptides. For one of the male-biased transcripts (SAGE_M_22_3), no potential coding sequence could be obtained and hence we assumed that it could be a non protein coding transcript.

Novel genes in D. pseudoobscura. List of novel genes obtained by analyzing SAGE, GLGI and 5'RACE with their protein length and chromosomal location. Black boxes represent coding regions, grey boxes represent non-coding regions and UTRs and white boxes represent introns. For the gene SAGE_M_71 (*), the chromosome location is determined based on its synteny in D. persimilis.
Cross-species conservation of novel genes
We took advantage of the 12 sequenced Drosophila genomes and performed TBLASTN searches with default parameters provided by flybase [22] to identify orthologs of the novel genes. Apart from D. persimilis, a very close relative of D. pseudoobscura, we did not detect any sequence similarity in the other species. Using PCR primers conserved between D. pseudoobscura and D. persimilis (See Additional file 1, Table 2), we successfully amplified the novel genes in D. miranda. Successful amplification of orthologs of the new, putatively protein coding genes from cDNA indicated their conservation in D. miranda. This implies that the novel genes arose before the split of D. pseudoobscura and D. miranda, which occurred about 2–5 MYA [23–25]. We assessed the protein coding potential of these short transcripts using the QRNA software, which distinguishes between coding alignments, conserved non-coding alignments and RNA secondary structure [26]. For none of the six transcripts did we find support for a protein coding potential (Table 2). However, as the sequences were short and the divergence between D. miranda and D. pseudoobscura is low, it is not clear how powerful this method is. Furthermore, it has been noted previously that failure of QRNA to identify a coding potential does not exclude the possibility of small ORFs [27]. Except for one (SAGE_M_79), all putatively protein coding genes had a ratio of synonymous to non-synonymous substitutions smaller than one, indicating purifying selection on a protein sequence. SAGE_M_79 contained only non-synonymous substitutions and no synonymous substitutions (Table 2). Supporting this hypothesis, the ratio of polymorphic non-synonymous substitutions to synonymous substitutions is also smaller than one for the polymorphic loci (Table 3). On the other hand, the likelihood ratio test (LRT), which tests if the dN/dS ratio is significantly lower than 1, was not significant for any of the loci except SAGE_M_71 (Table 2). However, it should be noted that the power of LRT is affected by sequence length, divergence and number of species [28] and may lead to a lower power in our case.
Evolution of novel genes
Recently evolved novel genes were shown to be under adaptive evolution in D. melanogaster lineage species [8, 9]. To understand the pattern of variation and to test the deviations from neutral expectations in the novel genes of D. pseudoobscura, we sequenced the protein coding genes in one D. pseudoobscura population from Mesa-Verde (Colorado, USA, n = 8). Neutrality tests like Tajima's D (Table 3), Fu & Li's D, Fu & Li's F and Fu's Fs did not show a significant deviation from neutral expectations. We also performed a McDonald-Kreitman test [29] using D. miranda, which basically assumes that under neutrality, the ratio of fixed differences between species to polymorphic differences within species should be similar for both the synonymous and non-synonymous sites. None of the novel loci showed significant deviations from neutral expectations (Table 4). It should be noted, however, that due to the short sequence of the loci analyzed the statistical power to detect selection is limited. A multi locus HKA test [30], which tests for the deviations between the polymorphism within species versus divergence between species across different loci was also not significant, suggesting that none of the genes deviates significantly from the remaining ones.
Discussion
In this report we showed that the extension of SAGE tags by GLGI is a powerful approach for the identification of novel genes.
Origin of the novel genes
Novel genes can be generated through different evolutionary trajectories: retrotransposition [31], gene/genome duplication [5, 32] and exon shuffling [7]. All these processes have in common that novel genes are built from existing genes or exons. Hence, it is expected that these building blocks should be detectable in the genome. We performed BLAST search of the novel genes against the D. pseudoobscura genome and detected only for one gene (SAGE_M_310) two BLAST hits. These two hits with complete sequence conservation were separated by 4.2-kilobase and the entire duplicated region spans approximately 1.1-kilobase with 92% identity. While we cannot rule out an assembly error, this observation suggests a recent duplication of the entire region encompassing the novel gene. Nevertheless, it is also apparent that both copies qualify as novel genes by our criteria.
