
Abstract
Background
Our understanding of the eukaryotic tree of life and the tremendous diversity of microbial eukaryotes is in flux as additional genes and diverse taxa are sampled for molecular analyses. Despite instability in many analyses, there is an increasing trend to classify eukaryotic diversity into six major supergroups: the 'Amoebozoa', 'Chromalveolata', 'Excavata', 'Opisthokonta', 'Plantae', and 'Rhizaria'. Previous molecular analyses have often suffered from either a broad taxon sampling using only single-gene data or have used multigene data with a limited sample of taxa. This study has two major aims: (1) to place taxa represented by 72 sequences, 61 of which have not been characterized previously, onto a well-sampled multigene genealogy, and (2) to evaluate the support for the six putative supergroups using two taxon-rich data sets and a variety of phylogenetic approaches.
Results
The inferred trees reveal strong support for many clades that also have defining ultrastructural or molecular characters. In contrast, we find limited to no support for most of the putative supergroups as only the 'Opisthokonta' receive strong support in our analyses. The supergroup 'Amoebozoa' has only moderate support, whereas the 'Chromalveolata', 'Excavata', 'Plantae', and 'Rhizaria' receive very limited or no support.
Conclusion
Our analytical approach substantiates the power of increased taxon sampling in placing diverse eukaryotic lineages within well-supported clades. At the same time, this study indicates that the six supergroup hypothesis of higher-level eukaryotic classification is likely premature. The use of a taxon-rich data set with 105 lineages, which still includes only a small fraction of the diversity of microbial eukaryotes, fails to resolve deeper phylogenetic relationships and reveals no support for four of the six proposed supergroups. Our analyses provide a point of departure for future taxon- and gene-rich analyses of the eukaryotic tree of life, which will be critical for resolving their phylogenetic interrelationships.
Similar content being viewed by others

Background
A major remaining gap in our knowledge of the tree of life is the uncertain relationships among eukaryotes, including the many divergent microbial lineages plus plants, animals and fungi. Microbial eukaryotes, often referred to as protists, are an eclectic assemblage of lineages that are defined as eukaryotes that are not plants, animals, or fungi [1]. Clearly, knowledge of the phylogenetic positions of protists is a key to understanding the origins of eukaryotes, and where the ancestries of plants, animals and fungi lie within these microbial groups.
During the 1970's and 1980's a revolution in understanding eukaryotic diversity occurred as a result of ultrastructural studies. These data [2, 3] demolished traditional classifications where algae, protozoa and fungi were considered discrete entities, and microbial eukaryotes were inappropriately lumped into one of four classes: amoebae, flagellates, ciliates, and sporozoans. Ultrastructural studies revealed distinct assemblages of organisms that are distinguished by their complement and organization of organelles, providing lineages with ultrastructural identities [1]. About 60 different, robust patterns of ultrastructural organization are recognized, but ~200 genera of uncertain affinities have yet to be examined [1, 4]. Determining relationships among groups using ultrastructure, however, has proven difficult, largely due to the lack of unambiguously homologous structures.
Early molecular analyses relied on comparisons of rDNAs from diverse protists and suggested that diplomonads, trichomonads, and microsporidia were basal lineages [5–7]. These analyses of rDNAs sequences also produced a topology with a base and crown (putatively recently radiated) lineages [8–11], which is now argued to be an artifact of long branch attraction. Given the well known limitations of single gene genealogies when inferring deep evolutionary relationships, the current trend is to focus on multigene datasets [12, 13]. However, taxon representation in many of these analyses is sparse. With such incomplete taxon sampling, distantly related groups may appear as sister taxa and many deep nodes are poorly supported [14].
The past decade has seen the emergence of six eukaryotic 'supergroups' that aim to portray evolutionary relationships between microbial and macrobial lineages. The supergroup concept is increasingly accepted as evidenced by several reviews [15, 16] and the recently proposed formal reclassification by the International Society of Protozoologists [17]. However, the support for supergroups is highly variable in the published literature [14].
The six putative supergroups have complex and often unstable histories. The supergroup 'Amoebozoa' was proposed in 1996 [18, 19] based largely on molecular genealogies. The controversial supergroup 'Chromalveolata' was proposed based on the assertion that the last common ancestor of the 'Chromista' (cryptophytes, haptophytes, stramenopiles) and the undisputed Alveolata (dinoflagellates, apicomplexans, ciliates) contained a common chlorophyll c-containing red algal plastid [20]. 'Excavata' is another controversial supergroup composed predominately of heterotrophic flagellates whose ancestor is postulated to have had a synapomorphy of a conserved ventral feeding groove [21]. The supergroup 'Opisthokonta' includes animals, fungi, and their microbial relatives, and is supported by many molecular genealogies [10]. The 'Opisthokonta' is united by the presence of a single posterior flagellum in many constituent lineages [22]. The supergroup 'Plantae' was erected as a Kingdom in 1981 [23] to unite the three lineages with double-membrane primary plastids: green algae (including land plants), rhodophytes, and glaucophytes. Finally, the 'Rhizaria' emerged from molecular data in 2002 to unite a heterogeneous group of flagellates and amoebae including: cercomonads, foraminifera, some of the diverse testate amoebae, and former members of the polyphyletic radiolaria [24].
We believe that comprehensive taxon sampling, coupled with gene-rich analyses, is critical for resolving accurate phylogenies [14]. This is particularly relevant for the eukaryotes where only a tiny fraction of the >200,000 species of microbial eukaryotes have thus far been characterized for any gene sequence, and over one-half of identified protists groups [1] have yet to be subjected to any molecular study. Misleading results can also arise if a study addressing "deeper" splits in the eukaryotic tree does not include a broad diversity of lineages, including members of all six putative supergroups [14]. This is because the addition of diverse lineages is critical to break long single branches that pose a significant problem for robust phylogenetic inference. We know that the lack of adequate sampling and the use of highly derived (e.g., parasitic) taxa have created unstable tree topologies and led to inaccurate statements of sister-group relationships (i.e., in the creation of the now-abandoned supergroup Archezoa, whose history is described in [25]). Yet only a handful of studies have been published that take a multigene taxon-rich approach for assessing the eukaryotic tree of life.
Here, we set out to accomplish two tasks: (1) place newly determined sequences from a diversity of microbial eukaryotes onto relatively well-sampled multigene eukaryote phylogenies, and (2) evaluate the support for the six supergroups. Our approach was to use phylogenetic analyses of four genes from two distinct taxon sets that included 61 newly-characterized sequences. The two taxon sets represent 1) 105 diverse eukaryotic lineages and 2) a reduced 92 taxon set in which long-branch taxa were removed. The four loci, SSU-rDNA, actin, alpha-tubulin, and beta-tubulin, have a rich history in eukaryotic phylogenetics [7, 12, 26]. These genes have been used for more intensive studies of some groups such as 'Amoebozoa' [27], 'Rhizaria' [28] and 'Opisthokonta' [29] as well as for the establishment of many of the proposed supergroups [14]. Yet, there are few studies in which a multigene data set has been combined with extensive taxon sampling from all six supergroups [30, 31].
