
Abstract
Background
Lutzomyia intermedia and Lutzomyia whitmani (Diptera: Psychodidae) are important and very closely related vector species of cutaneous leishmaniasis in Brazil, which are distinguishable by a few morphological differences. There is evidence of mitochondrial introgression between the two species but it is not clear whether gene flow also occurs in nuclear genes.
Results
We analyzed the molecular variation within the clock gene period (per) of these two species in five different localities in Eastern Brazil. AMOVA and Fst estimates showed no evidence for geographical differentiation within species. On the other hand, the values were highly significant for both analyses between species. The two species show no fixed differences and a higher number of shared polymorphisms compared to exclusive mutations. In addition, some haplotypes that are "typical" of one species were found in some individuals of the other species suggesting either the persistence of old polymorphisms or the occurrence of introgression. Two tests of gene flow, one based on linkage disequilibrium and a MCMC analysis based on coalescence, suggest that the two species might be exchanging alleles at the per locus.
Conclusion
Introgression might be occurring between L. intermedia and L. whitmani in period, a gene controlling behavioral rhythms in Drosophila. This result raises the question of whether similar phenomena are occurring at other loci controlling important aspects of behavior and vectorial capacity.
Similar content being viewed by others

Background
The Phlebotominae sand flies Lutzomyia intermedia Lutz & Neiva 1912 and Lutzomyia whitmani Antunes & Coutinho 1912 are vectors of cutaneous leishmaniasis in Brazil. These are closely related species that can be only distinguished by a few morphological differences [1] and both show high anthropophily and reported natural infections with Leishmania in different regions of Brazil [2].
Despite their importance as vectors, only a handful of studies have been carried out in these two species using molecular techniques [3–6]. One of the most important findings from an epidemiological perspective is the evidence obtained for introgression between the two species using mitochondrial DNA [4]. This was particularly interesting because apparently, only lineages of L. whitmani sympatric with L. intermedia have been involved in cutaneous leishmaniasis transmission in the peridomestic environment [4], which suggests that genes controlling aspects of vectorial capacity could be passing from one species to the other. In fact, mitochondrial introgression has been reported in other sand fly species [7, 8] suggesting that might be a common phenomenon in these insect vectors. However, because mitochondrial genes can introgress relatively easily between closely related species [9], it becomes important to examine whether introgression can occur with nuclear genes.
The Drosophila period (per) gene homologue was isolated in sand flies by Peixoto et al. [10]. This circadian clock gene was originally identified using mutagenesis by Konopka and Benzer [11], but is also known to control the differences in the "lovesong" rhythms between D. melanogaster and D. simulans [12], that are important to the sexual isolation between these two species [13–15]. In addition, per was implicated in the control of species-specific circadian mating rhythms in Drosophila and Bractocera, which might also constitute a reproductive isolation mechanism [16–18]. Thus per may possibly represent an example of a Drosophila speciation gene [19], and in fact it has been used as a molecular marker in a number of speciation and evolutionary studies, not only in Drosophila (reviewed in [20]) but also in other insects (e.g. [21]) including sand flies [22–24].
Because per controls the circadian clock in different insects [25], it is almost certainly involved in the rhythms of activity and biting of sand flies [26], which are very important to leishmaniasis transmission. In addition, per might be involved in reproductive isolation in sand flies, via mating rhythms, or via their "lovesongs" [2, 27]. per is thus a particularly interesting marker, among the few available, for an introgression analysis in L. intermedia and L. whitmani. Evidence for introgression in per might suggest that gene flow between these two vector species is occurring at other genes controlling important aspects of behavior and vectorial capacity. It might also suggest that per does not have a strong role in their reproductive isolation. In the current study, we analyzed the molecular variation within the per gene of L. intermedia and L. whitmani in five different localities in Eastern Brazil.
Results
Polymorphism and divergence between L. intermedia and L. whitmani
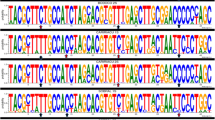
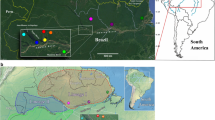
A total of 68 sequences from L. intermedia and 53 from L. whitmani homologue to a fragment of the period gene were analyzed from populations of five localities in Eastern Brazil (Fig 1). The alignment of 72 variable sites is shown in Fig 2. Although most of the changes are either synonymous or occur within the 58 bp intron, non-synonymous substitutions are observed causing 9 amino acid differences among the sequences (Fig 2).