Given that we lack support for the origin of the described novel genes from already existing ones, we favour the hypothesis that they are derived from previously non-coding regions. Recently, several cases of such novel genes derived from non-coding sequences were described in other Drosophila species [8, 9]. Like in our study, the functional evidence came from gene expression in the focal species. In order to obtain additional support for functionality of these transcripts, we also tested for gene expression in a related species and found all of the novel genes also to be transcribed in D. miranda. But, we cannot exclude that these transcripts are non-coding or the regions are spuriously expressed in the closely related species. However, such small ORFs were previously reported in Drosophila and other species and are known to be functional [33–36]. The generation of novel genes by mutation appears a very unlikely event and the chance of generating a functional gene by random mutations decreases with the length of a gene. Hence, it is particularly interesting that all the novel genes identified in this study and previous ones [8, 9] tend to be very short. Once more novel genes become available in combination with information about their date of origin, it will be possible to infer the probability to generate novel genes by random mutations. In particular, we will gain more insight if some sequences are more prone to develop into functional genes.
Novel genes are male-biased
In this report we searched for novel genes among SAGE tags showing either a strong male or female expression bias. Despite that we screened a considerably larger number of female-biased SAGE tags, the majority of the novel genes had a male-biased gene expression pattern. This pattern is consistent with previous results [8, 12, 37–39]. It appears to be a general trend that many novel genes possess functional significance in males relative to females, but the reason for this trend is still not completely clear. Possible reasons include a higher transcription rate in male germline, greater functional pleiotropy of genes expressed in females and/or sexual competition [40]. Alternatively, there could be more testis specific promoter sequences in the genome.
Non-preferential X chromosomal location of novel genes
There has been considerable controversy about the genomic location of male-biased genes. Earlier theoretical predictions [41] as well as expression studies in mice [39] suggested that recessive genes conferring an advantage to males should be located on the X-chromosome. Expression studies in Drosophila [42] and C. elegans [43] showed, however, that male biased genes are preferentially located on the autosomes. This discrepancy could be explained by X-chromosome inactivation during spermatogenesis [44–46]. The preferential X-linkage of novel genes, which show a male expression pattern [8] requires further explanation. It has been suggested that genes conferring an advantage to males first originate on the X-chromosome and move to an autosomal location later in evolution [42, 47]. Hence, it is conceivable that these novel male-biased genes do not serve functions that are essential during male spermatogenesis. Once such novel genes become essential, they could move to the autosomes. It was also suggested that mutations generating de novo genes might occur more often on the X chromosome or fix more readily [8]. The novel genes we identified in this analysis can be dated from a minimum of 2–5 MYA (D. pseudoobscura and D. miranda divergence time [23–25]) to a maximum of 13–15 MYA (divergence of obscura group [25, 48]). It is possible that the novel genes identified in this study may be older compared to the novel genes reported by Levine et al. [8], which was 2.5 MYA and if this is true, then these genes might have already moved to autosomes [42]. More data are required to see if the discrepancy between our study and the results of Levine et al. [8] are related to the difference in age or could be simply attributed to sampling effects due to the small number of genes in both studies.
High incidence of novel genes in D. pseudoobscura
The discovery of previously uncharacterized transcripts is an observation common to many SAGE and EST sequencing experiments [11, 39, 49, 50]. Nevertheless, the identification of evolutionary novelties requires also the absence of sequence conservation in related species. This approach was pioneered by Schmid and Tautz [51], who studied the conservation of cDNAs across various species using hybridization to genomic Southern blots. More recently, Levine et al. [8] specifically searched for lineage specific genes by BLAST search of all cDNA sequences available in D. melanogaster against D. yakuba, D. erecta and D. ananassae. A similar approach was also pursued for D. yakuba by BLAST search of ESTs against the genomic sequences of D. yakuba, D. melanogaster, D. erecta and D. ananassae [9]. In this study, we focused on novel genes in D. pseudoobscura. Our previous SAGE analysis indicated that about 12% of the genes surveyed showed a significant sex bias. Of these, 73% could not be mapped to the D. pseudoobscura transcriptome and the majority of them possessed female-biased gene expression. This discrepancy could arise from a high number of novel female-biased genes in D. pseudoobscura. Contrary to our expectations and in agreement with the other studies we observed a high number of male-biased novel genes compared to female-biased novel genes. This result could be the outcome of different rates of evolution of male and female-biased genes [15, 18] in D. pseudoobscura.