Our work contrasts with many past efforts that have used either single-gene data with a broad taxon sampling [8–11], or multigene data with a limited number of taxa [12, 13, 26, 32]. We performed individual and concatenated analyses of four genes. To assess rate heterogeneity and possible lateral gene transfers, we analyzed each gene individually prior to concatenation and then applied a variety of phylogenetic inference methods with both DNA and the inferred protein sequences. Use of a concatenated data set greatly reduces phylogenetic error in simulation studies [33] and the large number of characters that we have obtained for this study is expected to improve the accuracy of resulting phylogenetic trees [34].
Seventy-two sequences were characterized for this study, the bulk of which are newly-characterized (47 sequences) or were previously characterized from other strains (14 sequences), were available as ESTs in public databases (1 sequence) or are previously published and confirmed here (10 sequences; see Additional file 1). These sequences include representatives of all six 'Chromalveolata' groups thereby sampling a sizable fraction of the diversity in this supergroup. This is critical with respect to overall eukaryotic diversity because 'Chromalveolata' contain about one-half of the recognized species of protists and algae [35]. In addition, eight of the ten 'Excavata' lineages were included in our study. Finally, we also add genes from several lineages within the 'Rhizaria', another poorly supported eukaryotic supergroup [14].
Results and Discussion
Sampling strategy
The 105-taxon dataset was chosen because the included taxa: (1) contained at least three of the four targeted genes; and (2) represented the known breadth of eukaryotic diversity (see additional file 1). These 105 diverse lineages represent 26 well-established eukaryotic groups as well as those of uncertain affiliation (e.g., Ancyromonas, Malawimonas, Stephanopogon), and contained members of all six putative supergroups (see Additional file 1). As discussed below, a second data set containing 92 taxa was generated by removing known problematic taxa including long branched ciliates [36], foraminifera [37, 38], Giardia [39], plus several others (see Additional file 1).
To assess the impact of evolutionary models and phylogenetic methods, both data sets (105 and 92 taxa) were analyzed under five combinations of data and methods: (1) a four-gene data set (SSU-rDNA, actin, alpha-, and beta-tubulin) as nucleotides excluding third codon positions using RAxML, (2) a four-gene data set as nucleotides excluding third codon positions using MrBayes, (3) a data set of mixed nucleotide (SSU) and amino acid sequences using MrBayes, (4) a three-gene data set (actin, alpha-, and beta-tubulin) as amino acids using MrBayes, and (5) a three-gene data set (actin, alpha-, and beta-tubulin) as amino acids using PHYML (Figs. 1, 2, 3 and see Additional files 2, 3, 4, 5, 6, 7, 8, 9). We also analyzed individual gene data sets to identify taxa that are characterized by high sequence divergence (i.e., a long-branch) or had an unexpected position in the phylogeny (see Additional files 10, 11, 12). Overall, there is some heterogeneity in support for hypotheses among models and algorithms but the major trends discussed below were consistent across the different analyses (summarized in Fig. 1).

Overall evaluation of support for nodes in 105 and 92 taxon sets. Support for nodes using different taxon datasets, algorithms and evolutionary models. The top portion lists taxa with clear ultrastructural identities [1] whereas taxa below the line represent putative eukaryotic supergroups. As described in the text, two datasets were examined (105 and 92 taxa). R:n = RAxML analysis of 4 genes as nucleotides, with third codon positions removed; B:n = Bayesian analysis of 4 genes as nucleotides, with third codon positions removed; B:S-A = Bayesian analysis of the SSU-rDNA gene and the remaining three genes as amino acids; B:A = Bayesian analysis of three genes as amino acids; PhyML = Likelihood analysis under PHYML of three genes as amino acids. The 'Rhizaria' are indicated by grey boxes for the 92 taxon analysis as there were insufficient taxa to address this hypothesis; instead, we report support for the nested group 'Cercozoa.'

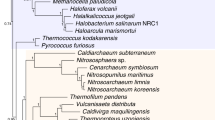
Eukaryotic phylogeny based on 105 taxon set. (A) Phylogeny of the major eukaryotic groups inferred from a Bayesian analysis of the combined SSU-rDNA and amino acid sequences of actin, alpha-tubulin, and beta-tubulin from 105 taxa. This is the 50% majority rule consensus tree with average branch lengths that were calculated from the Bayesian posterior tree distribution. Results of a RAxML bootstrap analysis are shown above the branches. Node thickness indicates >95% Bayesian posterior probability support, as indicated in the key. The branch lengths are proportional to the number of substitutions per site. The six eukaryotic supergroups are shown in different colors (see key). (B) Schematic phylogeny summarizing the results of Hackett et al. 2007. The thickest branches define clades that received ≥ 90% maximum likelihood bootstrap support, whereas the relatively thinner branches provide 80 – 89% bootstrap support. Stars indicate taxa that were characterized in this study (see additional file 1 for details).

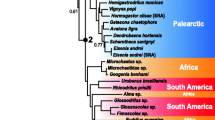
Eukaryotic phylogeny based on 92 taxon set. Phylogeny of the major eukaryotic groups inferred from a Bayesian analysis of the combined SSU-rDNA and amino acid sequences of actin, alpha-tubulin, and beta-tubulin from 92 taxa. Other notes as in Figure 2.
Placement of newly-determined sequences
We determined the position of newly characterized taxa using both the 105 (Fig. 2) and 92 (Fig. 3) taxon data sets. To simplify comparisons, all of the genealogies are drawn rooted with 'Opisthokonta', as hypothesized by Arisue et al. [40] and are generally concordant with previous studies using a similar set of genes [12, 41]. We sampled additional genes from several members of the putative 'Rhizaria' including both the SSU rRNA gene and protein coding genes for a cercomonad Cercomonas sp. (ATCC PRA-21) and the dimorphid Dimorpha sp. (ATCC PRA-54), and protein coding genes for the thaumatomonad Thaumatomonas seravini (ATCC 50636; see Additional file 1). Relationships among these taxa generally agree with the proposed phylogeny of core 'Cercozoa' [42, 43].
All four genes were analyzed for two amoeboid taxa – the myxogastrid Hyperamoeba sp. (ATCC PRA-39) and a Thecamoeba-like lineage (ATCC PRA-35) that is currently being described (Nerad et al., personal communication). Hyperamoeba sp. is sister to Physarum in all analyses except in the beta-tubulin protein tree (see Additional file 12). The Physarum + Hyperamoeba sister group relationship was predicted on the basis of morphology [44]. The Thecamoeba-like lineage falls close to the mycetozoa plus Entamoebida. In contrast to published hypotheses [12, 45], we do not find support for the monophyly of dictyostelids plus myxogastrids with our taxon and gene sampling. This finding is further supported by a well-sampled multigene analysis that included several previously-uncharacterized 'Amoebozoa' taxa [46].
