
Abstract
Background
Previous phylogenetic analyses of African elephants have included limited numbers of forest elephant samples. A large-scale assessment of mitochondrial DNA diversity in forest elephant populations here reveals a more complex evolutionary history in African elephants as a whole than two-taxon models assume.
Results
We analysed hypervariable region 1 of the mitochondrial control region for 71 new central African forest elephants and the mitochondrial cytochrome b gene from 28 new samples and compare these sequences to other African elephant data. We find that central African forest elephant populations fall into at least two lineages and that west African elephants (both forest and savannah) share their mitochondrial history almost exclusively with central African forest elephants. We also find that central African forest populations show lower genetic diversity than those in savannahs, and infer a recent population expansion.
Conclusion
Our data do not support the separation of African elephants into two evolutionary lineages. The demographic history of African elephants seems more complex, with a combination of multiple refugial mitochondrial lineages and recurrent hybridization among them rendering a simple forest/savannah elephant split inapplicable to modern African elephant populations.
Similar content being viewed by others
Background
The taxonomic status of the African elephant (Loxodonta africana) has been debated since the turn of the 20th century [1] and up to 22 subspecies have been described [2]. However, modern taxonomy refers to two types, with their names reflecting the habitat in which they are found, namely the larger savannah (Loxodonta africana africana) (Blumenbach 1797) and the smaller forest (Loxodonta africana cyclotis) (Matschie 1900) elephants. It has become increasingly established in the literature that forest and savannah elephants are distinct species (L. Africana and L. cyclotis) [3–7], with recent publications considering their datasets in the light of this concept. The most persuasive genetic basis for a two-taxon model originates from a series of studies exploring patterns of differentiation at nuclear loci, culminating in a study using male inherited Y-chr, andbi-parentally inherited X-chr sequences [6] that concluded "there was a deep and almost complete separation between African forest and African savannah elephants." In this study, divergent nuclear DNA sequences segregated with either forest or savannah elephant morphological types. There were, however, a number of exceptions, including a forest elephant from Garamba in the Democratic Republic of Congo (DRC, where forest and savannah populations are sympatric) that had nuclear sequences typical of savannah elephants and two savannah elephants from Cameroon (at the limit of the forest-savannah transition zone) that had nuclear sequences typical of forest elephants [6]. The study estimated the divergence between the savannah and forest elephants to be 3 million years. The two-taxon argument has also been used to explain data from two nuclear microsatellite DNA [5, 7] and one morphological study [8, 9]. However, subsequently Debruyne [10] performed a morphometric analysis of museum elephant skulls, and found evidence for a continuum between two morphotypes, suggesting that, despite historical separation that promoted subdivision, these two forms freely interbreed wherever their ranges intersect.
Molecular studies using mitochondrial (mt) DNA [10, 11] including data from the study by Roca et al [6] have pointed to a more complex scenario for African elephants. Debruyne [10] examined several thousand base pairs of mtDNA from elephants across Africa and although he also reported two highly divergent molecular clades, these did not conform to the morphological delineations of cyclotis and africana. He interpreted these results as a consequence of incomplete isolation between forest and savannah African elephant populations, followed by recurrent and ongoing introgression between the two forms. Roca et al. [6] obtained very similar mitochondrial results but explained the non-concordance between mitochondrial and nuclear markers as a result of cytonuclear genomic disassociation such that the mitochondrial tree did not reflect the species tree. The mtDNA results observed were explained as having arisen during episodes of backcrossing between successive generations of savannah males with forest females, leading to half of extant savannah elephants surveyed possessing 'forest' typical mitochondrial haplotypes but almost exclusively 'savannah' nuclear X and Y-chromosomal DNA. Eggert et al. [11] (in addition to Nyakaana et al.'s mitochondrial sequences [12]) included samples from west Africa and found a more complex picture using mtDNA and nuclear microsatellites, suggesting that western savannah and forest elephants formed a potential third Loxodonta taxonomic unit. Finally, Roca et al.[13]recently revisited the question with a statistical re-analysis of eight morphological and genetic datasets (nuclear and mitochondrial) including their own and those of Eggert et al.[11] and Debruyne [10] and reconfirmed their initial interpretation of a two taxon model with cyto-nuclear genomic dissociation.