In this study, we showed that 18% of the unmapped SAGE tags originate from putatively novel genes that arose in the obscura group. Hence, among the sex-biased genes approximately 2% are novel. In a cDNA library from D. pseudoobscura females we observed about 4% novel genes. This high incidence of novel genes in D. pseudoobscura underlines the need to supplement available genomic sequences with a thorough characterization of their transcriptome. In wake of the recent advances in the sequencing technology, we anticipate that this is already in close reach.
Conclusion
We identified eight novel genes with sex-biased gene expression in D. pseudoobscura from unmapped SAGE tags using the GLGI method. In agreement with previous results from the other Drosophila species, a majority of these novel genes are male-biased in gene expression. Interestingly, we found no significant excess of X-linked novel genes. Overall, our data show that a considerable number of novel sex-biased genes exists in D. pseudoobscura that could not be predicted based on D. melanogaster gene models, which underlines the need to supplement available genomic sequences with a thorough characterization of their transcriptome.
Methods
GLGI analysis
D. pseudoobscura SAGE tags were obtained from Metta et al. [19]. The GLGI procedure for the 3' extension of the SAGE tags was performed according to Chen et al. [52]. In brief, total RNA was extracted separately from 15 male and 15 female virgin flies using Trizol (Invitrogen, Carlsbad, CA) and treated with DNAseI (MBI Fermentas) to digest the genomic DNA residues. PolyA mRNA was isolated using a 5' biotinylated and anchored oligo dT primer (5' biotin-ATCTAGAGCGGCCGC(T)16V) and streptavidin beads (Dynal). Double strand cDNA was synthesized using the "Double strand cDNA synthesis kit" (Invitrogen, Carlsbad, CA) and digested with the NlaIII isoschizomer Hin1II (MBI Fermentas). The 3' end fragments of the digested cDNA was recovered with Dynal beads. PCR amplification was performed using SAGE tag as 5' primer and anchoring sequence to the polyA was used as 3' primer. The PCR product was then cloned into TA vector (Invitrogen, Carlsbad, CA) and sequenced.
Modified SAGE-GLGI method for detecting novel genes
To verify the consistency of the abundance of novel genes, we also performed an independent analysis, which is a modification of the SAGE-GLGI approach. We used mRNA isolated from virgin females. Double stranded cDNA was digested with NlaIII and an adapter (5'-GCCTCCCTCGCGCCATCAGCATG-3' and 5'-CTGATGGCGCGAGGGAGGC-3') was ligated to the 5' end of the digested cDNA fragments. The resulting template was amplified using an adapter specific primer and the primer anchoring to oligo dT. The products were cloned into TA vector and sequenced using M13 primers.
Bioinformatics
BLASTN search was performed using the D. pseudoobscura database to identify the location of the GLGI (and modified SAGE-GLGI) fragments in the genome. As the D. pseudoobscura genome is only coarsely annotated we re-annotated the D. pseudoobscura genome for the regions of interest. We noted that a GeneWise analysis using the D. melanogaster gene model often results in a more complete protein prediction than the one available in the annotation database [22]. Hence we first performed GeneWise using those D. melanogaster genes that mapped in the proximity of the GLGI fragments to test if the D. pseudoobscura gene prediction may have been incomplete and the GLGI fragment matches to the extended protein coding region. As neither our GeneWise annotations nor the available annotation of D. pseudoobscura contained UTR sequences for most genes, we also used ab initio gene predictions. Specifically, we submitted the genomic region spanning the 5' part of the closest gene in the correct orientation and the genomic region matching to the GLGI fragment to a Genescan prediction. The coding potential of the transcripts was assessed using QRNA program [26].
Structure of novel genes
To identify the 5' boundaries of the novel GLGI sequences, we performed 5' RACE (rapid amplification of cDNA ends) using GeneRacer RACE ready cDNA kit (Invitrogen, Carlsbad, CA). The 5' RACE analysis was performed on an independent extraction of total RNA.