Map of Southeastern Brazil. The five localities (Afonso Claudio, Corte de Pedra, Ilhéus, Jacarepaguá and Posse) where the sand flies were collected are indicated on the map. Approximated distances between localities are also indicated.

Alignment of L. intermedia and L. whitmani per DNA and predicted amino acid sequences (variable sites only). IPO – L. intermedia from Posse; IAC – L. intermedia from Afonso Claudio; ICP L. intermedia from Corte de Pedra; IJC – L. intermedia from Jacarepaguá; WPO – L. whitmani from Posse; WAC – L. whitmani from Afonso Claudio; WCP – L. whitmani from Corte de Pedra; WIL – L. whitmani from Ilhéus.
Table 1 shows the number of sequences of each population of the two species, the number of polymorphic sites (S) and the estimates of molecular polymorphism θ (based on the total number of mutations) and π. Table 1 also shows the Tajima's [28] and Fu & Li's [29] statistics. Within each species, all populations present similar levels of polymorphism with the exception of L. whitmani from Ilhéus, which seems to be less polymorphic than the others. This population was also the only one presenting a significant value in the Fu & Li test but only at the 5% level. Finally, the last column of Table 1 presents the recombination estimator γ [30] indicating that both species show evidence of intragenic recombination in the per gene.
To investigate the level of intra and interspecific differences, initially an AMOVA was carried out as shown in Table 2. The results show a non-significant within species and a significant between species molecular variation at the per locus. Table 3 shows a more detailed analysis of the intraspecific differentiation among populations of L. intermedia and L. whitmani. None of the pairwise and overall fixation indexes (Fst) are significant in the case of L. intermedia and only one (Posse × Ilhéus) has a borderline significant value in L. whitmani. The results therefore show that no significant geographical heterogeneity was detected among the populations of the two species. The estimated number of migrants per generation, based on the overall Fst values, is 20.683 for L. intermedia and 23.125 for L. whitmani.
Table 4 shows measures for DNA divergence between species (Dxy and Da), as well as the Fst and Nm values considering each species as a unique population. Dxy is the average number of nucleotide substitutions per site between alleles from two different populations and Da is the number of net nucleotide substitutions between two populations. Table 4 also shows the number of polymorphisms exclusive for each species (Sint and Swhit), the number of shared polymorphisms (Ss) and the number of fixed differences (Sf) between species. As one can note, there is a high number of shared polymorphisms between species, and no fixed differences between them suggesting either the persistence of ancestral polymorphisms or the occurrence of introgression. In fact, there is one shared haplotype between the two species (IPO13, WPO10 and WPO19) and three L. whitmani sequences (WAC02, WPO13 and WPO14) which show only one nucleotide difference to "typical" L. intermedia haplotypes (see also below).
Genealogy of period sequences
A phylogenetic analysis of the period gene sequences from L. intermedia and L. whitmani was carried out with the Minimum Evolution method using the Kimura 2-parameter distance (Fig 3). A sequence from L. umbratilis, a related species from the same subgenus Nyssomyia, was used as outgroup [24]. The tree shows L. intermedia and L. whitmani as non-monophyletic. However, despite the low bootstrap values, which are below 50% in most cases, there is a large group that contains most L. intermedia sequences and a second large group with most L. whitmani sequences. A few other sequences are clustered outside these two main groups. It is interesting to note that there are three L. whitmani alleles (WAC2, WPO13 and WPO14) inside L. intermedia main group, as well as one L. intermedia allele (ICP16) inside the L. whitmani main group. In addition, a second L. intermedia allele (IPO13) is a shared haplotype between the two species as mentioned above. Again, the results suggest either the persistence of ancestral polymorphisms or the occurrence of introgression between the two species. Very similar results were obtained using the maximum likelihood algorithm as implemented in PAUP 4.0b10 software [31] (data not shown).

Minimum Evolution tree. A Minimum Evolution tree of the period gene sequences of Fig 2 using Close-Neighbor-Interchange Heuristic Search with an initial tree obtained by Neighbor-joining method, Kimura 2-parameter distance and 1000 bootstrap replications. L. intermedia sequences in blue circles and L. whitmani sequences in red squares. Putative introgressed sequences are highlighted with larger fonts.