We also included additional genes from several ciliates (Nyctotherus ovalis, Metopus palaeformis and Chilodonella uncinata). In many of the analyses, either there is a spurious relationship between ciliates and the Heterolobosea (e.g., see Additional files 2 and 3) or the ciliates are polyphyletic (see Additional files 4 and 5). As has been shown in previous work, ciliates are marked by considerable heterogeneity in protein evolution as rates of evolution are highly variable among lineages [36, 47] and it is perhaps not surprising that increasing taxon sampling in this group fails to produce more stable trees. Use of the rDNA plus amino acid data for both 105 and 92 taxon data sets however recovers trees that support the expected monophyly of each of the lineages, ciliates, alveolates, and Heterolobosea (Figs. 1, 2, 3).
Furthermore, we characterized additional loci from two enigmatic taxa, the flagellate Ancyromonas sigmoides (ATCC 50267; species identity from [48]) and Stephanopogon apogon (ATCC 50096). In all our analyses, S. apogon shows a sister group relationship to the Heterolobosea (Figs. 1, 2, 3). Stephanopogon apogon has been suggested to be sister to the Euglenozoa based on mitochondrial cristae morphology and similarity in nuclear division profiles [49, 50]. Ancyromonas sigmoides is a small, heterotrophic gliding flagellate with one recurrent and one anterior flagellum and flat mitochondrial cristae [1]. The phylogenetic position of A. sigmoides varies depending on which phylogenetic method is used. This is the case in other analyses of a subset of genes [14], suggesting that either there is inadequate sampling of this lineage or too few data to resolve its position, or that Ancyromonas represents a novel lineage of eukaryotes [51].
We also sampled representatives of all six chromalveolate phyla including multiple members from each phylum. Together, our analysis included new data for 12 taxa out of 43 chromalveolates in the tree. These 12 taxa were placed in their expected positions among the 6 different 'Chromalveolata' phyla, and all received moderate to strong support (i.e., apicomplexans, cryptophytes, haptophytes, dinoflagellates, stramenopiles) except for ciliates (see Figs. 1, 2). Within the 'Plantae' the 4-gene data support strongly the early divergence of Mesostigma viride within the streptophyte branch of the Viridiplantae. This is consistent with a recent multigene analysis of nuclear, chloroplast, and mitochondrial data; the 'Plantae' phyla Glaucophyta and Viridiplantae received strong bootstrap and Bayesian support whereas there was moderate support for Rhodophyta monophyly [52].
Evaluation of supergroups
Putative eukaryotic supergroups receive mixed support in our analyses of the 105-taxon dataset. To improve our understanding of supergroup support, we compare these results with a recent studies including one [30] that used a concatenated alignment of 16-proteins but fewer (46) taxa (see inset in Fig. 1). In the present analyses, the supergroup 'Opisthokonta', receives high bootstrap support (>95%) and a significant posterior probability (1.00) under several of the different models and algorithms (R:n, B:S-A, B-A; Fig. 1). The relationship of at least some members of this supergroup emerged in previously published rDNA [10] and multigene analyses [12, 30, 41]. In addition, there are two compelling synapomorphies for this group: (1) the presence of a single flagellum in flagellate members of this group, with the flagellum 'posterior' in that it beats from base to tip and projects behind swimming cells, and (2) a unique amino acid insertion in those members that contain a canonical EF-1α gene [53] (some members of the 'Opisthokonta' have an EF-like protein and not EF-1α [54]). The inclusion of animals and fungi within 'Opisthokonta' refutes the monophyly of animals plus plants that has been suggested in some recent studies [55].
The putative supergroup 'Amoebozoa' receives high support only under a limited number of models and algorithms, including strong support (posterior probability = 1.00) under Bayesian analyses of amino acid sequences (B:A) and of SSU-rDNA as nucleotides plus amino acid sequences (B:S-A). However, this supergroup is poorly supported or not monophyletic in the three other analyses (Fig. 1). This is consistent with the fact that 'Amoebozoa' is defined largely by molecular phylogenies and lacks any clearly defined ultrastructural synapomorphies. In addition, our analyses fail to provide support for the 'unikont hypothesis', which argues for the monophyly of the 'Amoebozoa' plus 'Opisthokonta' [12, 15, 16, 56–58]. The lack of support for 'unikonts' may reflect insufficient phylogenetic signal in our data sets. Alternatively, the hypothesized 'unikont' monophyly may be an artifact of limited taxon sampling in previous multigene studies.
The 'Rhizaria' receives only limited support (e.g., bootstrap support under RAxML <50% with the nucleotide data [R:n] and 6% under PHYML; Fig. 1). The 'Rhizaria' are supported by some published molecular phylogenies [42, 59, 60], but not by others [14]. The core Cercozoa show a sister-group relationship to the stramenopiles in all multigene analysis (Fig 2, 3, S1-S8), but without significant posterior probability or bootstrap support. This result is consistent with a recent multigene (85-protein) phylogenetic study from a limited number of taxa [30] that supports the sister relationship of Reticulomyxa (Foraminifera) plus Bigelowiella (chlorarachniophyte) with stramenopiles [41]. The relationship between 'Rhizaria' and Stramenopiles suggested by Hackett et al. (2007) has strong bootstrap and Bayesian support in their analyses and is the significantly favored topology using the approximately unbiased (AU) test. An independent study using a larger data set of nearly 30,000 amino acid positions also reported a specific relationship between 'Rhizaria' and 'Chromalveolates' [61]. This intriguing result needs to be tested using additional analyses that include more extensive taxon sampling.
The remaining three putative supergroups – 'Chromalveolata', 'Plantae', and 'Excavata' – are not found to be monophyletic (Fig. 1). In these analyses as in many others, members of the putative 'Excavata' are non-monophyletic [14]. This putative supergroup contains lineages whose ancestor is postulated to have had a distinctive feeding groove [1, 24, 62]. Here, we find two subclades of 'Excavata', albeit with mixed support (Fig. 1). The first group is consistent with the hypothesized 'Fornicata' [17, 58]: Diplomonadida plus Carpediemonas. The second includes the Heterolobosea plus Euglenozoa, which share 'discoidal' mitochondria cristae and have been recovered in other multigene phylogenies [12, 14]. The phylogenetic position of the putative 'Excavata' lineage Malawimonas is unstable in our analyses (Figs. 2, 3, see Additional files 2, 3, 4, 5, 6, 7, 8, 9) and more data are needed to test its relationship to other excavates.
The supergroup 'Plantae' – Rhodophyta (red algae), Glaucophyta, and Viridiplantae (green algae and land plants) – is consistently polyphyletic (Fig. 2, 3). The case for 'Plantae' monophyly is largely based on plastid encoded genes [63, 64], plus recent evidence from some nuclear encoded proteins that are plastid targeted [65, 66] and nuclear genes that encode cytosolic proteins [30, 57, 67]. Other lines of evidence for 'Plantae' monophyly come from analysis of the plastid machinery including plastid targeted metabolite translocator genes [68] and the shared protein import system embedded in the organelle membranes of 'Plantae' members (Tic-Toc system, [69]). Therefore, supergroup 'Plantae' may be monophyletic even though our present analysis lacks resolution with regard to this group. The 'Plantae' however remains controversial because its monophyly is not supported by several other multigene data sets using nuclear loci, thus retaining the possibility that this supergroup may be paraphyletic or polyphyletic [70–72].