Evolution of novel genes
Eight individuals of D. pseudoobscura from Mesa-Verde (Colarado, USA) were sequenced for the five novel genes putatively coding for a protein (see Additional File 1, Table 2 for the primer sequences). Orthologous regions in D. persimilis were obtained from flybase [22]. One individual of D. miranda, a close relative of D. pseudoobscura, was obtained from the Tucson stock center (stock number: 14011-0101.08) and sequenced for these loci at both genomic as well as transcriptomic level. A typical cycling profile consisted of 3 min denaturation at 94°C followed by 35 cycles of 94°C for 40 sec, 50°C for 50 sec, and extension at 72°C for 1 min. PCR products were sequenced for both strands using DYEnamic ET Terminator Sequencing Kit (GE Health care Bio-Sciences AB, Sweden) according to manufacturer's instructions. The extension products were purified with Sephadex G-50 fine (GE Health care Bio-Sciences AB, Sweden) and separated on a MegaBACE 500 automated capillary sequencer. Forward and reverse strands were assembled using CodonCode Aligner version 2.0.1 [53]. The sequences were aligned using clustalW [54] and standard neutrality tests and McDonald-Kreitman test were performed using DnaSP version 4.10 [55]. The pair wise synonymous and non synonymous substitutions between D. pseudoobscura and D. miranda were obtained using yn00 program of PAML [56]. Likelihood ratio test was performed using codeml program of PAML using three species tree to test if the dN/dS ratio is significantly lower than 1. Multi locus HKA test [30] was performed using HKA program written by Jody Hey [57]. All the sequences were submitted to GenBank (accession numbers EU379026–EU379076).
Abbreviations
- SAGE:
-
serial analysis of gene expression
- GLGI:
-
generation of longer cDNA fragments from serial analysis of gene expression tags for gene identification
- RACE:
-
rapid amplification of cDNA ends
- UTR:
-
untranslated region
- ORF:
-
open reading frame
- MYA:
-
million years ago.
References
Ferea TL, Botstein D, Brown PO, Rosenzweig RF: Systematic changes in gene expression patterns following adaptive evolution in yeast. Proc Natl Acad Sci U S A. 1999, 96: 9721-9726. 10.1073/pnas.96.17.9721.
Wray GA: The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 2007, 8: 206-216. 10.1038/nrg2063.
Hoekstra HE, Coyne JA: The locus of evolution: evo devo and the genetics of adaptation. Evolution. 2007, 61 (5): 995-1016. 10.1111/j.1558-5646.2007.00105.x.
Long M, Betran E, Thornton K, Wang W: The origin of new genes: glimpses from the young and old. Nat Rev Genet. 2003, 4: 865-875. 10.1038/nrg1204.
Ohno S: Evolution by gene duplication. 1970, New York, Springer-Verlag
Patthy L: Protein Evolution by Exon-shuffling. 1995, New York, Springer-Verlag
Patthy L: Genome evolution and the evolution of exon-shuffling--a review. Gene. 1999, 238: 103-114. 10.1016/S0378-1119(99)00228-0.
Levine MT, Jones CD, Kern AD, Lindfors HA, Begun DJ: Novel genes derived from noncoding DNA in Drosophila melanogaster are frequently X-linked and exhibit testis-biased expression. Proc Natl Acad Sci U S A. 2006, 103: 9935-9939. 10.1073/pnas.0509809103.
Begun DJ, Lindfors HA, Kern AD, Jones CD: Evidence for de novo evolution of testis-expressed genes in the Drosophila yakuba/Drosophila erecta clade. Genetics. 2007, 176: 1131-1137. 10.1534/genetics.106.069245.
Richards M, Tan SP, Chan WK, Bongso A: Reverse serial analysis of gene expression (SAGE) characterization of orphan SAGE tags from human embryonic stem cells identifies the presence of novel transcripts and antisense transcription of key pluripotency genes. Stem Cells. 2006, 24: 1162-1173. 10.1634/stemcells.2005-0304.
Chen J, Sun M, Lee S, Zhou G, Rowley JD, Wang SM: Identifying novel transcripts and novel genes in the human genome by using novel SAGE tags. Proc Natl Acad Sci U S A. 2002, 99: 12257-12262. 10.1073/pnas.192436499.