The above-mentioned studies largely share a pronounced lack of forest elephant data. The nuclear DNA studies [4, 11] featured limited sampling from central African forest elephants. Despite describing a narrow hybrid zone between the two elephant types, only one population located in this zone (Garamba, (DRC)) was included and none from elsewhere in DRC or from west central Africa were examined. Elsewhere, Debruyne [10] included elephants from across DRC in his study but was again limited by sample size. The study by Eggert et al. [11] was limited by the inclusion of only two populations of Central African forest elephants, both from the edge of the forest range in Cameroon which may conceivably have influenced their conclusion of the genetic uniqueness of forest and western elephants. To date, no study has addressed the partitioning of genetic diversity in the equatorial forests of Africa. Further, the potential effect of Pleistocene forest refugia was partially addressed by Eggert et al.[11] and also previously reported as having a major influence on large mammal distribution and range dynamics [14–18] has yet to be addressed in African elephants. Here we report results from the most extensive sample of forest elephants to date, from the core of their range, and compare these results with previously published mitchondrial DNA sequences for savannah elephants from east and southern Africa and populations from west Africa and DRC.
We examined the phylogeographic history, population structure and past demography of African elephants using patterns of molecular diversity for the mtDNA control region and cytochrome b gene. Since mtDNA is maternally inherited, this marker provides a female-biased view of population history and structure. We included the most variable mtDNA segment, the hypervariable region 1 (HVR1) of the control region since it has a high rate of nucleotide change, allowing recently diverged lineages to be distinguished [19–21]. This segment is equivalent to data previously published by Eggert et al. [11] and Debruyne [10], allowing us to examine forest elephant sequences within the context of a sample set with the largest geographic coverage. We could not use Roca's mtDNA sequences as he studied a different fragment (ND5 instead of control region).
Results
Central forest samples
We sequenced 316 bp of HVR1 of the control region from 71 samples and 396 bp of the cytochrome b from 28. No nuclear copies of mitochondrial DNA (Numts) were detected for either sequence.
Combined sequences
Genetic diversity
For HVR1, we analysed 189 sequences from 66 sites across Africa in both forest and savannah elephants (Figure 1). Of these 102 were from forest elephants (71 samples from the present study and 31 from Genbank) and 87 savannah elephants (all from Genbank). The combined dataset comprised eighty-eight haplotypes (33 and 51 from forest and savannah elephants, respectively) and four haplotypes found in both types. Of the 21 central African forest elephant haplotypes identified in this study, 17 were novel (Genbank accessions EU096114 – EU096130). Mean nucleotide diversity (π) for HVR1 sequences for all African elephants was 0.030 (SD = 0.015), while mean haplotype diversity (h) was 0.985 (SD = 0.003). When haplotypes were divided into forest and savannah, based on prior designation, the forest population π was 0.022 (SD = 0.11), significantly lower than for savannah elephants (0.034, SD = 0.017; p < 0.001). The mean haplotype diversity for forest and savannah populations was 0.960 (SD = 0.007) and 0.986 (SD = 0.004), respectively. The lowest nucleotide diversity of all groupings was for the new central African forest samples in this study (0.013, SD = 0.007), while haplotype diversity was 0.947 (SD = 0.009).

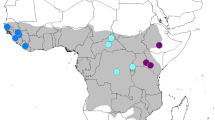
Map of Africa showing approximate sampling sites from previous mtDNA studies combined with those from this study. The green, yellow and blue dots are sampling sites from Nyakaana et al. [12], Eggert et al. [11] and Debruyne [10], respectively. The red dots are the sites from this study.