We find no support for the putative supergroup 'Chromalveolata', despite the addition of numerous species from this lineage; i.e., 43 putative members in the 105 taxon dataset. The chromalveolate hypothesis unites the chlorophyll c-containing photosynthetic eukaryotes and their relatives and includes the cryptophytes, haptophytes, stramenopiles, apicomplexa, dinoflagellates, and ciliates [20]. The common origin of the plastid in chromalveolates, like in the Plantae, is supported by plastid multigene analyses [63, 64, 73], trees inferred from plastid-targeted proteins such as GAPDH and FBA [74–76] and plastid translocator genes (for apicomplexans, haptophytes, and stramenopiles, [68]).
Relationships among 'Chromalveolata' were recently tested using a 16-nuclear protein dataset that provided moderate bootstrap support for 'Chromalveolata' monophyly when including 'Rhizaria' (see inset in Fig. 1; [30]). However, most nuclear (host) trees using single and multigene analyses provide limited or no support for the monophyly of this supergroup (reviewed in [14]). Most clearly, our trees as well as recent published studies [30] refute the 'Chromista' hypothesis because we find no support for the monophyly of haptophytes plus stramenopiles plus cryptophytes, as is found in some plastid gene trees [54]. Instead, our 92 taxon tree (Fig. 3) supports the monophyly of stramenopiles and Alveolata (ciliates, apicomplexa, and dinoflagellates) that is consistent with the results of other studies [12, 30]. Given that 'Chromista' is invalid then the 'Chromalveolata' hypothesis as proposed by Cavalier-Smith [20] is also falsified by our study.
Impact of Taxon Sampling
We assessed how the removal of known problematic taxa affected the support for clades with ultrastructural identities and for putative supergroups using the reduced 92-taxon dataset (Figs. 1, 3, see Additional files 6, 7, 8, 9). Although we see an increase in support for clades with ultrastructural identities, the reduced taxon dataset shows little improvement for most supergroups. For example, there is an increase in support for groups such as the Heterolobosea (i.e., posterior probability support increases from 0.77 to full support under a B-n analysis; Fig. 1) and red algae (i.e., posterior probability support increases from 0.69 to 0.85 in B-n analysis; Fig 1.) There is also an increase in support for two supergroups, 'Amoebozoa' and 'Opisthokonta,' in our 92-taxon analysis (Fig. 1). For example, posterior probability support for the 'Amoebozoa' increases from <0.50 to 0.85 in the B-n analysis. A result of removing the long-branch Foraminifera to generate our 92 taxon set is that we no longer can assess the 'Rhizaria' because the remaining members represent only the subclade 'Cercozoa', as indicated in Figure 1; this subclade does show robust support with the 92 taxon data set (Fig. 1). Removal of problematic taxa does not provide any support for three supergroups, 'Chromalveolata,' 'Excavata,' and 'Plantae.'
Explanations for the limited support or lack of monophyly of the supergroups (with the exception of 'Opisthokonta' and to a lesser extent 'Amoebozoa') include: (1) taxon and gene sampling is too limited to support these deep relationships and (2) these putative supergroups do not reflect accurately deep relationships within eukaryotes. Disentangling these alternatives will require the use of both a broad taxon sampling, as used here, combined with greater sequence data.
Conclusion
Intriguingly, the level of support in these analyses of four genes, including numerous newly-characterized sequences, matches what emerged from a review of the literature on molecular phylogenetic analyses of eukaryotes in general [14]. In both the analyses presented here and in our synthesis of the literature [14] the 'Opisthokonta' receive relatively strong support, the 'Amoebozoa' receive low to moderate support, and the remaining four supergroups ('Excavata', 'Rhizaria', 'Plantae' and 'Chromalveolata') are unsupported. As discussed above, the core 'Cercozoa' within 'Rhizaria' do show a sister-group relationship to stramenopiles, though our trees provide only Bayesian support for this result (see Fig. 3). This association of some 'Rhizaria' with some members of the 'Chromalveolata' calls into question the taxonomic validity of these two supergroups.
As we are using the same set of genes that are present in many other analyses (including some of those used to establish the putative supergroups), there is some circularity in the comparison between our and previously published analyses. Hence, assessment of potential supergroups must await analyses of novel gene data sets sampled from many taxa, in particular including enigmatic taxa such as Ancyromonas and Malawimonas that could potentially form independent lineages. Ultimately, resolving deep nodes will require the use of multigene alignments incorporating a wide diversity of taxa combined with the identification of robust ultrastructural or molecular synapomorphies for proposed clades.
Methods
Cultures and molecular methods
One hundred and five species from all six eukaryotic supergroups were used in this study. We obtained cultures from the American Type Culture Collection (ATCC), the Provasoli-Guillard National Center for Culture of Marine Phytoplankton (CCMP) and the Culture Collection of Algae at the University of Texas at Austin (UTEX). Cells were frozen in liquid nitrogen and ground with glass beads using a glass rod and/or Mini-BeadBeater™ (Biospec Products, Inc., Bartlesville, OK, USA). Total genomic DNA was extracted using the DNeasy Plant Mini Kit (Qiagen, Santa Clarita, CA, USA). Some DNA samples were obtained directly from the American Type Culture Collection (ATCC).
Primers for SSU-rDNA genes are from Medlin et al. [77] with three additional primers that were used to generate overlapping sequences from each clone as described in Snoeyenbos-West et al. [78]. Other primers were designed for actin, alpha-tubulin and beta-tubulin from broad eukaryotic alignments of these genes. PCR amplification was carried out using the following primers: actin, AAC TGG GAY GAY ATG GAR AAG AT and ATC CAC ATY TGY TGG AAN GT; beta-tubulin, GGT GCT GGT AAY AAY TGR GC and ACC AGG TCG TTC ATR TTN GA; alpha tubulin, initial PCR with CTA GGC AAY GCN TGY TGG GA and CAT GCC TTC NCC NAC RTA CC reamplified with nested primers TTG TAC TGC YTN GAR CAY G and AC GTA CCA GTG NAC RAA NGC. Phusion DNA Polymerase, a strict proofreading enzyme, was used to amplify our genes of interest and we have used the Lucigen PCRSmart, Novagen Perfectly Blunt, Invitrogen Zero Blunt TOPO, Invitrogen TOPO TA cloning kits. Sequencing of cloned plasmid DNA was done using vector- or gene-specific primers and the BigDye™ terminator kit (PE-Applied Biosystems, Norwalk, CT, USA). Sequences were run on an ABI 3100 automated sequencer. We have fully sequenced 2–4 clones of each gene for each organism and surveyed up to 8 clones per taxon in order to detect potential paralogs.