As mentioned before, there is evidence of intragenic recombination in the per gene fragment of both species (see Table 1) and for that reason the bifurcating tree shown in Fig 3 has to be viewed with caution, as different regions of the gene might have different phylogenetic histories [32]. Therefore, we constructed Minimum Evolution trees with the two most polymorphic non-recombining blocks of the per gene fragment identified using the Hudson and Kaplan [33] method available in the DNAsp 4.1 program [34]. We did not observed major changes in the genealogy of the L. intermedia and L. whitmani per sequences, especially regarding the five haplotypes (ICP16, IPO13, WAC2, WPO13 and WPO14) that clearly cluster with sequences of the other species (data not shown).
Finally, a haplotype network was estimated from per sequences using statistical parsimony, as described by Templeton et al. [35] and implemented in the TCS1.21 software [36] (Fig 4). A small number of ambiguities were resolved as suggested by Crandall and Templeton [37]. The haplotype network shows connections between sequences from each species, separating most of the sequences of L. intermedia and L. whitmani in two groups. No intraspecific geographical structuring was found. Once again, some of the L. whitmani sequences (WAC2, WAC10, WPO13 and WPO14) appear more closely related to L. intermedia haplotypes. In addition, one L. intermedia allele (ICP16) is connected by a small number of mutations to some of the main L. whitmani haplotypes and IPO13 is a shared haplotype between the two species. These results confirm the same putative introgressed sequences indicated by the phylogenetic reconstructions.

Statistical parsimony network. Each population is represented by a different color: PO – Posse, AC – Afonso Claudio, CP – Corte de Pedra, JC – Jacarepaguá, IL – Ilhéus. Each circle corresponds to a unique haplotype and is proportional to the number of sequences. The diagram on the left indicates the number of sequences depending on the circle size. Each cross bar represents one nucleotide substitution between two observed haplotypes. Putative introgressed sequences are indicated by their names.
LD test of introgression
We tested the hypothesis of gene flow between L. intermedia and L. whitmani using a method based on linkage disequilibrium (LD) developed by Machado et al. [38]. In this test, x is the difference between the average LD found among all pairs of shared polymorphisms (DSS) between the two species and the average LD among all pairs of sites for which one member is a shared polymorphism and the other is an exclusive polymorphism (DSX). In case of gene flow x should tend to be positive [see [38] for more details].
Because of limitations on the total number of sequences that could be handled by the WH program we could not perform the simulations with all sequences. Therefore, we carried out the LD test of introgression between each pair of sympatric populations of L. intermedia and L. whitmani from the localities of Posse, Afonso Claudio and Corte de Pedra. The input files were prepared using the values of recombination and linkage disequilibrium calculated by the SITES program [30] for each population (data not shown). Although no significant values were found for the smaller samples of Afonso Claudio and Corte de Pedra, the results (Table 5) present evidence for introgression in the period gene in both directions (from L. intermedia to L. whitmani and vice-versa) in the locality of Posse.
Isolation with Migration model
To further examine the gene flow between L. intermedia and L. whitmani we used the IM software [39]. The Isolation with Migration model has six demographic parameters that include two migration rates, one for each population. The IM software estimates the posterior probability for each of the model parameters, fitting the Isolation with Migration model to the data. One of the assumptions of this model is that the loci studied do not have internal recombination. Therefore, we identified four different non-recombining blocks of our fragment of per, which were then treated as different loci in the analysis. The four-gametes test [33] implemented in DnaSP4.1 was used for the identification of possible recombination events. Since the program estimates parameters for a pair of closely related populations or species, all sequences of each species were used in the analysis as a single population. We performed MCMC runs using the IM software with different seed numbers, in order to guarantee convergence of the sample.
Maximum likelihood estimates of migration parameters revealed a non-zero value for both species, m1 = 1.398 and m2 = 1.014 (m1 – from L. whitmani towards L. intermedia; m2 – from L. whitmani towards L. intermedia). Fig 5 shows the posterior distributions for migration rates and reveals a null probability for the absence of migration from L. whitmani towards L. intermedia. In addition, the absence of migration in the opposite direction is not included in the 95% confidence interval (values range from 0.222 to 8.898), thus supporting the presence of migration in both directions. The conversion of the migration rate estimate to population migration rate per generation (m1 and m2) is not accurate when the population size is based on a single locus. However, the average of the migrant number per generation for both species was very close to the Nm estimate based on Fst values (Nm ~0.49 in Table 4, m1 ~0.52 and m2 ~0.34).