Long M, Langley CH: Natural selection and the origin of jingwei, a chimeric processed functional gene in Drosophila. Science. 1993, 260: 91-95. 10.1126/science.7682012.
Wang W, Zhang J, Alvarez C, Llopart A, Long M: The origin of the jingwei gene and the complex modular structure of its parental gene, yellow emperor, in Drosophila melanogaster. Mol Biol Evol. 2000, 17: 1294-1301.
Ranz JM, Castillo-Davis CI, Meiklejohn CD, Hartl DL: Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science. 2003, 300: 1742-1745. 10.1126/science.1085881.
Musters H, Huntley MA, Singh RS: A genomic comparison of faster-sex, faster-X, and faster-male evolution between Drosophila melanogaster and Drosophila pseudoobscura. J Mol Evol. 2006, 62: 693-700. 10.1007/s00239-005-0165-5.
Meiklejohn CD, Parsch J, Ranz JM, Hartl DL: Rapid evolution of male-biased gene expression in Drosophila. Proc Natl Acad Sci U S A. 2003, 100: 9894-9899. 10.1073/pnas.1630690100.
Zhang Z, Hambuch TM, Parsch J: Molecular evolution of sex-biased genes in Drosophila. Mol Biol Evol. 2004, 21: 2130-2139. 10.1093/molbev/msh223.
Haerty W, Jagadeeshan S, Kulathinal RJ, Wong A, Ravi Ram K, Sirot LK, Levesque L, Artieri CG, Wolfner MF, Civetta A, Singh RS: Evolution in the fast lane: rapidly evolving sex-related genes in Drosophila. Genetics. 2007, 177: 1321-1335. 10.1534/genetics.107.078865.
Metta M, Gudavalli R, Gibert JM, Schlotterer C: No accelerated rate of protein evolution in male-biased Drosophila pseudoobscura genes. Genetics. 2006, 174: 411-420. 10.1534/genetics.106.057414.
Silva AP, Chen J, Carraro DM, Wang SM, Camargo AA: Generation of longer 3' cDNA fragments from massively parallel signature sequencing tags. Nucleic Acids Res. 2004, 32: e94-10.1093/nar/gnh095.
The NCBI database. [http://www.ncbi.nlm.nih.gov]
Flybase. [http://www.flybase.org]
Wang RL, Hey J: The speciation history of Drosophila pseudoobscura and close relatives: inferences from DNA sequence variation at the period locus. Genetics. 1996, 144: 1113-1126.
Wang RL, Wakeley J, Hey J: Gene flow and natural selection in the origin of Drosophila pseudoobscura and close relatives. Genetics. 1997, 147: 1091-1106.
Gao JJ, Watabe HA, Aotsuka T, Pang JF, Zhang YP: Molecular phylogeny of the Drosophila obscura species group, with emphasis on the Old World species. BMC Evol Biol. 2007, 7: 87-10.1186/1471-2148-7-87.
Rivas E, Eddy SR: Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics. 2001, 2: 8-10.1186/1471-2105-2-8.
Rivas E, Klein RJ, Jones TA, Eddy SR: Computational identification of noncoding RNAs in E. coli by comparative genomics. Curr Biol. 2001, 11: 1369-1373. 10.1016/S0960-9822(01)00401-8.
Anisimova M, Bielawski JP, Yang Z: Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol. 2001, 18: 1585-1592.
McDonald JH, Kreitman M: Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991, 351: 652-654. 10.1038/351652a0.
Hudson RR, Kreitman M, Aguade M: A test of neutral molecular evolution based on nucleotide data. Genetics. 1987, 116: 153-159.
Betran E, Wang W, Jin L, Long M: Evolution of the phosphoglycerate mutase processed gene in human and chimpanzee revealing the origin of a new primate gene. Mol Biol Evol. 2002, 19: 654-663.
Wang W, Yu H, Long M: Duplication-degeneration as a mechanism of gene fission and the origin of new genes in Drosophila species. Nat Genet. 2004, 36: 523-527. 10.1038/ng1338.