For cytochrome b, 100 sequences were analysed, 28 from this study, 27 provided by SN and 45 from Genbank. Forty-four haplotypes were identified including three and 22 new forest and savannah elephant sequences, respectively (Genbank accessions EU115995 – EU116019). Of the 44 haplotypes, 32 were found in savannah elephants and 10 in forest elephants, with two haplotypes found in both. Mean π for cytochrome b was 0.023 (0.012) for all elephants. When forest and savannah elephants were subdivided, π was again significantly lower for forest populations (0.009, SD = 0.005) than for savannah populations (0.026, SD = 0.013; p < 0.001). These results contrast with the study of Roca et al. (2005) who reported 15 haplotypes for 281 elephants at the mitochondrial ND5 locus and described low genetic diversity as being typical for savannah elephants.
Population structure
The median joining networks for the HVR1 and cytochrome b sequences (Figures 2 and 3, respectively), exhibit patterns consistent with a complex demographic history. The HVR1 pattern is more complex (comprising four haplogroups – here labelled HVR1 Haplogroup I, II, III and IV) than for cytochrome b (three haplogroups -labelled Cytb Haplogroup I, II and III). Haplotype designations for this and previous studies for both sequences are found in Table 1 (HVR) and Table 2 (cyt b). For the HVR1 region, the most obvious feature is that central African forest elephants (excluding those from DRC) fall into two separate groups (HVR1 Haplogroups I and II) with little geographic structuring, consisting of 19 (HVR1 Haplogroup I) and 20 (HVR1 Haplogroup II) haplotypes with variable frequencies. Only two forest elephants from DRC, share the same haplotype with other forest elephants in HVR1 Haplogroup II. The remaining seven DRC forest elephant haplotypes (all south-east of the Congo River), group with sequences in HVR1 Haplogroup III (which additionally comprises savannah elephants from eastern and southern Africa and one savannah elephant from Cameroon). The other striking feature is that for West African elephants (from Eggert et al 2002, see Table 1 for haplotype designations), both forest and savannah types possess haplotypes found almost exclusively within the same haplogroup as central African forest elephants (HVR1 Haplogroups I and II). Twenty-five out of 26 haplotypes from west Africa are more closely related to central Forest elephants from Gabon, Congo and CAR. A single western savannah sequence (H15) can be found in HVR1 Haplogroup IV grouping with savannah elephants from eastern, southern and central Africa. Analysis of Molecular Variance (AMOVA) of HVR1 sequences revealed a non-significant (p = 0.065) genetic structure (18.62% variation among populations) when populations were grouped according to geographic distribution (west, central, east and southern Africa).

Median-joining networks for African elephants HVR1 mtDNA haplotypes. Circle size is proportional to the haplotype frequency. The numbers on the connecting line determine the number of substitutions estimated by NETWORK V.4. 1. 1. 1. The entire list of haplotypes for HVR1 MJN can be found in Table 1.

Median-joining networks for African elephants cytochrome b mtDNA haplotypes. Circle size is proportional to the haplotype frequency. The numbers on the connecting line determine the number of substitutions estimated by NETWORK V.4. 1. 1. 1. The entire list of haplotypes for cytochrome b MJN can be found in Table 2.
As expected, Cytochrome b is less variable than HVR1. However, direct comparison between patterns obtained from both regions is compromised here due to a lack of equivalent individuals examined at both loci, specifically for savannah elephants. However the overall pattern when individuals from different populations were examined is consistent with the pattern obtained with HVR1, despite the resolution of only three haplogroups as opposed to four. Savannah elephant haplotypes fall into two distinct haplogroups (Cyt b Haplogroup II and III) as do forest elephant haplotypes (Cyt b Haplogroups I and II). Cytochrome b Haplogroup II, which is divided into two haplogroups for HVR1, is characterised by a network structure in which forest and savannah elephant samples are not overlaid (see Figure 3). Again all western elephants, both forest and savannah, cluster with central African forest elephants (Cyt b Haplogroup I).
Demographic history
When HVR1 sequences from forest and savannah elephants were examined separately, Fu's Fs was -14.2954 (P = 0.0021) and -24.4427 (P < 0.0001), respectively. Although significant values can indicate historical population expansion, the multimodal pattern (Figure 4) for the forest elephant groups suggests that these populations encompass several subgroups as indicated in the networks. When we examined each haplogroup separately for signatures of demographic change (Table 3), a smooth and predominantly unimodal pattern was observed for HVR1 Haplogroup I, indicating a recent demographic expansion (Figure 5), while HVR1 Haplogroups II, III and IV were more complex, including the presence of some divergent haplotypes.