Posterior distribution for migration estimates. Posterior or likelihood distributions for migration rate estimates under the "Isolation with Migration" model [39]; m1 is the migration rate estimate from L. whitmani towards L. intermedia (in blue) and m2 from L. intermedia towards L. whitmani (in red).
Discussion
There is some evidence that L. intermedia and L. whitmani might represent sibling-species complexes in Brazil. Lutzomyia neivai Pinto 1926, a sibling of L. intermedia is found in parts of Southern and Western Brazil and some other countries of South America [40]. The present study did not include populations of this species. In the case of L. whitmani, mitochondrial data [3, 6] indicates three main lineages in Brazil: an Amazonian group, a North-South group and a Northeast group. We did not find strong evidence of a geographical differentiation in the period gene among populations of L. whitmani although one of the pairwise Fst comparisons (Posse × Ilhéus) was significant at the 5% level.
When we compare L. intermedia and L. whitmani, we find a highly significant Fst value (0.3373), which is however smaller than that observed for the period gene between sympatric siblings of Lutzomyia longipalpis (Fst = 0.3952) [23], a complex of cryptic species that are vectors of American visceral leishmaniasis. Therefore, despite the presence of diagnostic morphological characters to identify L. intermedia and L. whitmani [1] the level of molecular divergence in period is not as high as the cryptic L. longipalpis siblings.
Even though it is hard to distinguish introgression from the persistence of ancestral polymorphisms, a test of gene flow based on the signature introgression leaves on the patterns of linkage disequilibrium [38] as well as simulations that fit the "Isolation with Migration" model to the data suggest that L. intermedia and L. whitmani might be exchanging alleles at the per locus. This is further supported by the presence of shared haplotypes between the two species in Posse and very similar sequences in all sympatric populations. There is mounting evidence that introgression plays a major role in the evolution of closely related insect vector species. Introgression among vectors may have important epidemiological consequences. Gene flow in loci that affect vectorial capacity, such as those controlling host preference and susceptibility to parasite infection, can change the transmission patterns and consequently make the disease control a harder task. Introgression of genes that control adaptation to particular types of environment can also have a major impact on the spread of vector-borne diseases as was proposed for the major African malaria vector Anopheles gambiae [41]. The same can be said about genes controlling insecticide resistance. For example, Weill et al. [42] found a kdr mutation responsible for pyrethroid resistance in the Mopti form of Anopheles gambiae, a normally susceptible taxon of this species complex. Sequence analysis reveals that this resistant allele probably originates through introgression from the Savanna form.
Although L. intermedia and L. whitmani are closely related and only distinguished by a few morphological differences, they do show differentiation in some other important traits. For example, in Posse, one of the localities we studied, the two species show differences in abundance during the year. L. intermedia is more abundant in the summer while L. whitmani is more frequent in the winter months [2]. They also show differences in microhabitat preferences, L. intermedia being more common in the peridomestic area while L. whitmani is found mainly in the surrounding forest [2]. In addition, the two species show marked differences in their tendencies to bite humans in the early morning, with L. whitmani showing higher feeding rates than L. intermedia [26]. Therefore, despite the evidence of introgression in the period gene in this locality, there are important ecological and behavioral differences between the two species in Posse suggesting that gene flow is probably rather limited in loci controlling these traits. Hence, it is yet not clear whether introgression has played an important role in the evolution of L. intermedia and L. whitmani. Further work with other genes might help clarify the issue.
Conclusion
Evidence for introgression between L. intermedia and L. whitmani obtained using mitochondrial DNA [4] seems to be corroborated by our data on the period gene, a nuclear marker. Nevertheless, considering that period is potentially involved in reproductive isolation and might be, therefore, less prone to introgression than the "average" gene [43], it is possible that much higher levels of gene flow between the two species occur at other genes. It might, on the other hand, suggest that this behavioral gene, or at least the fragment we analyzed, did not play a role in speciation between L. intermedia and L. whitmani. In fact the same has been suggested for some Drosophila species [44] despite per's role controlling lovesong and mating rhythm differences between D. melanogaster and D. simulans [13–16].