Data analysis
To align SSU-rDNA sequences, we used HMMER v2.1.4 [79] whereas protein-coding genes were aligned by Clustal W [80]. For the SSU-rDNA alignment, we aligned the sequences using HMMER while incorporating secondary structure. These sequences were downloaded from The European Ribosomal Database [81]. The resulting alignment was further edited manually in MacClade v4.05 [82]. Protein coding genes were aligned as amino acids using Clustal W [80] as implemented in DNAstar's Lasergene software and manually adjusted in MacClade v4.05 [82]. For the phylogenetic analysis, we restricted our analysis to unambiguously aligned regions for which we were confident in positional homology as assessed by eye. For a subset of our analyses, we tried two different masks (conservative vs. liberal) of ambiguous positions and found no significant differences in inferences from topologies and support (data not shown).
Genealogies were inferred using MrBayes [83], RAxML [84] and PHYML [85]. Bayesian analyses were performed with the parallel version of MrBayes 3.1.2 using the GTR+I+ Γ (for nucleotide) and RtREV (for amino acid) models of sequence evolution [86]. Four to 16 simultaneous MCMCMC chains were run for 4 million generations sampling every 100 generations. Stationarity in likelihood scores was determined by plotting the -1nL against the generation. All trees below the observed stationarity level were discarded, resulting in a 'burnin' that comprised 25% of the posterior distribution of trees. The 50% majority-rule consensus tree was determined to calculate the posterior probabilities for each node. RAxML was run for 100 iterations using GTRGAMMA model for nucleotide data and PROTGAMMA with matrix RtREV for amino acid data. The datasets were partitioned to allow RaxML to assign different parameters for each gene. One hundred replicates for bootstrap analyses were run in RAxML and PHYML, and a 50% majority rule consensus was calculated to determine the support values for each node. MrModelTest [87] and ProtTest 1.3 [88] were used to select the appropriate model of sequences evolution for the nucleotides and amino acid data, respectively.
References
Patterson DJ: The Diversity of Eukaryotes. Am Nat. 1999, 154 (S4): S96-S124. 10.1086/303287.
Melkonian M, Andersen RA, Schnepf E: The Cytoskeleton of Flagellate Protists. 1991, New York , Springer-Verlag
Wetherbee R, Andersen RA, Pickett-Heaps J: Protistan Cell Surfaces. 1994, New York , Springer-Verlag
Patterson DJ: Changing views of protistan systematics: the taxonomy of protozoa - an overview. An Illustrated Guide to the Protozoa. Edited by: Lee JJ. 2002, Lawrence, Kansas , Society of Protozoologists, 2nd edition
Leipe DD, Gunderson JH, Nerad TA, Sogin ML: Small subunit ribosomal RNA of Hexamita inflata and the quest for the first branch of the eukaryotic tree. Mol Biochem Parasitol. 1993, 59: 41-48. 10.1016/0166-6851(93)90005-I.
Vossbrinck CR, Maddox JV, Friedman S, Debrunner-Vossbrinck BA, Woese CR: Ribosomal RNA sequence suggests microsporidia are extremely ancient eukaryotes. Nature. 1987, 326 (6111): 411-414. 10.1038/326411a0.
Sogin ML, Gunderson JH, Elwood HJ, Alonso RA, Peattie DA: Phylogenetic meaning of the kingdom concept: an unusual ribosomal RNA from Giardia lamblia. Science. 1989, 243 (4887): 75-77. 10.1126/science.2911720.
Edman JC, Kovacs JA, Masur H, Santi DV, Elwood HJ, Sogin ML: Ribosomal RNA sequence shows Pneumocystis carinii to be a member of the fungi. Nature. 1988, 334 (6182): 519-522. 10.1038/334519a0.
Gajadhar AA, Marquardt WC, Hall R, Gunderson J, Ariztia-Carmona EV, Sogin ML: Ribosomal RNA sequences of Sarcocystis muris, Theileria annulata and Crypthecodinium cohnii reveal evolutionary relationships among apicomplexans, dinoflagellates, and ciliates. Mol Biochem Parasitol. 1991, 45 (1): 147-154. 10.1016/0166-6851(91)90036-6.
Wainright PO, Hinkle G, Sogin ML, Stickel SK: Monophyletic origins of the metazoa: an evolutionary link with fungi. Science. 1993, 260 (5106): 340-342. 10.1126/science.8469985.
Sogin ML, Silberman JD: Evolution of the protists and protistan parasites from the perspective of molecular systematics. Int J Parasitol. 1998, 28 (1): 11-20. 10.1016/S0020-7519(97)00181-1.
Baldauf SL, Roger AJ, Wenk-Siefert I, Doolittle WF: A kingdom-level phylogeny of eukaryotes based on combined protein data. Science. 2000, 290 (5493): 972-977. 10.1126/science.290.5493.972.
Bapteste E, Brinkmann H, Lee JA, Moore DV, Sensen CW, Gordon P, Durufle L, Gaasterland T, Lopez P, Muller M, Philippe H: The analysis of 100 genes supports the grouping of three highly divergent amoebae: Dictyostelium, Entamoeba, and Mastigamoeba. Proc Natl Acad Sci U S A. 2002, 99 (3): 1414-1419. 10.1073/pnas.032662799.
Parfrey LW, Barbero E, Lasser E, Dunthorn M, Bhattacharya D, Patterson DJ, Katz LA: Evaluating support for the current classification of eukaryotic diversity. PLoS Genet. 2006, 2 (12): e220-10.1371/journal.pgen.0020220.
Baldauf SL: The deep roots of eukaryotes. Science. 2003, 300 (5626): 1703-1706. 10.1126/science.1085544.
Keeling PJ, Burger G, Durnford DG, Lang BF, Lee RW, Pearlman RE, Roger AJ, Gray MW: The tree of eukaryotes. Trends Ecol Evol. 2005, 20 (12): 670-676. 10.1016/j.tree.2005.09.005.
Adl SM, Simpson AG, Farmer MA, Andersen RA, Anderson OR, Barta JR, Bowser SS, Brugerolle G, Fensome RA, Fredericq S, James TY, Karpov S, Kugrens P, Krug J, Lane CE, Lewis LA, Lodge J, Lynn DH, Mann DG, McCourt RM, Mendoza L, Moestrup O, Mozley-Standridge SE, Nerad TA, Shearer CA, Smirnov AV, Spiegel FW, Taylor MF: The new higher level classification of eukaryotes with emphasis on the taxonomy of protists. J Eukaryot Microbiol. 2005, 52 (5): 399-451. 10.1111/j.1550-7408.2005.00053.x.
Parfrey LW, Barbero E, Lasser E, Dunthorn M, Bhattacharya D, Patterson DJ, Katz LA: Evaluating Support for the Current Classification of Eukaryotic Diversity. PLoS Genet. 2006, 2 (12): e220-10.1371/journal.pgen.0020220.
Cavalier-Smith T, Chao EE: Molecular phylogeny of the free-living archezoan Trepomonas agilis and the nature of the first eukaryote. J Mol Evol. 1996, 43 (6): 551-562. 10.1007/BF02202103.