Mismatch distribution of the HVR1 forest and savannah African elephants haplotypes.

Mismatch distribution of the HVR1 haplogroups of African elephants.
Discussion
In the light of the results obtained with the mitochondrial sequences used here, additional interpretations of the history of African elephants become evident and suggest that the conclusions drawn in previous studies may have been hampered by incomplete sample sets. Forest elephants have been affected by cyclical climatic changes that occurred over the last 2.6 million years as the colder drier periods experienced during Pleistocene glacial maxima are believed to have led to the repeated retraction of forest cover into refugial zones followed by re-expansion, fostering allopatric divergence between isolated populations [22] and secondary contact. The forest elephant range is therefore likely to have become centred around such refugia on several occasions. The dataset presented here raises the possibility of at least two different refugia in the central African region harbouring distinct elephant populations that diverged allopatrically. If this was the case, forest elephants possessing distinct mitochondrial genotypes are likely to have come into contact relatively rapidly after the end of the last glaciation (12,000 years BP), when the forests re-expanded [23]. Such a scenario might explain not only the two haplogroups present in forest elephants but also the lower nucleotide diversity that characterises elephant populations found in forest habitat.
This scenario might also explain the high microsatellite diversity reported for forest elephants [5]. If several populations diverged in isolation, accumulating different microsatellite profiles, and subsequently became sympatric as the forest expanded, the large single population that today comprises two central African forest elephant lineages might be expected to have engendered higher microsatellite diversity. Savannah populations, especially those in the south and east, would not have been affected by forest expansion since these areas remained unforested and thus habitat would not have been lost. Those savannah populations that may have been affected are those that may have occurred in areas that subsequently became forested. One explanation for the close genetic proximity between forest and savannah genotypes in DRC could be introgression between savannah haplotypes into forest genomes as forests expanded and savannah habitat was lost. Such introgression would be in the opposite direction to that proposed by Roca et al. [4, 6].
The results obtained for elephants in west and central Africa have strong implications for the division of elephants into forest and savannah species. These elephants are taxonomically indeterminate [24] and have been described as having an intermediate morphology [8]. Mitochondrially, West African elephants are found in the same haplogroups as the (two) forest elephant lineages of central Africa. If ancient female-mediated introgression between the two forms followed by backcrossing into savannah populations is the reason why western savannah elephants possess largely 'forest' haplotypes then nuclear markers at these loci should resemble predominantly those of southern and eastern savannah elephants today. Alternatively these elephant populations could be an example of protracted gene flow between two forms of elephant, which is ongoing (or was until recently) and that west African savannah elephants are not distinguishable at the genetic or morphological level from their forest counterparts (thus undermining the two-taxon model). A third explanation could be a 'second movement' of elephants out of the forest (from either west or central Africa) and into the savannah. There are insufficient data to determine whether there was a single movement from forest to savannah habitat or whether these were multiple events, precipitating the morphological changes observed today. Whatever the origin of the two types, our data would support continued extensive hybridisation between the two proposed forms.
Conclusion
Our mitochondrial analysis does not support the simple separation of modern African elephants into two groups. The evidence is most clear in west Africa where savannah elephants are indistinguishable at both the mitochondrial and morphological level from their forest counterparts. The two species model cannot be easily applied in this region and neither do west African elephants represent a third distinct entity. Central African elephant populations west of the Congo river also question the current classification. Forest elephants fall into two major groupings with mitochondrial DNA. Previous studies found two major groups for all African elephants, savannah and savannah/forest perhaps suggesting ancient introgression between forest females and savannah males in the past. However the inclusion of a larger central forest sample in this study would suggest that this explanation is too simple and that African elephants were subject to a more complex demographic history. Phylogenetic and phylogeographic reanalysis of species is important for many reasons but with the massive extinction of species in the wild in the last 50 years accurate descriptions are essential for management of wild resources. For elephants, the classification of species into savannah and forest may not reflect their evolutionary history but simply the habitat in which they currently exist. While ecotypic differentiation has been shown to be the predominant factor driving molecular divergence in one widely distributed African herbivore recently [25], this may not apply in elephants and if it does, may not conform to a simple forest versus savannah habitat driven divergence. To develop management strategies incorporating a simple forest/savannah model could be misleading until further lines of evidence give us a clearer picture of the origins and current conservation needs of elephants populations throughout the continent. Future studies should analyse nuclear DNA markers, including those which evolve rapidly, across the range of forest and savannah elephants and especially in transition zones to investigate this complex ongoing process further.