Although the evidence for introgression in the per gene between L. intermedia and L. whitmani is not overwhelming, it does indicate the need to extend this analysis to other loci in the future. We are currently isolating new molecular markers in the two species to carry out a multi-locus approach [39] that might help determining how much variation in gene flow and differentiation there is across the genome of these two very important leishmaniasis vectors.
Methods
Sand fly samples
Sand fly samples used in this work were all the F1 generation from wild collected females from the Brazilian localities of Posse (Petrópolis, Rio de Janeiro State, 22°30'S 43°10'W), Jacarepaguá (Rio de Janeiro, Rio de Janeiro State, 22°55'S 43°21'W), Afonso Claudio (Espírito Santo State, 20°04'S 41°07'W), Corte de Pedra (Presidente Tancredo Neves, Bahia State, 13°27'S 39°25'W) and Ilhéus (Bahia State, 14°50'S 39°06'W). L. intermedia and L. whitmani were identified according to Young and Duncan [1]. The progeny of each wild caught female was raised separately according to Souza et al. [45] and only one F1 male of each female was used for the molecular analysis, which included 68 individuals of L. intermedia (12 from Afonso Claudio, 18 from Posse, 20 from Corte de Pedra and 18 from Jacarepaguá) and 51 individuals of L. whitmani (12 from Afonso Claudio, 17 from Posse, 3 from Corte de Pedra and 19 from Ilhéus). Note that, although the distribution of the two species shows considerable overlap in Eastern Brazil, in many localities only one species is found or is far more abundant than the other. There are also seasonal and microhabitat differences in abundance between them in areas of sympatry [2].
DNA methods
Genomic DNA was prepared according to Jowett [46] with slight modifications and the PCR was carried out for 30 cycles at 95°C for 30 sec, 60°C for 30 sec and 72°C for 30 sec, using Abgene, Amersham Biosciences or Biotools reagents according to manufacturers directions. The per primer sequences are: 5llper2: 5'-AGCATCCTTTTGTAGCAAAC-3' (forward) and 3llper2: 5'-TCAGATGAACTCTTGCTGTC-3' (reverse). These primers amplify a 486 bp fragment of the sand fly per gene homologue that includes part of the PAS/CLD domain, an intron (58 bp) and the beginning of the perSdomain [24]. The amplified fragments were cloned using the pMOSBlue blunt ended cloning kit (Amersham Biosciences) and plasmid DNA preparation was carried out using the "Flexiprep" Kit (Amersham Biosciences). Cloned PCR fragments were sequenced at Fundação Oswaldo Cruz and at University of Leicester using ABI 377 sequencers. With the exception of two L. whitmani individuals from Corte de Pedra (see below), only one sequence of each sand fly (representing one of the two possible alleles) was used in the analysis but an average of three sequences per individual were obtained in order to check possible PCR induced mutations. In addition, PCR fragments were also sequenced directly in some cases for the same reason. In the case of the two L. whitmani mentioned above 6 and 9 clones were sequenced respectively from specimens WCP01 and WCP03 to determine both alleles simply to increase the size of this small sample.
Negative controls were performed for all amplification reactions. In addition, PCR, cloning and sequencing were repeated for two individuals to confirm putative introgressed sequences and to exclude the possibility that they were the result of PCR contamination. Finally, for at least two individuals with putative introgressed sequences, we could define the other allele from additional clones (not included in the analysis), which showed to be typical of the species, indicating no identification problems.
The sequences were submitted to GenBank (accession numbers AY927062 to AY927182).
Sequence analyses
The preliminary sequence editing was carried out using the Wisconsin Package Version 9.1, Genetics Computer Group (GCG), Madison, and ClustalX [47] was used to perform the multiple alignment. Analyses of population polymorphisms and differentiation between populations were carried out using DNAsp4.1 [34] and ProSeq [48] softwares, while Arlequin v. 2.0 [49] was used for an analysis of molecular variance (AMOVA) between populations. The Minimum Evolution phylogenetic tree was constructed using MEGA 3.1 software [50]. The haplotype network was estimated using TCS1.21 [36]. Recombination and linkage disequilibrium analyses were performed using the DNAsp4.1 and SITES program [30]. Linkage disequilibrium simulations were carried out by the WH program [51, 52] and Markov Chain Monte Carlo (MCMC) simulations of the isolation with migration model were performed using the algorithm implemented in the IM program [39].