Cavalier-Smith T: A revised six-kingdom system of life. Biological Reviews. 1998, 73 (3): 203-266. 10.1017/S0006323198005167.
Cavalier-Smith T: Principles of protein and lipid targeting in secondary symbiogenesis: Euglenoid, Dinoflagellate, and Sporozoan plastid origins and the Eukaryote family tree. J Eukaryot Microbiol. 1999, 46 (4): 347-366. 10.1111/j.1550-7408.1999.tb04614.x.
Simpson A, Patterson DJ: The ultrastructure of Carpediemonas membranifera (Eukaryota) with reference to the excavate hypothesis. Eur J Protistol. 1999, 35: 353-370.
Cavalier-Smith T: The origin of Fungi and pseudofungi. Evolutionary Biology of the Fungi. Edited by: Rayner ADM, Brasier CM, Moore D. 1987, Cambridge , Cambridge University Press, 339-353.
Cavalier-Smith T: Eukaryote kingdoms: seven or nine?. Biosystems. 1981, 14 (3-4): 461-481. 10.1016/0303-2647(81)90050-2.
Cavalier-Smith T: The phagotrophic origin of eukaryotes and phylogenetic classification of Protozoa. Int J Syst Evol Microbiol. 2002, 52 (Pt 2): 297-354.
Patterson DJ: The diversity of eukaryotes. Am Nat. 1999, 154: S96-S124. 10.1086/303287.
Harper JT, Waanders E, Keeling PJ: On the monophyly of chromalveolates using a six-protein phylogeny of eukaryotes. Int J Syst Evol Microbiol. 2005, 55 (Pt 1): 487-496. 10.1099/ijs.0.63216-0.
Nikolaev SI, Berney C, Petrov NB, Mylnikov AP, Fahrni JF, Pawlowski J: Phylogenetic position of Multicilia marina and the evolution of Amoebozoa. Int J Syst Evol Microbiol. 2006, 56: 1449-1458. 10.1099/ijs.0.63763-0.
Tekle YI, Grant J, Cole JC, Nerad TA, Anderson OR, Patterson DJ, Katz LA: A multigene analysis of Corallomyxa tenera sp. nov. suggests its membership in a dlade that includes Gromia, Haplosporidia and Foraminifera. Protist. 2007, 158 (4): 457-472. 10.1016/j.protis.2007.05.002.
Steenkamp ET, Wright J, Baldauf SL: The protistan origins of animals and fungi. Mol Biol Evol. 2006, 23 (1): 93-106. 10.1093/molbev/msj011.
Hackett JD, Yoon HS, Li S, Reyes-Prieto A, Rummele SE, Bhattacharya D: Phylogenomic analysis supports the monophyly of cryptophytes and haptophytes and the association of 'Rhizaria' with Chromalveolates. Mol Biol Evol. 2007
Burki F, Berney C, Pawlowski J: Phylogenetic position of Gromia oviformis Dujardin inferred from nuclear-encoded small subunit ribosomal DNA. Protist. 2002, 153 (3): 251-260. 10.1078/1434-4610-00102.
Edgcomb VP, Kysela DT, Teske A, de Vera Gomez A, Sogin ML: Benthic eukaryotic diversity in the Guaymas Basin hydrothermal vent environment. Proc Natl Acad Sci U S A. 2002, 99 (11): 7658-7662. 10.1073/pnas.062186399.
Rosenberg MS, Kumar S: Incomplete taxon sampling is not a problem for phylogenetic inference. Proc Natl Acad Sci U S A. 2001, 98 (19): 10751-10756. 10.1073/pnas.191248498.
Bininda-Emonds OR, Brady SG, Kim J, Sanderson MJ: Scaling of accuracy in extremely large phylogenetic trees. Pac Symp Biocomput. 2001, 547-558.
Cavalier-Smith T: Chromalveolate diversity and cell megaevolution: interplay of membranes, genomes and cytoskeleton. Organelles, Genomes and Eukaryotic Evolution. Edited by: Hirt RP, Horner D. 2004, London , Taylor and Francis, 71-103.
Zufall RA, McGrath CL, Muse SV, Katz LA: Genome architecture drives protein evolution in ciliates. Mol Biol Evol. 2006, 23 (9): 1681-1687. 10.1093/molbev/msl032.
Berney C, Pawlowski J: Revised small subunit rRNA analysis provides further evidence that Foraminifera are related to Cercozoa. J Mol Evol. 2003, 57 Suppl 1: S120-7. 10.1007/s00239-003-0015-2.
Habura A, Wegener L, Travis JL, Bowser SS: Structural and functional implications of an unusual foraminiferal beta-tubulin. Mol Biol Evol. 2005, 22 (10): 2000-2009. 10.1093/molbev/msi190.
Van de Peer Y, Baldauf SL, Doolittle WF, Meyer A: An updated and comprehensive rRNA phylogeny of (crown) eukaryotes based on rate-calibrated evolutionary distances. J Mol Evol. 2000, 51 (6): 565-576.
Arisue N, Hasegawa M, Hashimoto T: Root of the Eukaryota tree as inferred from combined maximum likelihood analyses of multiple molecular sequence data. Mol Biol Evol. 2005, 22 (3): 409-420. 10.1093/molbev/msi023.
Burki F, Pawlowski J: Monophyly of Rhizaria and multigene phylogeny of unicellular bikonts. Mol Biol Evol. 2006, 23 (10): 1922-1930. 10.1093/molbev/msl055.
Nikolaev SI, Berney C, Fahrni JF, Bolivar I, Polet S, Mylnikov AP, Aleshin VV, Petrov NB, Pawlowski J: The twilight of Heliozoa and rise of Rhizaria, an emerging supergroup of amoeboid eukaryotes. Proc Natl Acad Sci U S A. 2004, 101 (21): 8066-8071. 10.1073/pnas.0308602101.
Bass D, Moreira D, Lopez-Garcia P, Polet S, Chao EE, von der Heyden S, Pawlowski J, Cavalier-Smith T: Polyubiquitin insertions and the phylogeny of Cercozoa and Rhizaria. Protist. 2005, 156 (2): 149-161. 10.1016/j.protis.2005.03.001.
Walker G, Silberman JD, Karpov SA, Preisfeld A, Foster P, Frolov AO, Novozhilov Y, Sogin ML: An ultrastructural and molecular study of Hyperamoeba dachnaya, n. sp., and its relationship to the mycetozoan slime moulds. Europ J Protistol. 2003, 39 (3): 319-336. 10.1078/0932-4739-00906.
Tekle YI, Grant JR, Anderson OR, Cole JC, Nerad TA, Patterson DJ, Katz LA: Phylogenetic placement of diverse amoebae inferred from multigene analyses and assessment of clade stability within 'Amoebozoa’ upon removal of varying rate classes of SSU-rDNA. Mol Phylogenet Evol. 2007, 10.1016/j.ympev.2007.11.015.