Methods
Sampling and laboratory procedures
Elephant sequences from 66 sites across Africa were incorporated (Figure 1). New forest elephant samples (HVR1 mtDNA: n = 71; Cyt b mtDNA: n = 28) were obtained using feces from 12 sites in the central African forest block (red dots, Figure 1).
Samples were stored in RNAlater (Ambion RNA later® and Qiagen RNA later ™) or silica gel, and DNA was extracted from these using the Qiagen DNA stool mini kit (Qiagen, Hilden, Germany) kit following the manufacturer's protocol.
An approximately 630 bp fragment of mitochondrial DNA was amplified, encompassing the 3' end of the cytochrome b gene, transfer RNAs (Threonine, Proline) and 358 bp of the control region. The control region section was amplified in 71 samples using primers MDL3 and MDL5 [26]. Primers AFDL1 and AFDL2 (situated 400 bp from the 3'end of the cytochrome b gene through to the 5' end of the control region), and AFDL3 and AFDL4 (situated 377 bp from the 3' end of tRNA proline to the 5'end of the control region) were employed to gain overlapping sequence for some degraded samples [11]. A 494 bp fragment of cytochrome b was analysed separately with 28 sequences using the primers L15024 and H15516 [3]. Amplifications were performed in 50 μl containing 50 mM KCl, 10 mM Tris-HCl, 1.5 mM Mg2+, 200 μmol of each dNTP, 0.2 μmol of each primer, 1.5 U Taq DNA polymerase (Qiagen) and approx. 10 ng of genomic DNA. Thirty to 40 cycles were carried out using a Perkin-Elmer Cetus 9600 or 9700 DNA thermocycler with denaturation at 94°C for 45s, annealing at 63°C for 45s, and extension at 72°C for 45s. PCR products were purified using the Qiagen PCR purification kit and subsequently sequenced commercially (Macrogen, Korea).
Analysis of genetic diversity and differentiation
Forward and reverse sequences for each individual and the consensus sequences for all individuals were aligned using SEQUENCHER (Gene Codes Corporation 1998, version 3.1.1) and rechecked by eye. Genetic diversity for all geographic locations was estimated using haplotype h and nucleotide π diversities as implemented in Arlequin ver. 3.0 [27]. Paired t-tests were carried out to assess whether there was significant difference in nucleotide diversity between forest and savannah elephants. Genetic differentiation between pairs of populations was tested using the exact test using 10,000 Markov chain steps, as implemented in ARLEQUIN ver. 3.0, and this program was also employed for nested analysis of molecular variance (AMOVA) to test for patterns of spatial genetic structure. The dataset was divided in forest and savannah groupings and then four regional populations were defined (west, central, east and south). Using AMOVA the correlation among genotype distances is used as an F-statistic analog (Phi) at various hierarchical levels.
Weighted maximum likelihood distances [28] were used to derive a median joining network (MJN) with the program NETWORK V4.1.1.1. Haplotype networks may more effectively portray the relationships among sequences for populations than maximum likelihood or maximum parsimony which are the traditional methods developed to define interspecific relationships, leading to poor resolution at the population level [29].
Analysis of population demography
Tests were performed to detect evidence of past demographic change. We used ARLEQUIN ver.3.0 to perform a pairwise mismatch distribution, comparing the distribution of the observed pairwise nucleotide site differences with the expected distribution in an expanding population [30]. In a single origin, demographically expanding population, mismatches should follow a unimodal Poisson distribution whereas in populations at demographic equilibrium or with sub-groups, the distribution is usually multimodal. We tested the goodness-of-fit of the observed data to a simulated model of expansion with the sum of square deviations (SSD) and the Harpending's raggedness index r, using ARLEQUIN.