References
Young DG, Duncan MA: Guide to the identification and geographic distribution of Lutzomyia sand flies in Mexico, the West Indies, Central and South America (Diptera:Psychodidae). Mem Amer Ent Inst. 1994
Souza NA, Andrade-Coelho CA, Vilela ML, Peixoto AA, Rangel EF: Seasonality of Lutzomyia intermedia and Lutzomyia whitmani (Diptera: Psychodidae: Phlebotominae), occurring sympatrically in area of cutaneous leishmaniasis in the State of Rio de Janeiro, Brazil. Mem Inst Oswaldo Cruz. 2002, 97: 759-765.
Ishikawa EA, Ready PD, de Souza AA, Day JC, Rangel EF, Davies CR, Shaw JJ: A mitochondrial DNA phylogeny indicates close relationships between populations of Lutzomyia whitmani (Diptera: Psychodidae, Phlebotominae) from the rain-forest regions of Amazonia and northeast Brazil. Mem Inst Oswaldo Cruz. 1999, 94: 339-345. 10.1590/S0074-02761999000300010.
Marcondes CB, Day JC, Ready PD: Introgression between Lutzomyia intermedia and both Lu. neivai and Lu. whitmani, and their roles as vectors of Leishmania braziliensis. Trans R Soc Trop Med Hyg. 1997, 91: 725-726. 10.1016/S0035-9203(97)90540-X.
Rangel EF, Lainson R, Souza AA, Ready P, Azevedo AC: Variation between geographical populations of Lutzomyia (Nyssomyia) whitmani (Antunes & Coutinho, 1939) sensu lato (Diptera:Psychodidae:Phlebotominae) in Brazil. Mem Inst Oswaldo Cruz. 1996, 91: 43-50.
Ready PD, Day JC, Souza AA, Rangel EF, Davies CR: Mitochondrial DNA characterization of populations of Lutzomyia whitmani (Diptera:Psychodidae) incriminated in the peri-domestic and silvatic transmission of Leishmania species in Brazil. Bull Ent Res. 1997, 87: 187-195.
Testa JM, Montoya-Lerma J, Cadena H, Oviedo M, Ready PD: Molecular identification of vectors of Leishmania in Colombia: mitochondrial introgression in the Lutzomyia townsendi series. Acta Trop. 2002, 84: 205-218. 10.1016/S0001-706X(02)00187-0.
Pesson B, Ready JS, Benabdennbi I, Martin-Sanchez J, Esseghir S, Cadi-Soussi M, Morillas-Marquez F, Ready PD: Sandflies of the Phlebotomus perniciosus complex: mitochondrial introgression and a new sibling species of P. longicuspis in the Moroccan Rif. Med Vet Entomol. 2004, 18: 25-37. 10.1111/j.0269-283x.2004.0471.x.
Ballard JW, Whitlock MC: The incomplete natural history of mitochondria. Mol Ecol. 2004, 13: 729-744. 10.1046/j.1365-294X.2003.02063.x.
Peixoto AA, Gomes CA, de Amoretty PR, Lins RM, Meireles-Filho AC, de Souza NA, Kyriacou CP: New molecular markers for phlebotomine sand flies. Int J Parasitol. 2001, 31: 635-639.
Konopka RJ, Benzer S: Clock mutants of Drosophila melanogaster. Proc Natl Acad Sci USA. 1971, 68: 2112-2116. 10.1073/pnas.68.9.2112.
Wheeler DA, Kyriacou CP, Greenacre ML, Yu Q, Rutila JE, Rosbash M, Hall JC: Molecular transfer of a species-specific behavior from Drosophila simulans to Drosophila melanogaster. Science. 1991, 251: 1082-1085. 10.1126/science.1900131.
Kyriacou CP, Hall JC: The function of courtship song rhythms in Drosophila. Anim Behav. 1982, 30: 794-801. 10.1016/S0003-3472(82)80152-8.
Kyriacou CP, Hall JC: Interspecific genetic control of courtship song production and reception in Drosophila. Science. 1986, 232: 494-497. 10.1126/science.3083506.
Ritchie MG, Halsey EJ, Gleason JM: Drosophila song as a species-specific mating signal and the behavioural importance of Kyriacou & Hall cycles in D. melanogaster song. Anim Behav. 1999, 58: 649-657. 10.1006/anbe.1999.1167.
Sakai T, Ishida N: Circadian rhythms of female mating activity governed by clock genes in Drosophila. Proc Natl Acad Sci USA. 2001, 98: 9221-9225. 10.1073/pnas.151443298.