Katz LA, Bornstein JG, Lasek-Nesselquist E, Muse SV: Dramatic diversity of ciliate histone H4 genes revealed by comparisons of patterns of substitutions and paralog divergences among eukaryotes. Mol Biol Evol. 2004, 21 (3): 555-562. 10.1093/molbev/msh048.
Atkins MS, Teske AP, Anderson OR: A survey of flagellate diversity at four deep-sea hydrothermal vents in the Eastern Pacific Ocean using structural and molecular approaches (vol 47, pg 400, 2000). J Eukaryot Microbiol. 2000, 47 (5): 492-492. 10.1111/j.1550-7408.2000.tb00078.x.
Patterson DJ, Brugerolle G: The ultrastructural identity of Stephanopogon apogon and the relatedness of the genus to other kinds of protists. Eur J Protistol. 1988, 23 (3): 279-290.
Cavalier-Smith T: Kingdom Protozoa and its 18 phyla. Micro Rev. 1993, 57 (4): 953-994.
Atkins MS, McArthur AG, Teske AP: Ancyromonadida: a new phylogenetic lineage among the protozoa closely related to the common ancestor of metazoans, fungi, and choanoflagellates (Opisthokonta). J Mol Evol. 2000, 51 (3): 278-285.
Rodriguez-Ezpeleta N, Brinkmann H, Burey SC, Roure B, Burger G, Loffelhardt W, Bohnert HJ, Philippe H, Lang BF: Monophyly of primary photosynthetic eukaryotes: Green plants, red algae, and glaucophytes. Curr Biol. 2005, 15 (14): 1325-1330. 10.1016/j.cub.2005.06.040.
Baldauf SL, Palmer JD: Animals and fungi are each other's closest relatives: congruent evidence from multiple proteins. Proc Natl Acad Sci U S A. 1993, 90 (24): 11558-11562. 10.1073/pnas.90.24.11558.
Keeling PJ, Inagaki Y: A class of eukaryotic GTPase with a punctate distribution suggesting multiple functional replacements of translation elongation factor 1alpha. Proc Natl Acad Sci U S A. 2004, 101 (43): 15380-15385. 10.1073/pnas.0404505101.
Philip GK, Creevey CJ, McInerney JO: The Opisthokonta and the Ecdysozoa may not be clades: stronger support for the grouping of plant and animal than for animal and fungi and stronger support for the Coelomata than Ecdysozoa. Mol Biol Evol. 2005, 22 (5): 1175-1184. 10.1093/molbev/msi102.
Stechmann A, Cavalier-Smith T: Rooting the eukaryote tree by using a derived gene fusion. Science. 2002, 297 (5578): 89-91. 10.1126/science.1071196.
Rodriguez-Ezpeleta N, Brinkmann H, Burey SC, Roure B, Burger G, Loffelhardt W, Bohnert HJ, Philippe H, Lang BF: Monophyly of primary photosynthetic eukaryotes: green plants, red algae, and glaucophytes. Curr Biol. 2005, 15 (14): 1325-1330. 10.1016/j.cub.2005.06.040.
Simpson AG, Inagaki Y, Roger AJ: Comprehensive multigene phylogenies of excavate protists reveal the evolutionary positions of "primitive" eukaryotes. Mol Biol Evol. 2006, 23 (3): 615-625. 10.1093/molbev/msj068.
Keeling PJ: Foraminifera and Cercozoa are related in actin phylogeny: two orphans find a home?. Mol Biol Evol. 2001, 18 (8): 1551-1557.
Archibald JM, Longet D, Pawlowski J, Keeling PJ: A novel polyubiquitin structure in cercozoa and foraminifera: evidence for a new eukaryotic supergroup. Mol Biol Evol. 2003, 20 (1): 62-66. 10.1093/molbev/msg006.
Burki F, Shalchian-Tabrizi K, Minge M, Skjaeveland A, Nikolaev SI, Jakobsen KS, Pawlowski J: Phylogenomics reshuffles the eukaryotic supergroups. PLOS ONE. 2007, 2: e790-10.1371/journal.pone.0000790.
Simpson AG: Cytoskeletal organization, phylogenetic affinities and systematics in the contentious taxon Excavata (Eukaryota). Int J Syst Evol Microbiol. 2003, 53 (Pt 6): 1759-1777. 10.1099/ijs.0.02578-0.
Yoon HS, Hackett JD, Ciniglia C, Pinto G, Bhattacharya D: A molecular timeline for the origin of photosynthetic eukaryotes. Mol Biol Evol. 2004, 21 (5): 809-818. 10.1093/molbev/msh075.
Yoon HS, Hackett JD, Pinto G, Bhattacharya D: The single, ancient origin of chromist plastids. Proc Natl Acad Sci USA. 2002, 99 (24): 15507-15512. 10.1073/pnas.242379899.
Li S, Nosenko T, Hackett JD, Bhattacharya D: Phylogenomic analysis identifies red algal genes of endosymbiotic origin in the chromalveolates. Mol Biol Evol. 2006, 23 (3): 663-674. 10.1093/molbev/msj075.
Nosenko T, Lidie KL, Van Dolah FM, Lindquist E, Cheng JF, Bhattacharya D: Chimeric plastid proteome in the florida "red tide" dinoflagellate Karenia brevis. Mol Biol Evol. 2006, 23 (11): 2026-2038. 10.1093/molbev/msl074.
Moreira D, Le Guyader H, Phillippe H: The origin of red algae and the evolution of chloroplasts. Nature. 2000, 405 (6782): 69-72. 10.1038/35011054.
Weber AP, Linka M, Bhattacharya D: Single, ancient origin of a plastid metabolite translocator family in Plantae from an endomembrane-derived ancestor. Eukaryot Cell. 2006, 5 (3): 609-612. 10.1128/EC.5.3.609-612.2006.
Reumann S, Inoue K, Keegstra K: Evolution of the general protein import pathway of plastids (review). Mol Membr Biol. 2005, 22 (1-2): 73-86. 10.1080/09687860500041916.
Stiller JW, Harrell L: The largest subunit of RNA polymerase II from the Glaucocystophyta: functional constraint and short-branch exclusion in deep eukaryotic phylogeny. BMC Evol Biol. 2005, 5: 71-10.1186/1471-2148-5-71.
Stiller JW, Riley J, Hall BD: Are red algae plants? A critical evaluation of three key molecular data sets. J Mol Evol. 2001, 52 (6): 527-539.
Nozaki H, Matsuzaki M, Takahara M, Misumi O, Kuroiwa H, Hasegawa M, Shin IT, Kohara Y, Ogasawara N, Kuroiwa T: The phylogenetic position of red algae revealed by multiple nuclear genes from mitochondria-containing eukaryotes and an alternative hypothesis on the origin of plastids. J Mol Evol. 2003, 56 (4): 485-497. 10.1007/s00239-002-2419-9.
Yoon HS, Hackett JD, Van Dolah FM, Nosenko T, Lidie KL, Bhattacharya D: Tertiary endosymbiosis driven genome evolution in dinoflagellate algae. Mol Biol Evol. 2005, 22: 1299-1308. 10.1093/molbev/msi118.