Population history was also inferred using Fu's FS test of neutrality [31] as implemented in ARLEQUIN. We chose this test because it is the most powerful coalescent-based neutrality test for detecting population growth for larger sample sizes.
References
Matschie P: Uber geographische Abarten des Afrikanischen elephantens. Sitzber. Ges. Naturforsch Fr, Berlin. 1900, 8: 189-197.
Krumbiegel I: Von neuen und unentdeckten Tierarten. 1950
Barriel V, Thuet E, Tassy P: Molecular phylogeny of Elephantidae. Extreme divergence of the extant forest African elephant. C R Acad Sci III. 1999, 322 (6): 447-454.
Roca AL, Georgiadis N, Pecon-Slattery J, O'Brien SJ: Genetic evidence for two species of elephant in Africa. Science. 2001, 293 (24 August 2001): 1473-1477. 10.1126/science.1059936.
Comstock KE, Georgiadis N, Pecon-Slattery J, Roca AL, Ostrander EA, O'Brien SJ, Wasser SK: Patterns of molecular variation among African elephant populations. Mol Ecol. 2002, 11: 2489-2498. 10.1046/j.1365-294X.2002.01615.x.
Roca AL, Georgiadis N, O'Brien SJ: Cytonuclear genomic dissociation in African elephant species. Nature Genetics. 2005, 37: 96-100.
Wasser SK, Shedlock AM, Comstock KE, Ostrander EA, Mutayoba B: Assigning African elephant DNA to geographic region of origin: applications to the ivory trade. PNAS. 2004, 101: 14847-14852. 10.1073/pnas.0403170101.
Groves CP: What are the elephants of West Africa?. Elephant. 2000, 2: 7-8.
Grubb P, Groves CP, Dudley JP, Shoshani J: Living African elephants belong to two specices: Loxodonta africana (Blumenbach, 1797) and Loxodonta cyclotis (Matschie, 1900). Elephant. 2000, 2: 1-4.
Debruyne R: A case study of apparent conflict between molecular phylogenies: the interrelationships of African elephants. Cladistics. 2005, 21: 31-50.
Eggert LS, Rasner CA, Woodruff DS: The evolution and phylogeography of the African elephant inferred from mitochondrial DNA sequence and nuclear microsatellite markers. Proc R Soc Lond B. 2002, 269: 1993-2006. 10.1098/rspb.2002.2070.
Nyakaana S, Arctander P, Siegismund HR: Population structure of the African savannah elephant inferred from mitochondrial control region sequences and nuclear microsatellite loci. Heredity. 2002, 89: 90-98. 10.1038/sj.hdy.6800110.
Roca AL, Georgiadis N, O'Brien SJ: Cyto-nuclear genomic dissociation and the African elephant species question. Quaternary Intl. 2007, 169-170: 4-16. 10.1016/j.quaint.2006.08.008.
Arctander P, Johansen C, Coutellec-Vreto MA: Phylogeography of three closely related African bovids. Mol Biol Evol. 1999, 16: 1724-1739.
Clifford S, Anthony N M, Bawe-Johnson M, Abernethy K A, Tutin C E G, White L J T, Bermejo M, Goldsmith M L, Mcfarland K, Jeffery K J, Bruford M W, J WE: Mitochondrial DNA phylogeography of western lowland gorillas (Gorilla gorilla gorilla). Mol Ecol. 2004, 13: 1551-1565. 10.1111/j.1365-294X.2004.02140.x.
Flagstad O, Roed K: Refugial origins of reindeer (Rangifer Tarandus L.) inferred from mitochondrial DNA sequences . Evolution. 2003, 57: 658-670.
Hewitt G: Some genetic consequences of ice ages and their role in divergence and speciation. Biological Journal of the Linnean Society. 1996, 58: 247-276. 10.1006/bijl.1996.0035.
Tosi AJ, Detwiler KM, Disotell TR: Y-chromosomal markers suitable for non-invasive studies of guenon hybridization. Int Jour Prim. 2005, 26: 685-696. 10.1007/s10764-005-4374-y.