Miyatake T, Matsumoto A, Matsuyama T, Ueda HR, Toyosato T, Tanimura T: The period gene and allochronic reproductive isolation in Bactrocera cucurbitae. Proc Biol Sci. 2002, 269: 2467-2472. 10.1098/rspb.2002.2152.
Tauber E, Roe H, Costa R, Hennessy JM, Kyriacou CP: Temporal mating isolation driven by a behavioral gene in Drosophila. Curr Biol. 2003, 13: 140-145. 10.1016/S0960-9822(03)00004-6.
Coyne JA: Genetics and speciation. Nature. 1992, 355: 511-515. 10.1038/355511a0.
Peixoto AA: Evolutionary behavioral genetics in Drosophila. Adv Genet. 2002, 47: 117-150.
Regier JC, Fang QQ, Mitter C, Peigler RS, Friedlander TP, Solis MA: Evolution and phylogenetic utility of the period gene in Lepidoptera. Mol Biol Evol. 1998, 15: 1172-1182.
Bauzer LG, Souza NA, Ward RD, Kyriacou CP, Peixoto AA: The period gene and genetic differentiation between three Brazilian populations of Lutzomyia longipalpis. Insect Mol Biol. 2002, 11: 315-323. 10.1046/j.1365-2583.2002.00340.x.
Bauzer LG, Gesto JS, Souza NA, Ward RD, Hamilton JG, Kyriacou CP, Peixoto AA: Molecular divergence in the period gene between two putative sympatric species of the Lutzomyia longipalpis complex. Mol Biol Evol. 2002, 19: 1624-1627.
Mazzoni CJ, Gomes CA, Souza NA, de Queiroz RG, Justiniano SC, Ward RD, Kyriacou CP, Peixoto AA: Molecular evolution of the period gene in sandflies. J Mol Evol. 2002, 55: 553-562. 10.1007/s00239-002-2351-z.
Hall JC: Genetics and molecular biology of rhythms in Drosophila and other insects. Adv Genet. 2003, 48: 1-280.
Souza NA, Andrade-Coelho CA, Peixoto AA, Rangel EF: Nocturnal activity rhythms of Lutzomyia intermedia and Lutzomyia whitmani (Diptera: Psychodidae) in a transmission area of American cutaneous leishmaniasis in Rio de Janeiro State, Brazil. J Med Entomol. 2005, 42: 986-992.
Souza NA, Vigoder FM, Araki AS, Ward RD, Kyriacou CP, Peixoto AA: Analysis of the copulatory courtship songs of Lutzomyia longipalpis in six populations from Brazil. J Med Entomol. 2004, 41: 906-913.
Tajima F: Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989, 123: 585-595.
Fu YX, Li WH: Statistical tests of neutrality of mutations. Genetics. 1993, 133: 693-709.
Hey J, Wakeley J: A coalescent estimator of the population recombination rate. Genetics. 1997, 145: 833-846.
Swofford DL: PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. 2001, Sinauer Associates, Sunderland, Massachusetts
Schierup MH, Hein J: Consequences of recombination on traditional phylogenetic analysis. Genetics. 2000, 156: 879-891.
Hudson RR, Kaplan NL: Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics. 1985, 111: 147-164.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R: DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003, 19: 2496-2497. 10.1093/bioinformatics/btg359.
Templeton AR, Crandall KA, Sing CF: A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA-sequence data III Cladogram estimation. Genetics. 1992, 132: 619-633.
Clement M, Posada D, Crandall KA: TCS: a computer program to estimate gene genealogies. Molecular Ecology. 2000, 9: 1657-1660. 10.1046/j.1365-294x.2000.01020.x.
Crandall KA, Templeton AR: Empirical tests of some predictions from coalescent theory with applications to intraspecific phylogeny reconstruction. Genetics. 1993, 134: 959-969.
Machado CA, Kliman RM, Markert JA, Hey J: Inferring the history of speciation from multilocus DNA sequence data: the case of Drosophila pseudoobscura and close relatives. Mol Biol Evol. 2002, 19: 472-488.
Hey J, Nielsen R: Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004, 167: 747-760. 10.1534/genetics.103.024182.