Fast NM, Kissinger JC, Roos DS, Keeling PJ: Nuclear-encoded, plastid-targeted genes suggest a single common origin for apicomplexan and dinoflagellate plastids. Mol Biol Evol. 2001, 18 (3): 418-426.
Harper JT, Keeling PJ: Nucleus-encoded, plastid-targeted glyceraldehyde-3-phosphate dehydrogenase (GAPDH) indicates a single origin for chromalveolate plastids. Mol Biol Evol. 2003, 20 (10): 1730-1735. 10.1093/molbev/msg195.
Patron NJ, Rogers MB, Keeling PJ: Gene replacement of fructose-1,6-bisphosphate aldolase supports the hypothesis of a single photosynthetic ancestor of chromalveolates. Eukaryot Cell. 2004, 3 (5): 1169-1175. 10.1128/EC.3.5.1169-1175.2004.
Medlin L, Elwood HJ, Stickel S, Sogin ML: The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions. Gene. 1988, 71 (2): 491-499. 10.1016/0378-1119(88)90066-2.
Snoeyenbos-West OL, Salcedo T, McManus GB, Katz LA: Insights into the diversity of choreotrich and oligotrich ciliates (Class: Spirotrichea) based on genealogical analyses of multiple loci. Int J Syst Evol Microbiol. 2002, 52 (Pt 5): 1901-1913. 10.1099/ijs.0.02114-0.
Eddy SR: HMMER: Profile hidden markov models for biological sequence analysis. 2001, [http://hmmer.janelia.org/]
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22 (22): 4673-4680. 10.1093/nar/22.22.4673.
Wuyts J, Perriere G, Van De Peer Y: The European ribosomal RNA database. Nucleic Acids Res. 2004, 32 (Database issue): D101-3. 10.1093/nar/gkh065.
Maddison DR, Maddison WP: MacClade. 2005, Sunderland , Sinauer, 4.05
Huelsenbeck JP, Ronquist F: MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001, 17 (8): 754-755. 10.1093/bioinformatics/17.8.754.
Stamatakis A, Ludwig T, Meier H: RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005, 21 (4): 456-463. 10.1093/bioinformatics/bti191.
Guindon S, Gascuel O: A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, 52 (5): 696-704. 10.1080/10635150390235520.
Ronquist F, Huelsenbeck JP: MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19 (12): 1572-1574. 10.1093/bioinformatics/btg180.
Nylander JA: MrModelTest. 2004, Upsalla , Distributed by the author. Evolutionary Biology Centre, Uppsala University
Abascal F, Zardoya R, Posada D: ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005, 21 (9): 2104-2105. 10.1093/bioinformatics/bti263.
Acknowledgements
This project was made possible by a collaborative grant from the National Science Foundation Assembling the Tree of Life program (EF 04-31117) that was awarded to L.A.K., D.B., J.L., D.J.P., and to the ATCC.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
HSY: led on data collection and analysis, contributed to writing; JG: led on data collection and analysis, contributed to writing; YIT: contributed to data collection and analysis, contributed to writing; MW: contributed to data collection; BCC: contributed to data collection; JCC: contributed to data collection; JML: contributed to data collection; DJP: contributed to data interpretation and writing;
DB: oversaw data collection and analysis, contributed to writing and revision; LAK: oversaw project (data collection, data analysis) and led on much of the writing.
Hwan Su Yoon, Jessica Grant contributed equally to this work.
Electronic supplementary material
12862_2007_583_MOESM2_ESM.PDF
Additional file 2: Figure 4. Likelihood analysis of the 105-taxon data set of SSU-rDNA + nucleotide sequences of actin, alpha-tubulin and beta-tubulin performed with RaxML. See text and Figure 2 for additional notes. (PDF 939 KB)
12862_2007_583_MOESM3_ESM.PDF
Additional file 3: Figure 5. Bayesian analyses of the 105-taxon data set of SSU-rDNA + nucleotide sequences of actin, alpha-tubulin and beta-tubulin performed with MrBayes. See text and Figure 2 for additional notes. (PDF 2 MB)
12862_2007_583_MOESM4_ESM.PDF
Additional file 4: Figure 6. Bayesian analyses of the 105-taxon data set of amino acid sequences of actin, alpha-tubulin and beta-tubulin performed with MrBayes. See text and Figure 2 for additional notes. (PDF 649 KB)
12862_2007_583_MOESM5_ESM.PDF
Additional file 5: Figure 7. Likelihood analysis of the 105-taxon data set of amino acid sequences of actin, alpha-tubulin and beta-tubulin performed with PhyML. See text and Figure 2 for additional notes. (PDF 617 KB)
12862_2007_583_MOESM6_ESM.PDF
Additional file 6: Figure 8. Likelihood analysis of the 92-taxon data set of SSU-rDNA + nucleotide sequences of actin, alpha-tubulin and beta-tubulin performed with RaxML. See text and Figure 2 for additional notes. (PDF 897 KB)
12862_2007_583_MOESM7_ESM.PDF
Additional file 7: Figure 9. Bayesian analyses of the 92-taxon data set of SSU-rDNA + nucleotide sequences of actin, alpha-tubulin and beta-tubulin performed with MrBayes. See text and Figure 2 for additional notes. (PDF 370 KB)
12862_2007_583_MOESM8_ESM.PDF
Additional file 8: Figure 10. Bayesian analyses of the 92-taxon data set of amino acid sequences of actin, alpha-tubulin and beta-tubulin performed with MrBayes. See text and Figure 2 for additional notes. (PDF 623 KB)
12862_2007_583_MOESM9_ESM.PDF
Additional file 9: Figure 11. Likelihood analysis of the 92-taxon data set of amino acid sequences of actin, alpha-tubulin and beta-tubulin performed with PhyML. See text and Figure 2 for additional notes. (PDF 620 KB)
12862_2007_583_MOESM10_ESM.PDF
Additional file 10: Figure 12. Bayesian analysis of 93 actin amino acid sequences that are in the 105 multigene taxon analysis, performed with MrBayes. See text and Figure 2 for additional notes. (PDF 532 KB)
12862_2007_583_MOESM11_ESM.PDF
Additional file 11: Figure 13. Bayesian analysis of 96 alpha-tubulin amino acid sequences that are in the 105 multigene taxon analysis, performed with MrBayes. See text and Figure 2 for additional notes. (PDF 542 KB)
12862_2007_583_MOESM12_ESM.PDF
Additional file 12: Figure 14. Bayesian analysis of 98 beta-tubulin amino acid sequences that are in the 105 multigene taxon analysis, performed with MrBayes. See text and Figure 2 for additional notes. (PDF 541 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Yoon, H.S., Grant, J., Tekle, Y.I. et al. Broadly sampled multigene trees of eukaryotes. BMC Evol Biol 8, 14 (2008). https://doi.org/10.1186/1471-2148-8-14
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-8-14