Kondo T, Hashimoto Y, Kato K, Inagaki S, Hayashi S, Kageyama Y: Small peptide regulators of actin-based cell morphogenesis encoded by a polycistronic mRNA. Nat Cell Biol. 2007, 9: 660-665. 10.1038/ncb1595.
Savard J, Marques-Souza H, Aranda M, Tautz D: A segmentation gene in tribolium produces a polycistronic mRNA that codes for multiple conserved peptides. Cell. 2006, 126: 559-569. 10.1016/j.cell.2006.05.053.
Galindo MI, Pueyo JI, Fouix S, Bishop SA, Couso JP: Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biol. 2007, 5: e106-10.1371/journal.pbio.0050106.
Tupy JL, Bailey AM, Dailey G, Evans-Holm M, Siebel CW, Misra S, Celniker SE, Rubin GM: Identification of putative noncoding polyadenylated transcripts in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2005, 102: 5495-5500. 10.1073/pnas.0501422102.
Nurminsky DI, Nurminskaya MV, De Aguiar D, Hartl DL: Selective sweep of a newly evolved sperm-specific gene in Drosophila. Nature. 1998, 396: 572-575. 10.1038/25126.
Betran E, Long M: Dntf-2r, a young Drosophila retroposed gene with specific male expression under positive Darwinian selection. Genetics. 2003, 164: 977-988.
Wang PJ, McCarrey JR, Yang F, Page DC: An abundance of X-linked genes expressed in spermatogonia. Nat Genet. 2001, 27: 422-426. 10.1038/86927.
Zhang Y, Sturgill D, Parisi M, Kumar S, Oliver B: Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature. 2007, 450: 233-237. 10.1038/nature06323.
Rice WR: Sex-Chromosomes and the Evolution of Sexual Dimorphism. Evolution. 1984, 38: 735-742. 10.2307/2408385.
Parisi M, Nuttall R, Naiman D, Bouffard G, Malley J, Andrews J, Eastman S, Oliver B: Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science. 2003, 299: 697-700. 10.1126/science.1079190.
Reinke V, Smith HE, Nance J, Wang J, Van Doren C, Begley R, Jones SJ, Davis EB, Scherer S, Ward S, Kim SK: A global profile of germline gene expression in C. elegans. Mol Cell. 2000, 6: 605-616. 10.1016/S1097-2765(00)00059-9.
Lifschytz E, Lindsley DL: The role of X-chromosome inactivation during spermatogenesis. Proc Natl Acad Sci U S A. 1972, 69: 182-186. 10.1073/pnas.69.1.182.
Kelly WG, Schaner CE, Dernburg AF, Lee MH, Kim SK, Villeneuve AM, Reinke V: X-chromosome silencing in the germline of C. elegans. Development. 2002, 129: 479-492.
Wu CI, Xu EY: Sexual antagonism and X inactivation--the SAXI hypothesis. Trends Genet. 2003, 19: 243-247. 10.1016/S0168-9525(03)00058-1.
Schlötterer C: Genetics. Where do male genes live?. Science. 2003, 299: 670-671. 10.1126/science.1082030.
Barrio E, Latorre A, Moya A, Ayala FJ: Phylogenetic reconstruction of the Drosophila obscura group, on the basis of mitochondrial DNA. Mol Biol Evol. 1992, 9: 621-635.
Gorski SM, Chittaranjan S, Pleasance ED, Freeman JD, Anderson CL, Varhol RJ, Coughlin SM, Zuyderduyn SD, Jones SJ, Marra MA: A SAGE approach to discovery of genes involved in autophagic cell death. Curr Biol. 2003, 13: 358-363. 10.1016/S0960-9822(03)00082-4.
Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, Yamanaka I, Kiyosawa H, Yagi K, Tomaru Y, Hasegawa Y, Nogami A, Schonbach C, Gojobori T, Baldarelli R, Hill DP, Bult C, Hume DA, Quackenbush J, Schriml LM, Kanapin A, Matsuda H, Batalov S, Beisel KW, Blake JA, Bradt D, Brusic V, Chothia C, Corbani LE, Cousins S, Dalla E, Dragani TA, Fletcher CF, Forrest A, Frazer KS, Gaasterland T, Gariboldi M, Gissi C, Godzik A, Gough J, Grimmond S, Gustincich S, Hirokawa N, Jackson IJ, Jarvis ED, Kanai A, Kawaji H, Kawasawa Y, Kedzierski RM, King BL, Konagaya A, Kurochkin IV, Lee Y, Lenhard B, Lyons PA, Maglott DR, Maltais L, Marchionni L, McKenzie L, Miki H, Nagashima T, Numata K, Okido T, Pavan WJ, Pertea G, Pesole G, Petrovsky N, Pillai R, Pontius JU, Qi D, Ramachandran S, Ravasi T, Reed JC, Reed DJ, Reid J, Ring BZ, Ringwald M, Sandelin A, Schneider C, Semple CA, Setou M, Shimada K, Sultana R, Takenaka Y, Taylor MS, Teasdale RD, Tomita M, Verardo R, Wagner L, Wahlestedt C, Wang Y, Watanabe Y, Wells C, Wilming LG, Wynshaw-Boris A, Yanagisawa M, Yang I, Yang L, Yuan Z, Zavolan M, Zhu Y, Zimmer A, Carninci P, Hayatsu N, Hirozane-Kishikawa T, Konno H, Nakamura M, Sakazume N, Sato K, Shiraki T, Waki K, Kawai J, Aizawa K, Arakawa T, Fukuda S, Hara A, Hashizume W, Imotani K, Ishii Y, Itoh M, Kagawa I, Miyazaki A, Sakai K, Sasaki D, Shibata K, Shinagawa A, Yasunishi A, Yoshino M, Waterston R, Lander ES, Rogers J, Birney E, Hayashizaki Y: Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002, 420: 563-573. 10.1038/nature01266.
Schmid KJ, Tautz D: A screen for fast evolving genes from Drosophila. Proc Natl Acad Sci U S A. 1997, 94: 9746-9750. 10.1073/pnas.94.18.9746.
Chen J, Lee S, Zhou G, Wang SM: High-throughput GLGI procedure for converting a large number of serial analysis of gene expression tag sequences into 3' complementary DNAs. Genes Chromosomes Cancer. 2002, 33: 252-261. 10.1002/gcc.10017.
CodonCode Aligner. [http://www.codoncode.com]
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22: 4673-4680. 10.1093/nar/22.22.4673.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R: DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003, 19: 2496-2497. 10.1093/bioinformatics/btg359.
Yang Z: PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007, 24: 1586-1591. 10.1093/molbev/msm088.
HKA Program. [http://lifesci.rutgers.edu/~heylab/index.html]
Acknowledgements
We thank V. Nolte for assistance with the GLGI experiments. We are thankful to the CS lab members for continuous support and discussion. Special thanks to S. Schaeffer for sharing the D. pseudoobscura population. This work has been supported by Fonds zur Förderung der wissenschaftlichen Forschung (FWF) grants (No. P17005, P17373) to CS. MM is supported by a Doktoratsstipendium of the Veterinärmedizinischen Universität Wien.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
MM carried out the experiments, analyzed the data and drafted the preliminary manuscript. CS conceived of the study, and participated in its design and coordination and drafted the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12862_2008_751_MOESM1_ESM.doc
Additional file 1: 1a – List of female-biased tags with successful GLGI amplification. 1b – List of male-biased tags with successful GLGI amplification. †The gene is not predicted in the database of D. pseudoobscura. "Within the gene" indicates that the tag is located within the transcript but as it belongs to a mitochondrial gene or an unpredicted gene, it was not mapped previously. "Splice form" indicates that the SAGE tag is falling within an intron of a gene and so it is likely to be a splice variant. "Anti-sense" indicates that the GLGI sequence is matched in the anti-sense direction of the predicted gene. *This tag did not give any hit in D. pseudoobscura genome assembly but exists in the trace archive. The chromosome location is determined based on the sequence synteny in D. persimilis. "Sequencing artefact" indicates that the tag is falling within the gene but the region is having no sequence coverage (represented by 'n' in the database). 2 – Primers used for cross-species amplification as well as population analysis of D. pseudoobscura . (DOC 54 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Metta, M., Schlötterer, C. Male-biased genes are overrepresented among novel Drosophila pseudoobscurasex-biased genes. BMC Evol Biol 8, 182 (2008). https://doi.org/10.1186/1471-2148-8-182
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-8-182