Douzery E, Randi E: The mitochondrial control region of cervidae: evolutionary patterns and phylogenetic content. Mol Biol Evol. 1997, 14: 1154-1166.
Flagstad O, Syvertsen P, Stenseth N, Jakobsen K: Environmental change and rates of evolution: the phylogeographic pattern within the hartebeest complex as related to climatic variation. Proc R Soc Lond B. 2001, 268: 667-677. 10.1098/rspb.2000.1416.
Van Hooft W, Groen A, Prins H: Phylogeography of the African buffalo based on mitochondrial and Y-chromosomal loci: pleistocene origin and population expansion of the Cape buffalo subspecies. Mol Ecol. 2002, 11: 267-279. 10.1046/j.1365-294X.2002.01429.x.
Grubb P: Endemism in African rain forest mammals. In African rain forest ecology and conservation. Edited by: Weber W, White LJT, Vedder A, Naughton-Treves L. 2001, New Haven , Yale University Press, 88-100.
Elenga H, Peyron O, Bonnefille R: Pollen-based biome reconstruction for southern Europe and Africa 18,000 yr BP. J Biogeog. 2000, 27: 621-634. 10.1046/j.1365-2699.2000.00430.x.
Frade F: Ordre des proboscidiens (Proboscidea Illiger, 1811). In Traite de zoologie. 1955, Paris , Masson & Cie, 715-875. Grasse, P.-P.
Moodley Y, Bruford MW: Molecular Biogeography: Towards an Integrated Framework for pan-African Conservation. PloS One. 2007
Fernando P, Pfrender ME, Encalada SE, Lande R: Mitochondrial DNA variation, phylogeography and population structure of the Asian elephant. Heredity. 2000, 84: 362-372. 10.1046/j.1365-2540.2000.00674.x.
Excoffier L, Laval G, Schneider S: Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online. 2005, 1: 47-50.
Bandelt HJ, Forster P, Rohl A: Median Joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999, 16: 37-48.
Posada D, Crandall KA: Intraspecific gene gealogies: trees grafting into networks. Trends Ecol Evol. 2001, 16: 37-45. 10.1016/S0169-5347(00)02026-7.
Rogers A, Harpending H: Population growth makes waves in the distribution of pairwise genetics differences. Mol Biol Evol. 1992, 9: 552-569.
Fu YX: Statistical test of neutrality of mutations against population growth, hitchhicking and backgroung selection. Genetics. 1997, 147: 915-925.
Acknowledgements
This work was supported by a Darwin Initiative for the Survival of Species Scholarship, US Fish and Wildlife Service, Royal Society, British Ecological Society, Tusk Trust and FORINFO (French Ministry of Foreign Affairs). We are grateful to F. Maisels, A. Turkalo, E. Stokes, S. Blake, the whole team of the Wildlife Conservation Society in Gabon, CAR and Republic of Congo and HELP Congo for their support in collecting samples. Many thanks to L. Momont, S. Lahm, K. Abernethy, P. Henschel, M. E. Bechem, G. Abitsi, R. Aba'A Nseme, N. Bout, H. Ontsana, E. Spotswood, N. Anthony for samples and M.F. Jalil for help with analysis. We thank L. Chikhi for his helpful comments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
MBJ carried out the molecular genetic studies, analyzed the data and drafted the manuscript as part of her PhD dissertation.
SLC made substantive contributions data analysis and interpretation and helped to draft the manuscript.
SN provided cytochrome b sequences for savannah elephants from Ghana and provided comments on the manuscript.
BC and LJTW participated in the design of the study.
BG, EJW and MWB conceived and initiated the study, participated in its coordination, advised on data analysis and helped to draft the manuscript and revise it critically. MWB made substantial text contributions, especially during the review process.
All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Johnson, M.B., Clifford, S.L., Goossens, B. et al. Complex phylogeographic history of central African forest elephants and its implications for taxonomy. BMC Evol Biol 7, 244 (2007). https://doi.org/10.1186/1471-2148-7-244
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-7-244