Marcondes CB, Lozovei AL, Vilela JH: [Geographic distribution of phlebotomine sandflies of the Lutzomyia intermedia (Lutz & Leiva, 1912) complex (Diptera, Psychodidae)]. Rev Soc Bras Med Trop. 1998, 31: 51-58.
Besansky NJ, Krzywinski J, Lehmann T, Simard F, Kern M, Mukabayire O, Fontenille D, Toure Y, Sagnon N: Semipermeable species boundaries between Anopheles gambiae and Anopheles arabiensis: evidence from multilocus DNA sequence variation. Proc Natl Acad Sci USA. 2003, 100: 10818-10823. 10.1073/pnas.1434337100.
Weill M, Chandre F, Brengues C, Manguin S, Akogbeto M, Pasteur N, Guillet P, Raymond M: The kdr mutation occurs in the Mopti form of Anopheles gambiae s.s. through introgression. Insect Mol Biol. 2000, 9: 451-455. 10.1046/j.1365-2583.2000.00206.x.
Ting CT, Tsaur SC, Wu CI: The phylogeny of closely related species as revealed by the genealogy of a speciation gene, Odysseus. Proc Natl Acad Sci USA. 2000, 97: 5313-5316. 10.1073/pnas.090541597.
Ford MJ, Yoon CK, Aquadro CF: Molecular evolution of the period gene in Drosophila athabasca. Mol Biol Evol. 1994, 11: 169-182.
Souza NA, Andrade-Coelho CA, Barbosa AF, Vilela ML, Rangel EF, Deane MP: The influence of sugars and amino acids on the blood-feeding behaviour, oviposition and longevity of laboratory colony of Lutzomyia longipalpis (Lutz & Neiva, 1912) (Diptera: Psychodidae, Phlebotominae). Mem Inst Oswaldo Cruz. 1995, 90: 751-757.
Jowett T: Preparation of nucleic acids. Drosophila: A practical approach. Edited by: Roberts D. 1998, Oxford: IRL press, 347-371.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25: 4876-4882. 10.1093/nar/25.24.4876.
Filatov DA, Charlesworth D: DNA polymorphism, haplotype structure and balancing selection in the Leavenworthia PgiC locus. Genetics. 1999, 153: 1423-1434.
Schneider S, Roessli D, Excoffier L: Arlequin ver. 2000: A software for population genetic data analysis. 2000, Genetics and Biometry Laboratory, University of Geneva, Switzerland
Kumar S, Tamura K, Nei M: MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Briefings in Bioinformatics. 2004, 5: 150-163. 10.1093/bib/5.2.150.
Wakeley J, Hey J: Estimating ancestral population parameters. Genetics. 1997, 145: 847-855.
Wang RL, Wakeley J, Hey J: Gene flow and natural selection in the origin of Drosophila pseudoobscura and close relatives. Genetics. 1997, 147: 1091-1106.
Hudson RR, Slatkin M, Maddison WP: Estimation of levels of gene flow from DNA sequence data. Genetics. 1992, 132: 583-589.
Nei M: Molecular Evolutionary Genetics. New York. 1987
Jukes TH, Cantor CR: Evolution of protein molecules. Mammalian Protein Metabolism. Edited by: Munro HN. 1969, New York: Academic Press, 21-132.
Acknowledgements
We would like to thank Paulo de Amoretty and Caroline Assucena for their technical assistance, Robson Costa da Silva and Karen Garner for their help with the DNA sequencing. The sand flies used in this work were obtained thanks to the help of Aloisio Falqueto (Universidade Federal do Espirito Santo), Paulo Raimundo Barbosa (Secretária de Saúde do Estado da Bahia, Ilhéus) and Luís Henrique Costa (Fiocruz). This work was funded by the Howard Hughes Medical Institute and UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases (TDR) with additional support from Faperj, Fiocruz and CNPq. CPK acknowledges a Royal Society Wolfson Research Merit Award.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
CJM generate and analyzed all the data and drafted the manuscript. NAS and CAC collected and maintained sand fly samples. CPK helped to write the manuscript and supervised CJM during her stay in Leicester. AAP is the principal investigator, participated in its design and coordination, and helped to write the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Mazzoni, C.J., Souza, N.A., Andrade-Coelho, C. et al. Molecular polymorphism, differentiation and introgression in the period gene between Lutzomyia intermedia and Lutzomyia whitmani. BMC Evol Biol 6, 85 (2006). https://doi.org/10.1186/1471-2148-6-85
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-6-85